Peculiarities of Gene Expression of the EcoRII Modification-Restriction System
N. N. Matvienko,1 L. A. Zheleznaya,2 E. E. Chernyshova,3 Ya. I. Buryanov,1 and N. I. Matvienko3,4
1Branch of Shemyakin--Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia.2Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia.
3Institute of Protein Research, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 924-0493; E-mail: nikmatv@sun.ipr.serpukhov.su
4To whom correspondence should be addressed.
Submitted June 3, 1997.
The restriction-modification genes of the EcoRII system have been cloned into plasmids under control of phage-specific promoters T7 and SP6. The transcription was induced by cell infection with the recombinant M13 phages with the corresponding genes of phage RNA-polymerases under control of the plac-promoter in the presence of IPTG. The induction yields significant amounts of EcoRII DNA-methylase for both phage-specific promoters. In both cases no increase in EcoRII endonuclease expression could be achieved. We hypothesize that the expression of the endonuclease gene is regulated on the translational level.
KEY WORDS: restriction-modification, EcoRII.
To perform modification-restriction functions the corresponding systems have to be strictly regulated because the appearance of DNA sites without protection by methylation could lead to cell suicide. The strict regulation of modification-restriction systems is especially important when genetic elements are introduced de novo. In this case all chromosomic DNA sites of a new host have to be methylated before the endonuclease activity appears in the cell (see for review [1]).
The EcoRII system is encoded by the endonuclease and DNA-methylase genes which are oriented in opposite direction and whose transcription is initiated from their own promoters [2-6]. It has been recently shown that the DNA-methylase gene is under autogenic control. At the defined enzyme concentration DNA-methylase binds on its own promoter preventing the methylase gene transcription [7-9]. The mechanism of regulation of the endonuclease gene is unknown; however, it is obvious that endonuclease activity should appear later than DNA-methylase in the course of cell transformation with the plasmid encoding the EcoRII system.
In this work we present data supporting the proposal of a translational control of the endonuclease gene expression.
MATERIALS AND METHODS
We used the recombinant plasmid pUC128 constructed by V. Kosykh and Ya. Buryanov [10] with the genes of the modification-restriction system of the native plasmid N3 cloned in XbaI and PstI restriction sites. Plasmids pGEM1, pGEM2, and pGEM9Zf(-) were from Promega (USA). M13 phage with the T7 polymerase gene was constructed by Studier and co-authors [11] and with SP6 RNA-polymerase was constructed by Zheleznaya and co-authors [12]. Strains E. coli XL1 Blue (recA1, endA1, gyrA96, thi, hsdR17 (r-km-k), supE44, relA1, lac/F´::Tn10, proA+B+, lacIqdelta(lacZ)M15) were used for transformation [13]. To cleave and methylate in vitro we usedphage lambdaCI857S7 isolated after induction of lysogenic E. coli dam-dcm- strain constructed earlier [14].
Induction of the Cloned Genes of the EcoRII System. Overnight cultures of E. coli XL1 (pGEM1-RII) or E. coli XL1 (pGEM2-RII) grown on 2YT medium (bactotrypton Difco, yeast extract Difco, and NaCl at concentrations 16, 10, and 5 g/liter, respectively) in the presence of ampicillin (100 µg/ml) and tetracycline (10 µg/ml) were 100-fold diluted with the same medium containing the antibiotics and grown at 37°C with intense aeration until A590 = 0.6 corresponding to 2·108 cells per ml. The cultures were divided into 5 tubes of 10 ml each. The first tube was used as a control with no addition; 140 µl of M13T7pol phage was added to the second tube; 140 µl of the phage and 20 µl of IPTG stock solution (40 µg/ml DMSO) were added to the third tube; 140 µl of M13SP6pol phage was added to the forth tube; 140 µl of the phage and 20 µl of IPTG stock solution (40 µg/ml DMSO) were added to the fifth tube. Both phages were diluted to titer 7·1011 per ml; thus, the multiplicity of infection was about 5 for each phage. After 3 h aeration at 37°C, the absorbance at 590 nm has been measured, and the cells were precipitated by centrifugation at 6000g for 10 min. The pellet was homogenized in 2 ml of a lysis buffer (200 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 7 mM beta-mercaptoethanol, 0.1 mg/ml lysozyme), and the cells were disrupted by sonication for 3 min (3 × 30 sec with 30 sec breaks) using an UZDN-A ultrasonic disintegrator (Russia). The mixture was centrifuged at 20,000g for 20 min, and supernatant aliquots were used in SDS-PAGE and for endonuclease and DNA-methylase activity measurements.
