Effect of Asp-97->Glu Substitution on the pH Dependence of Catalysis by Inorganic Pyrophosphatase of Escherichia coli
I. P. Fabrichniy,1 R. Lahti,2 and A. A. Baykov1,3
1Belozersky Institute of Physico-Chemical Biology and School of Chemistry, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: abaykov@protchem.genebee.msu.su2Department of Biochemistry and Food Chemistry, University of Turku, Turku, FIN-20500, Finland; fax: (358-2) 633-6860; E-mail: reila@utu.fi
3To whom correspondence should be addressed.
Submitted April 10, 1997; revision submitted May 26, 1997.
Substitution of Glu for the evolutionarily conserved Asp-97 in the active site of Escherichia coli inorganic pyrophosphatase increases the apparent pKa of the essential acidic group controlling the catalytic constant for pyrophosphate hydrolysis. In combination with previously reported data, this fact suggests that activity decreases in alkaline medium because of decreased rate of the release of the first molecule of the product phosphate from the active site.
KEY WORDS: pyrophosphatase, site-directed mutagenesis, pH dependence, kinetics.
Inorganic pyrophosphatases are essential for metabolism and catalyze hydrolysis and synthesis of the phosphoanhydride bond. The main substrate of this enzyme, pyrophosphate, is a by-product of many ATP-dependent reactions, such as protein, nucleic acid, and lipid syntheses; its hydrolysis pulls the equilibria of these reactions in the direction of synthesis [1].
Pyrophosphatases are metal-activated enzymes: they absolutely require divalent metal ions for catalysis, with Mg2+ conferring the highest activity [2]. Being the simplest phosphoryl-transfer enzymes, pyrophosphatases are a convenient model for the studies of the whole class of such enzymes [3].
Escherichia coli pyrophosphatase (E-PPase), a typical prokaryotic pyrophosphatase, is a hexamer of identical subunits, ~20 kD each [4]. E-PPase is quite stable over a wide pH range. PPi hydrolysis proceeds in four steps: substrate (Mg2PPi complex) binding, its hydrolysis and successive release of two resulting phosphate molecules (Scheme 1).
The value of n (the number of metal ions on the enzyme) varies from 1 to 3 depending on medium Mg2+ concentration [5]. The mechanism of the enzyme has been under study in several laboratories [3, 6-8]. Based on the three-dimensional structure of PPase, determined to 2.2 . [6, 8], conservative amino acid residues which form the active site have been identified. Mutant forms of E-PPase wherein all these residues have been substituted have been prepared [9, 10], and some of them have been characterized in detail [11-13].Scheme 1
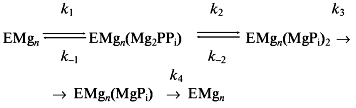
Detailed kinetic analysis of E-PPase has revealed two basic and one acidic groups which determine the bell-shaped dependence of the catalytic constant on pH. One of the basic groups controls bound PPi hydrolysis. Its pKa is < 6.5 in the wild-type enzyme but is increased by several units on active site residue substitutions [9, 11, 13]. There are strong arguments that this group is a water molecule (hydroxide ion) which is bound simultaneously to two Mg2+ ions on the enzyme [6]. The second basic group of unknown nature controls the release of the second phosphate molecule (k4) [5].
The catalytic step requiring the acidic group, whose protonation in the alkaline medium is responsible for activity decrease, as well as its nature still remain to be elucidated. As a first approach to this problem, we determined the effect of the D97E substitution in the active site of E-PPase on the pH dependence of catalysis in the alkaline pH range.
MATERIALS AND METHODS
The mutant form of E. coli PPase carrying the D97E substitution was obtained as previously described [14]. The enzyme preparation was nearly homogeneous according to electrophoresis in the presence of sodium dodecyl sulfate. The concentrations of E-PPase solutions were calculated in terms of monomer concentrations using the molecular weight of 20 kD and A1%1cm,280 = 11.8 [14]. Stock enzyme solution (0.03-1 mg/ml) was made in 0.15 M Tris-HCl buffer (pH 7.2) containing 2 mM MgCl2, bovine serum albumin (0.8 mg/ml), and 40 µM EGTA and kept at room temperature. Components of the buffer systems were obtained from Sigma (USA).
