Telomere Functions. A Review
E. V. Kurenova1 and J. M. Mason1,2
1Laboratory of Molecular Genetics, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, 27709-2233 USA; fax: (919) 541-7593; E-mail: masonj@niehs.nih.gov, kurenova@niehs.nih.gov2To whom correspondence should be addressed.
Submitted August 3, 1997.
Telomeres are structurally and functionally complex. They consist of an array of simple DNA repeats at the extreme end of the chromosome, with a more complex array of repeats adjacent to it. A large number of proteins have been identified that bind to the telomeric DNA repeats or to the protein complexes that are built at the chromosome end. Telomeres tend to form associations with each other. These associations have been implicated in the formation of nuclear domains that may be important for transcriptional regulation, for sister chromatid pairing at mitosis, and for homologous meiotic synapsis. Telomeric chromosome ends do not cause delays in cell cycle progression, nor are they subject to DNA repair as are broken chromosome ends. Telomeres also provide a separate mechanism for adding additional copies of the telomeric DNA to chromosome ends. This is needed to counterbalance the loss of DNA sequences from chromosome ends due to incomplete DNA replication. The components that participate in the latter mechanism and this process have been characterized in detail; the other functions of telomeres are less well understood but are the subjects of active investigation.
KEY WORDS: telomere, Drosophila melanogaster, yeast, chromosomes, transcription, replication, nuclear architecture, silencing, chromosome stability, mitosis, meiosis, chromosome structure, telomerase, heterochromatin, position effect, Tetrahymena.
Telomeres are nucleoprotein structures on the ends of linear chromosomes. They are important for chromosome stability, nuclear architecture, and certain chromosome movements. The telomere ends are different from broken ends. This is important because chromosome breaks delay progression through the cell cycle and are subject to DNA repair mechanisms, which may result in exonucleolytic attack or ligation to other chromosomal fragments. The latter may result in dicentric or ring chromosomes, or to translocations or deletions. Given that DNA polymerase cannot replicate a linear chromosome completely, a special mechanism is required to maintain chromosome ends. In most organisms this is accomplished by telomerase, a reverse transcriptase with an internal RNA template. Under some circumstances, however, telomere length may be maintained by recombination or transposition.
In many cells chromosome arms are sequestered within the nucleus, and they may be aligned from telomere to centromere. This arrangement may be maintained in part by associations of telomeres with each other and with the nuclear envelope. There is reason to believe that the resulting nuclear domains are important for establishing or maintaining chromatin structure and transcriptional activity. Dramatic changes in chromosomal position within the nucleus may occur, for example, at the beginning of meiosis. Telomeres may play a profound role in these movements which are critical for meiotic recombination and segregation. Here we present an overview of the telomere role in the biology of the cell with an emphasis on nuclear architecture and transcription silencing.
Telomere Elongation
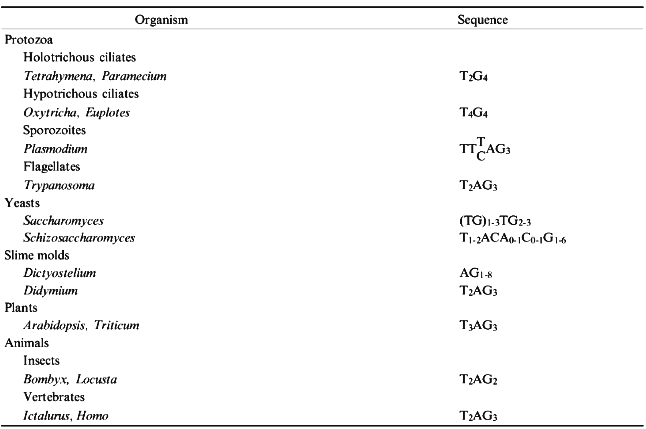
In most species DNA sequences at the chromosome tips consist of a simple repeat array in which the repeat unit is generally 5-8 bp in length. The array tends to be GC-rich, with a strand bias (Table 1), and the G-rich strand is oriented with its 3´ end to the end of the chromosome. Because DNA polymerase synthesizes in only one direction and requires a primer, the lagging strand during DNA replication cannot be replicated completely. Even if replication of the lagging strand is initiated at the chromosome tip, removal of the RNA primer will result in a 3´ overhang. In addition, it now appears that a 3´ overhang is added to the leading strand as well [1].