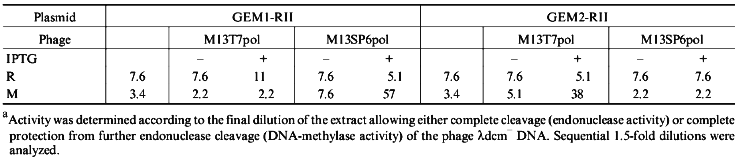
Determination of Endonuclease and DNA-Methylase Activities. One unit of EcoRII endonuclease activity is defined as the amount of enzyme necessary for the complete cleavage of 1 µg of phage lambdadcm- DNA at 37°C for 1 h in 50 µl of the reaction buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl2). One unit of EcoRII DNA-methylase has to protect completely 1 µg of phage lambdadcm- DNA from endonuclease cleavage. Methylation was performed at 37°C in 30 µl of reaction mixture containing 1 µg of phage lambdadcm- DNA, the methylating buffer (30 mM Tris-HCl, pH 7.5, 8 mM EDTA, 5 mM beta-mercaptoethanol, 80 µM S-adenosylmethionine, SAM) and aliquots of the extract in sequential dilutions. A 1.5-fold dilution was performed with the methylating buffer without SAM. The reaction mixture was incubated for 1 h, then 25 mM MgCl2 and 5 units of EcoRII endonuclease were added. The incubation was continued for 1 h more, and then the products were analyzed by electrophoresis in 1% agarose gel. The final dilution of the extract allowing the complete protection from EcoRII cleavage was determined. Since the dilutions were 1.5-fold the accuracy of the activity determination for both DNA-methylase and endonuclease was ±50%. The experiments were repeated in triplicate and gave qualitatively similar results for the activity measurements in the extracts of induced and non-induced cells.
SDS-Polyacrylamide Gel Electrophoresis of the Proteins. In addition to the activity measurements, endonuclease and methylase proteins were tested by SDS-PAGE of the extract samples [15]. A 5% concentrating gel and a 12% separating gel were used. Molecular weight markers were from a preparation of muscle proteins produced by Dombi Company (Russia).
RESULTS AND DISCUSSION
To obtain a superproducer of the enzymes of the EcoRII modification-restriction system, we decided to use vectors with phage-specific promoters [11, 12]. In the case of these vectors the cloned genes can be induced by two methods. In the first the cloned gene is expressed due to induction of the endogenous phage-specific RNA-polymerase encoded either by a chromosome of the defective prophage or by a compatible plasmid. In the second method the phage RNA-polymerase gene in introduced into the cell by infecting with the recombinant lambda or M13 phages encoding the gene. The second method is preferred for clones in which the proteins are toxic for E. coli.
We chose pGEM9Zf(-) vector from Promega, because it allows both visual identification of recombinant clones and transcription of the integrated fragment in opposite directions depending on the type of phage RNA-polymerase, i.e., T7 or SPR. Phage RNA-polymerases were introduced using M13T7pol phage constructed by Studier et al. [11] or M13SP6pol phage constructed in this laboratory earlier [12]. Polymerase genes were under control of plac promoters in both phages and thus were induced by IPTG. Since the EcoRII system is characterized by convergent transcription of endonuclease and methylase genes, we planned to use the same recombinant strain to produce either methylase or endonuclease depending on the infecting phage selected, M13T7pol or M13SP6pol (Fig. 1).