Enzyme activity in hydrolysis was measured in a continuous way with an automatic phosphate analyzer [15]. The reaction mixture of 25 ml total volume contained buffer, MgCl2, and Na4P2O7. The concentrations of the two last components needed to maintain desired concentrations of free Mg2+ and Mg2PPi complex were calculated as previously described [16]. The reaction was started with 4-20 µl enzyme and carried out at 25°C. The following pH buffers were used (0.15-0.20 M ionic strength): imidazole-HCl (pH 6.5-6.8); Tris-HCl (pH 7.2-8.0); 2-amino-2-methyl-1,3-propanediol-HCl (pH 8.5-9.0); and 2-amino-2-methyl-1-propanol-HCl (pH 9.3-10.5). All solutions contained EGTA: 50 µM (pH 6.5-7.2); 10 µM (pH 8.5); or 1 µM (pH 9.3-10.5).
The catalytic constant (kh) and the Michaelis constant (Km) were estimated from dependences of the initial rate of hydrolysis on substrate (Mg2PPi) concentration, which was varied in the range of 0.2Km to 50Km at each set of pH and [Mg2+] values. Values of parameters in the equations were obtained with a program for nonlinear regression analysis, DNRP53 [17].
RESULTS
Values of kh were measured as a function of pH in the range 6.5-10.5 at two fixed free Mg2+ concentrations (0.2 and 20 mM), as well as a function of Mg2+ in the range of 0.1-50 mM at pH 7.2 and 10.0 (Fig. 1). This data set sufficed for determination of the pKa values for the basic and acidic groups controlling the catalytic step in two enzyme--substrate complexes--with 4 and 5 metal ions (Scheme 2).
An analogous approach was earlier used for mutant E-PPases carrying E20D [12], Y55F, and K104R [13] substitutions. According to this scheme, complexes HEMg2(Mg2PPi) and HEMg3(Mg2PPi), in which the basic group is deprotonated and the acidic group is protonated, are catalytically competent; the respective catalytic constants are kh(1) and kh(2). The other parameters refer to the dissociation constants for respective equilibria.Scheme 2
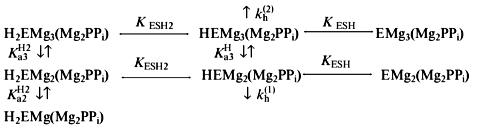
Scheme 2 is similar to the scheme previously derived for wild-type E-PPase [5] and is minimal in the sense that addition of other enzyme--substrate complex species does not make the fit to the experimental data better, whereas omission of any species makes it significantly worse. Values of parameters in Scheme 2, as determined by nonlinear regression with Eq. (1), are listed in Table 1. When comparing with wild-type E-PPase, one should keep in mind that only some of these parameters have been determined for it [5]. One can see that the D97E substitution markedly increases pKESH2 (by 2.1), pKESH and pK´ESH (by 1.0), as well as decreases kh(1) (by a factor of 5.3). The exact value of kh(2) for wild-type E-PPase is unknown but it can hardly exceed kh(1) [5]. Thus, the substitution affects catalysis with four metal ions more than that with five metal ions involved, which agrees with earlier findings [11].Fig. 1. Dependence of kh for D97E-PPase on pH and [Mg2+]. The curves were obtained with Eq. (1) using parameter values shown in Table 1.
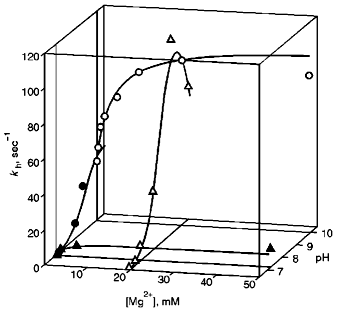
TABLE 1. Parameters of Scheme 2
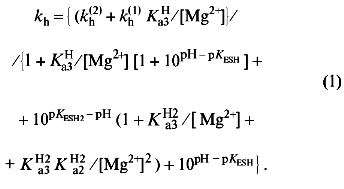

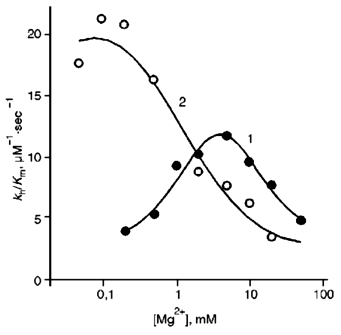
The dependences of the ratio kh/Km on Mg2+ concentration at pH 10.0 for both PPases are shown in Fig. 2. For E-PPase, the value of kh/Km is quite close to k1 over a wide range of pH because k-1 << kh [5, 11]. Since the D97E substitution does not affect Mg2+ binding at two high-affinity sites on the enzyme [11], the shift in the maximum on the curve for D97E-PPase to higher Mg2+ concentrations indicates that the mutant enzyme requires more metal ions for fast substrate binding.