TABLE 1. Telomeric DNA Sequences
The single-stranded overhang is a template for telomerase, a specialized reverse transcriptase with an internal RNA template. Telomerase has been reviewed extensively recently [2] and is not covered in detail here. Telomerase activity has been identified in a wide range of organisms, from protozoa and fungi to humans. The RNA template contains sequences that are complementary to the G-strand of the telomeric repeat. In Tetrahymena, for example, the template region of the RNA strand is CAACCCCAA [3]. Mutations in the most 5´ six residues of this sequence result in the predicted changes in the DNA sequence at telomeres [4, 5]. Similar results have been obtained in yeast [6, 7] and human cells [8]. Telomerase from Tetrahymena has two protein subunits [9]. The p95 subunit appears to be involved in DNA recognition [9, 10], while the p80 subunit is closely associated with the RNA template and may carry the catalytic activity [9].
A discontinuous mechanism of telomere synthesis has been proposed that involves sequence-specific binding to the telomeric overhang, polymerization, and translocation. In the first step, the 3´ end of the primer DNA aligns with the RNA template while upstream primer sequences bind to an anchor site on the enzyme. Then a short stretch of DNA is copied from the template. When the end of the template region is reached, the enzyme realigns the primer and template. Binding of the primer with the anchor site on the enzyme during translocation prevents dissociation and allows another round of polymerization [2].
Telomerase may also cleave nucleotides from the primer if it extends beyond the template domain of the RNA subunit [11-13]. This may be a proofreading function and alternatively, it may be used to identify potential telomeric sequences during de novo addition of new telomere sequence onto a broken chromosome end (see below). Although the latter is known to occur, it is a relatively rare event.
Most human somatic cells have undetectable levels of telomerase activity, and over time their telomeres shorten as a function of population growth. It has been proposed that once telomeres shorten below a critical length progression through the cell cycle is arrested. This may provide a mitotic clock that prevents unlimited growth of somatic cells [14]. The few cells that overcome this cell cycle delay can grow indefinitely and are considered immortalized. One of the changes necessary for immortalization is the ability to add telomeric repeats onto chromosome ends. This is usually achieved by re-activation of telomerase, and most tumors and immortal cell lines have stable telomere arrays and exhibit telomerase activity [4, 15]. Spontaneous immortalization can occur, however, in the absence of telomerase activity [16-18]. The mechanism responsible for the elongation of telomeres in the absence of telomerase is unknown, but recombination has been suggested as one possibility [19].
A few organisms do not exhibit telomerase activity and maintain telomere length by another mechanism [20]. The most notable of these is Drosophila melanogaster, which uses telomere-specific transposition of two families of non-LTR, polyadenylated, LINE-like retrotransposable elements, HeT-A and TART, to elongate chromosome ends [21]. Other species, including dipteran insects and alliaceous plants, lack telomerase activity, but the mechanisms they use for telomere maintenance are not known in detail. In most of these cases recombination has been proposed. Most of these species have relatively simple repeat arrays at their chromosome ends, and these arrays bear a passing resemblance to canonical telomeric arrays [20]. Again, D. melanogaster is the exception. It carries tandem arrays of truncated HeT-A and TART elements at its chromosome ends. Complete members of these families of elements are found only in telomere regions. The mechanism for targeting telomeres by these elements is not known. HeT-A elements do not code for a reverse transcription gene. Thus, this function must be provided extrinsically, and may be provided by the host to control the rate and/or the time of transposition [21]. LINEs are transcribed using an internal 5´ promoter or exogenous 5´ promoter at the site of insertion. HeT-A elements at the chromosome end often lose the 5´ end of the element because of incomplete DNA replication and do not have an exogenous promoter at the 5´ end. Their promoter is near the 3´ end. As they form tandem arrays the promoter on one element is used to transcribe the adjacent element [22]. Thus, although the position of HeT-A elements at chromosome ends present it with special problems, the general mechanism of transcription and transposition may be similar to other LINEs.
Capping of Broken Chromosome Ends
Early radiation experiments in D. melanogaster led to the recovery of numerous chromosomal rearrangements which involved two or more chromosome breaks, and the ends of the rearranged chromosomes all carried the material that originated at the ends of the untreated chromosomes. Simple, one-break deletions and inversions were not recovered (at the time), and this led to the prediction of the special structures, telomeres, that are required to stabilize, or cap, chromosome ends [23]. Similar results were obtained in many other species using many different clastogenic agents, including ionizing radiation and chemicals. The primary exceptions were circular chromosomes, which have no ends.
While spontaneous and mutagen induced tip deletions are rare, they have been observed. McClintock [24, 25] broke maize chromosomes at meiotic anaphase and found that broken chromosome ends that entered the endosperm were subject to a repair process that resulted in ligation of sister broken ends after replication and further anaphase bridges. However, broken chromosome ends that entered the embryo were stabilized. The breaks induced in this assay were produced randomly along a chromosome arm and were not analyzed molecularly. Recently, terminal deficiencies have been demonstrated in several species, including D. melanogaster, yeast, and humans. Chromosomes with putative terminal deficiencies are fairly frequent in some grasses. In situ hybridization using a plant telomere sequence as a probe revealed that the ends on the broken chromosomes in these plants have telomeric DNA [26, 27]. In most other species as well chromosomes and chromosome fragments can only be recovered if they have telomeric DNA sequences on their ends. Double-stranded breaks must be repaired, or the cell dies.