The level of DNA-methylase activity in E. coli XL1 Blue/pGEM9Zf(-)-RII cells induced by infection with M13T7pol phage in the presence of IPTG is increased significantly. This produces about 60 mg of homogeneous EcoRII methylase after three purification steps from 0.5 liter of E. coli culture (data not shown); this is close to the amount obtained in [16] using T7 promoter. However, there is nearly no increase in the EcoRII endonuclease activity upon cell transfection with M13SP6pol phage in the presence of IPTG. We suggest that IPTG addition opening the plac promoter which controls the opposite direction of transcription by bacterial RNA-polymerase could be the reason for inefficient expression of the endonuclease gene. The presence of antisense RNA of the EcoRII endonuclease gene can inhibit the enzyme synthesis.Fig. 1. Construction scheme of plasmids pGEM9Zf(-)-RII, pGEM1-RII, pGEM2-RII. R, the endonuclease gene; M, the methylase gene; plac, the lactose promoter; pT7 and pSP6, promoters of phage T7 and SP6, respectively.
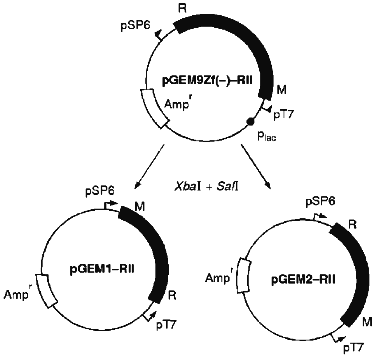
To answer this question, we cloned the genes of the EcoRII system from PGEM9Zf(-)-RII plasmid to pGEM1 and pGEM2 plasmids with the lack of plac promoter. Using both vectors we created a reciprocal situation when the methylase gene is under control of the SP6 promoter and the endonuclease gene is under control of T7 promoter in PGEM1 plasmid and vice versa in pGEM2 plasmid (Fig. 1). The experiments demonstrated that the synthesis of methylase was enhanced while the synthesis of endonuclease was not (Fig. 2, Table 1) independent of the plasmid used. Thus, the presence of plac promoter was not the reason for the absence of endonuclease induction. It is likely that the situation reflects the regulation peculiarities of the operon itself. It is difficult to imagine that the proteins of the EcoRII system, i.e., methylase, endonuclease, etc., could regulate the transcription from both phage-specific promoters. Thus, we suggest that the endonuclease expression could be regulated on the posttranscriptional level.
TABLE 1. Activitya (U/µl) of EcoRII Endonuclease (R) and Methylase (M) of the Cell Extracts of E. coli XL1 Blue/pGEM1-RII and pGEM2-RII Transformants without or with Phage M13T7pol or M13SP6pol Infection in the Absence (-) or Presence (+) of IPTG-Induced Phage RNA-PolymeraseFig. 2. SDS-PAGE analysis of expression levels of endonuclease and methylase: a) recombinant E. coli XL1 Blue (pGEM1-RII); b) recombinant E. coli XL1 Blue (pGEM2-RII). Molecular weight markers, lanes a8 and b1 (molecular weights in kD are shown on the left and on the right from the lanes). M, purified methylase (lanes a1, a9); R, purified endonuclease (lanes a2, a10, b7). Lanes: a3, b2) non-infected cultures; a4, a5) cultures with plasmid pGEM1-RII infected with M13T7pol phage in the absence and presence of IPTG, respectively; a6, a7) the same cultures infected with M13SP6pol phage in the absence and presence of IPTG, respectively; b3, b4) cultures with plasmid pGEM2-RII infected with M13T7pol phage in the absence and presence of IPTG, respectively; b5, b6) the same cultures infected with M13SP6pol phage in the absence and presence of IPTG, respectively.
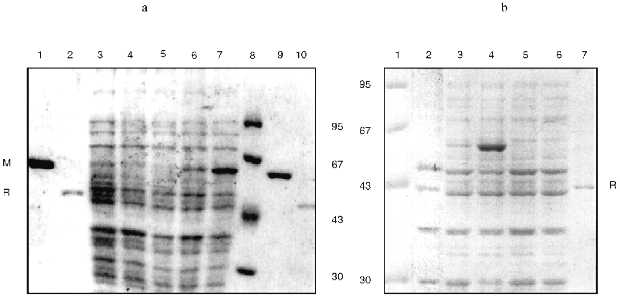
A number of possible regulatory mechanisms may occur.