Fig. 2. Dependence of kh/Km on [Mg2+] at pH 10: 1) D97E-PPase; 2) wild-type E-PPase.
DISCUSSION
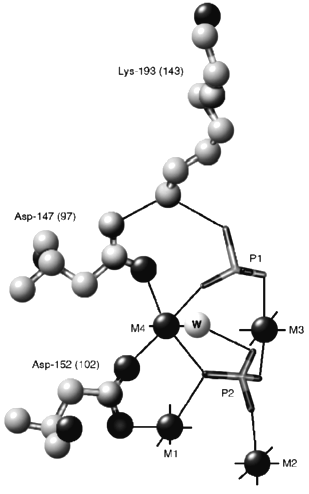
Asp-97 occupies one of the key positions in the complex system of hydrogen and ionic bonds connecting protein groups to activating metal ions and the substrate molecule in the PPase active site. The structure of an EMn4(Pi)2 complex of yeast PPase around Asp-147, which corresponds to Asp-97 of E-PPase [6], is shown in Fig. 3. The structure of the analogous complex of E-PPase still remains to be determined, but, based on the similarity of the active site structures in apo- [18, 19] and Mn-forms [6, 8] of the two enzymes, it cannot be much different. Asp-147 fixes in space positions of Lys-193 and the metal ion M4 which directly interact with two phosphate molecules and, supposedly, with the pyrophosphate molecule. Asn substitution for Asp-97 in E-PPase decreases kh 200-fold and the ratio kh/Km, which characterizes the rate of substrate addition to the enzyme [5], 2000-fold [7]. A milder Asp-97->Glu substitution used in this work exerts a markedly smaller effect [11] (Table 1), which facilitates functional characterization of the mutant enzyme. It has been previously shown that this substitution increases the pKa of the essential base by more than unity and increases the number of Mg2+ ions required for maximal activity from four to five per substrate molecule [11]. These conclusions have been fully confirmed in the present work.
The major new finding is that the substitution also greatly increases the pKa of the acidic group (pKESH, pK´ESH) governing kh in alkaline medium. As a consequence, the activity of D97E-PPase is decreased less with increasing pH compared with the wild-type E-PPase. In the presence of 20 mM Mg2+, kh values at pH 10.0 are, respectively, 81% (Fig. 1) and 5% [5] of maximal. The effect of D97E substitution on pKESH has not been detected previously because measurements were done in a narrower pH range and at a fixed Mg2+ concentration [7, 11]; this complicated the separation of the reaction routes involving four and five metal ions (Scheme 2).Fig. 3. Structure of the fragment of yeast PPase complexed with four Mn2+ ions and two Pi molecules around Asp-147, corresponding to Asp-97 of E. coli PPase [6]. M1-M4 are Mn2+ ions; P1 and P2 are phosphate molecules. Residue numbering corresponds to yeast PPase (without parentheses) and E. coli PPase (in parentheses).
Based on this observation, it is unlikely that the acidic group participates at step k2 (Scheme 1), for instance to protonate the leaving phosphate group [6]. The logic of our speculation is as follows. If the group under consideration is involved in one (or even two) of the three catalytic steps (k2, k3, and k4), its true pKa is smaller than estimated from the pH dependence of kh (pKESH, pK´ESH), the difference being greater the greater is the corresponding rate constant in comparison with the other two. But if the step at which the acidic group participates is the only rate-limiting one, the two pKa values would coincide. For wild-type E-PPase, values of k2 and k3 are similar over the pH range of 6.5-9.3 [5], whereas k4 is markedly greater. Therefore, the true pKa value for the acidic group in it is less than 10.04 and 9.5 in respective enzyme--substrate complexes (Table 1). For D97E-PPase, the k2 step is the only rate-limiting step, at least at pH 7.2-8.0 [11], which would result in decreased pKESH and pK´ESH in comparison with wild-type E-PPase, i.e., to an opposite effect, were the acidic group involved in the k2 step. Even if k2 for D97E-PPase increases in alkaline medium to the level corresponding to wild-type E-PPase, this cannot explain the observed increase in pKESH and pK´ESH. The acidic group most likely participates in removal of the first phosphate molecule (k3), as the rate of the subsequent step (k4) is increased rather than decreased with increasing pH in the range of 6.5-9.3 [5].