Different methods are used to characterize the formation of new telomeres [28, 29]. These are transformation of yeast cells with linear plasmids [30], analysis of programmed chromosome fragmentation [13, 31], integration of telomeric sequences into interstitial sites [32, 33], and characterization of broken chromosome ends [34, 35]. These various approaches give a reasonably coherent picture of de novo telomere formation, as outlined below.
Chromosome breaks delay the cell cycle, allowing time for repair. Cells with unrepaired chromosomes may return to mitotic growth after this delay, but the broken chromosome is often lost within a few cell cycles [36]. New telomere sequences may be added at the site of the break by telomerase [35, 37], but this depends on a short, 2-4 bp 3´ overhang at the tip that resembles telomeric DNA [38]. Telomerase has a high affinity for single-stranded primers corresponding to the G-rich telomeric sequence. Primers with multiple clusters of G-residues are optimal substrates for elongation by telomerase, because they are more likely to have a telomere nucleation site within a few hundred base pairs of the tip [39] that is recognized by the telomerase anchor site. Telomerase will also extend a 3´ overhang lacking any complementarity to the RNA template, provided a tract of G-residues is present on the primer to recruit enzyme [38, 40]. De novo telomere formation is very precise. Euplotes crassus macronuclear telomeres formed during developmentally programmed chromosome fragmentation always initiate with the sequence GGGGTTTT, one of the eight possible permutations of the telomeric sequence in this organism [41]. Similarly, in Saccharomyces cerevisiae new telomeres are initiated with a precise sequence [39], even though the sequence found on natural chromosome ends are variable (Table 1).
Although telomeric DNA sequences may be found in interstitial chromosomal sites [28], chromosome breakage can be induced by the integration of telomeric DNA into the genome [32, 42]. Telomere formation induced by this method is dependent on the integration of the telomeric "seed", although it may be located more than 100 bp from the new chromosome end [33, 39, 43]. In yeast the nucleation sequence need not be a perfect match to the telomeric sequence, but in human cells sequence identity is required [33]. This strict sequence requirement is imposed by the telomere binding factor TRF [33, 44] rather than telomerase.
An exception to the rule that all chromosomes must have telomeric DNA at the termini, as was mentioned above, is found in D. melanogaster. This species does not have canonical telomeric DNA that fits the pattern shown in Table 1, and broken chromosomes can be stabilized without the addition of telomeric DNA. A specific gene-mutator has been found in D. melanogaster that potentiates terminal deficiencies [45]. The broken chromosome ends are not associated with any specific DNA sequence. Instead, the chromosome may terminate with any sequence, even within the open reading frame of a gene [34, 46]. The telomeric component responsible for stabilizing broken chromosome ends is not known, although a protein with double-stranded end binding properties has been proposed [46]. Eventually, by a process of stochastic transposition, the broken chromosome ends acquire the retrotransposons found at natural termini [47].
Nuclear Architecture
Beyond their role in replication and capping, telomeres have been proposed to participate in meiotic chromosome pairing, meiotic and mitotic chromosome segregation, and in the organization of the nucleus. Observations of both somatic and meiotic cells suggest that the positions of telomeres within the nucleus is highly specific and dependent on interactions of telomeres with the nuclear envelope [48, 49]. A nonrandom arrangement of chromosomes with telomeres in close proximity to each other and to the nuclear envelope was first described by Rabl [50] in Amphibia. A Rabl orientation of chromosomes has since been observed during prophase in somatic cells of a variety of organisms. It is unclear whether the relationship between telomeres and the nuclear envelope is a passive consequence of anaphase chromosome orientation or the result of an active process that maintains an ordered nuclear architecture during interphase. Direct evidence that chromosomes are restricted to subdomains in interphase D. melanogaster nuclei comes from examination of the arrangement of polytene chromosomes. Individual chromosome arms never intertwine, and telomeres tend to cluster at the side of the nucleus opposite to the chromocenter [51, 52]. With probes that paint whole chromosomes it was shown that individual chromosomes occupy distinct positions [53, 54]. The organization of telomeres in embryonic diploid D. melanogaster nuclei has also been investigated; again a highly polarized configuration was revealed [53, 55, 56]. A peripheral localization of telomeres has been also reported in Trypanosoma [57], plant cells [58], budding yeast [59-62], and fission yeast [63].