1. EcoRII methylase, besides exhibiting DNA-methylase and DNA-binding activities, is able to bind specifically on the transcript of the endonuclease gene activating its translation.
2. EcoRII endonuclease binds specifically on the transcript of its own gene and inhibits the translation. Such a mechanism was postulated for TaqI endonuclease [17]. TaqI endonuclease expression is stimulated by deletion of the RNA loop with the stem sequence corresponding to the DNA recognition site from the endonuclease gene transcript. This observation suggested that TaqI endonuclease binds on this loop and stem and inhibits the translation of its own mRNA.
3. It is possible that EcoRII methylase is necessary for the correct folding of the endonuclease, protecting the enzyme from rapid proteolysis. A similar mechanism has been recently proposed to explain the peculiarities of EcoP15 system expression [18].
4. We cannot exclude the existence of unidentified regulatory elements of EcoRII operon analogous to c-peptide in PvuII, SmaI, EcoRV [19], and BamHI [20] operons or to w-peptide encoded by the inner sequence of the methylase gene of the PvuII system [21]. If this is the case for the observed phenomenon, we have to suggest that this peptide binding not only inhibits the endonuclease activity, but makes the endonuclease highly sensitive to proteolysis.
We plan to probe the mechanism in the coupled transcription-translation system in vitro.
This work was supported by grant No. 96-04-49050 from the Russian Foundation for Basic Research.
LITERATURE CITED
1.Bickle, T. A., and Kruger, D. H. (1993)
Microbiol. Rev., 50, 434-350.
2.Kosykh, V. G., Buryanov, Yu. I., and Baev, A. A.
(1980) Mol. Gen. Genet., 178, 717-718.
3.Kosykh, V. G., Solonin, A. S., Buryanov, Yu. I.,
and Baev, A. A. (1981) Biochim. Biophys. Acta, 655,
102-106.
4.Kosykh, V. G., Repik, A. V., Kaliman, A. V.,
Buryanov, Ya. I., and Baev, A. A. (1989) Dokl. Akad. Nauk SSSR,
308, 1497-1499.
5.Som, S., Bhagwat, A. S., and Friedman, S. (1987)
Nucleic Acids Res., 15, 313-331.
6.Bhagwat, A. S., Johnson, B., Weule, K., and
Roberts, R. R. (1990) J. Biol. Chem., 265, 767-773.
7.Friedman, S., and Som, S. (1993) J.
Bacteriol., 175, 6293-6298.
8.Som, S., and Friedman, S. (1993) EMBO J.,
12, 4297-4303.
9.Som, S., and Friedma, S. (1994) Nucleic Acids
Res., 22, 5347-5353.
10.Keen, N. T., Tamaki, S., Kobayshi, D., and
Trollinger, D. (1988) Gene, 70, 191-197.
11.Studier, F. W., Rosenberg, A. H., Dunn, J. J.,
and Dubendorff, J. W. (1990) Meth. Enzymol., 158,
60-89.
12.Zheleznaya, L. A., Savchenko, R. S., and
Matvienko, N. I. (1990) Nucleic Acids Res., 18,
4295.
13.Bullok, W. O., Fernandez, J. M., and Short, J. M.
(1987) BioTecniques, 5, 376-378.
14.Shapovalova, N. I., Zheleznaya, L. A., and
Matvienko, N. I. (1994) Biochemistry (Moscow), 59,
1730-1738 (Russ.).
15.Laemmli, U. K. (1970) Nature, 227,
680-685.
16.Som, S., and Friedman, S. (1990) J. Biol.
Chem., 265, 4278-4283.
17.Barany, F. (1988) Gene, 65,
167-177.
18.Redaschi, N., and Bickle, T. A. (1996) J. Mol.
Biol., 257, 790-803.
19.Tao, T., and Blumental, R. M. (1992) J.
Bacteriol., 174, 2295-2298.
20.Sohail, A., Ives, C. L., and Brooks, J. E. (1995)
Gene, 157, 227-228.
21.Adams, G. M., and Blumental, R. M. (1995)
Gene, 157, 193-199.