Thus, the substitution of Glu for the conserved Asp-97 residue in the active site of E-PPase increases the pKa of the essential acidic group in the enzyme, which makes unlikely participation of this group in hydrolysis of enzyme-bound PPi.
This work was supported by the Russian Foundation for Basic Research (grant 97-04-48487) and the program "Enzyme Engineering" (grant 1-42).
LITERATURE CITED
1.Kornberg, A. (1982) in Horizons in Biochemistry
(Kasha, H., and Pullman, P., eds.) Academic Press, New York, pp.
251-264.
2.Josse, J. (1966) J. Biol. Chem.,
241, 1938-1947.
3.Cooperman, B. S., Baykov, A. A., and Lahti, R.
(1992) Trends Biochem. Sci., 17, 262-266.
4.Wong, S. C. K., Hall, D. C., and Josse, J. (1970)
J. Biol. Chem., 245, 4335-4345.
5.Baykov, A. A., Hyytiä, T., Volk, S. E., Kasho,
V. N., Vener, A. V., Goldman, A., Lahti, R., and Cooperman, B. S.
(1996) Biochemistry, 35, 4655-4661.
6.Heikinheimo, P., Lehtonen, J., Baykov, A., Lahti,
R., Cooperman, B. S., and Goldman, A. (1996) Structure,
4, 1491-1508.
7.Avaeva, S., Ignatov, P., Kurilova, S., Nazarova,
T., Rodina, E., Vorobyeva, N., Oganessyan, V., and Haratyunyan, E.
(1996) FEBS Lett., 399, 99-102.
8.Harutyunyan, E. H., Oganessyan, V. Yu., Oganessyan,
N. N., Terzyan, S. S., Popov, A. N., Rubiskiy, S. B., Veinshtein, B.
K., Nazarova, T. I., Kurilova, S. A., Vorobyeva, N. N., and Avaeva, S.
M. (1996) Kristallografiya, 41, 84-96.
9.Salminen, T., Käpylä, J., Heikinheimo,
P., Kankare, J., Goldman, A., Heinonen, J., Baykov, A. A., Cooperman,
B. S., and Lahti, R. (1995) Biochemistry, 34,
782-791.
10.Avaeva, S. M., Rodina, E. V., Kurilova, S. A.,
Nazarova, T. I., and Vorobyeva, N. N. (1996) FEBS Lett.,
392, 91-94.
11.Käpylä, J., Hyytiä, T., Lahti, R.,
Goldman, A., Baykov, A. A., and Cooperman, B. S. (1995)
Biochemistry, 34, 792-800.
12.Volk, S. E., Dudarenkov, V. Yu.,
Käpylä, J., Kasho, V. N., Voloshina, O. A., Salminen, T.,
Goldman, A., Lahti, R., Baykov, A. A., and Cooperman, B. S. (1996)
Biochemistry, 35, 4662-4669.
13.Fabrichniy, I. P., Kasho V. N., Hyytiä, T.,
Salminen, T., Halonen, P., Dudarenkov, V. Yu., Heikinheimo, P.,
Chernyak, V. Ya., Goldman, A., Lahti, R., Cooperman, B. S., and Baykov,
A. A. (1997) Biochemistry, in press.
14.Lahti, R., Pohjanoksa, K., Pitkaranta, T.,
Heikinheimo, P., Salminen, T., Meyer, P., and Heinonen, J. (1990)
Biochemistry, 29, 5761-5766.
15.Baykov, A. A., and Avaeva, S. M. (1981) Anal.
Biochem., 116, 1-4.
16.Volk, S. E., Baykov, A. A., Duzhenko, V. S., and
Avaeva, S. M. (1982) Eur. J. Biochem., 125, 215-220.
17.Duggleby, R. (1984) Comput. Biol. Med.,
14, 447-455.
18.Kankare, J., Salminen, T., Lahti, R., Cooperman,
B. S., Baykov, A. A., and Goldman, A. (1996) Acta Crystallogr.,
D52, 551-563.
19.Terzyan, S. S., Voronova, A. A., Smirnova, E. A.,
Kuranova, I. P., Nekrasov, Yu. V., Harutyunyan, E. H., Veinshtein, B.
K., Hohne, W., and Hansen, G. (1984) Bioorg. Khim., 10,
1469-1482.