Telomeres interact not only with the nuclear envelope, but also with each other. For example, yeast cells immunostained with antibodies against Rap1p exhibit fewer spots than the expected number of telomeres, suggesting telomere clustering [60]. Rap1p is localized to the nuclear periphery [60] and colocalize with Sir3p, Sir4p, and with Y´ telomere-associated DNA [62, 64]. Genetic and biochemical evidence strongly suggest that Rap1p, Sir3p, and Sir4p form a multiprotein complex. Rap1p has a dispersed nuclear staining in sir3 and sir4 mutants; the normal focal pattern of Sir3p staining is diffuse in a sir4 mutant. Similarly, Sir4p staining is no longer punctate in a sir3 mutant. Telomeres, however, are still clustered in these mutants as detected by the telomere-associated DNA sequence Y´. One explanation is that proteins other than Sir3p and Sir4p may be involved in the localization of the telomeres [65]. According to the model of Maillet et al. [64] compartmentalization enables bifunctional proteins like Rap1p and Abf1p, which can both activate and repress transcription, to have both functions in the same nucleus. Additionally, peripheral localization of telomeres helps to anchor the chromosomes, preventing their reorganization during interphase [48].
Although telomeres themselves may not mediate chromosome segregation, the separation of telomeres during cell division creates a special problem for the segregational system [66]. Evidence that cis-acting functions are required for the separation of telomeres has been recently obtained for Tetrahymena. Altering the telomerase RNA template in Tetrahymena from GGGGTT to GGGGTTTT can create a block in anaphase chromosome separation in the micronucleus [67]. Cytological analysis revealed a failure of telomere separation of sister chromatids. Stretched chromosomes were often seen as one continuous fiber passing through the midzone of the spindle. The authors concluded that telomeres on sister chromatids are normally associated until metaphase and that the defective telomeres prevent telomere separation. Thus, a specific element of the telomere repeat may be required in cis to mediate chromatid separation.
Specific proteins are also required to mediate chromatid separation in D. melanogaster [68]. Mutations in the UbcD1 gene, which encodes a class I ubiquitin-conjugating enzyme, cause telomere--telomere attachments during both mitosis and male meiosis. In these mutants telomeres associate inappropriately with the telomeres of their sister chromatids and with telomeres of both homologous and nonhomologous chromosomes. The Sir4p of yeast telomeres also binds the deubiquitinating enzyme Ubp3p, suggesting that telomeric protein ubiquitination is a general phenomenon [69].
How sister chromatids adhere to one another until anaphase is unknown, but telomeres may be involved. Normal telomere--telomere associations, seen cytologically in a variety of organisms, could be mediated through single-stranded DNA tails [70] or telomere proteins. The latter include the TBP proteins of ciliates, the Rap1 or Sir proteins of yeast, and hTRF of humans [71, 72].There are three ways in which proteins could mediate telomere--telomere associations [49]. First, dimers or multimers of telomere-binding proteins might be able to associate simultaneously with two or more telomeres. Second, proteins may link telomeres indirectly, through a third element such as a component of the nuclear envelope. Several potential receptors for chromatin have been identified that localize specifically to the inner nuclear membrane during interphase [73]. Lamin-binding proteins LAP2 and LBR associate rapidly with chromosomes and the reassembling nuclear envelope during anaphase and telophase. LBR binds to two human homologs of the D. melanogaster heterochromatin protein HP1, which is also found at telomeres. The third variant suggests that telomere proteins might facilitate DNA--DNA interactions between telomeres. Oligonucleotides of single-stranded, G-rich, telomeric DNA from ciliates, vertebrates, and yeast form alternative DNA structures in vitro that depend on non-canonical base pairing of the guanines [74]. The beta-subunit of the Oxytricha telomere protein and Rap1p of yeast have the ability to fold or stabilize telomeric DNA in G-quartet structures that mediate telomere--telomere association in vitro [70]. Linear plasmids form telomere--telomere interactions in vitro, similar to those on molecules isolated from yeast, but only if their ends have a TG1-3 tail. Wellinger et al. [1] argue that telomere--telomere interactions involve duplex DNA held together by G:G base pairs, rather than a triple helix or G quartet. TG1-3 tails and telomere--telomere interactions were detected in vivo in strains lacking telomerase, suggesting that telomerase-independent mechanisms generate TG1-3 tails at the end of S-phase by cell cycle-regulated degradation of the C1-3A strand. The resulting single-stranded tails are a potential substrate for telomerase and other telomere-binding proteins.
Abundant cytological evidence suggests that the localization of telomeres during meiosis is distinct from that observed in somatic nuclei. In meiotic cells telomeres cluster at zygotene to form a so-called "bouquet". The bouquet arrangement of chromosomes has been noted in a variety of organisms [48]. This nuclear arrangement is likely to be functionally linked to the process of homologous pairing and synapsis. During the period in which telomeres form a cluster, there is also evidence that they are tightly attached to the nuclear envelope and possibly interact with the cytoskeleton. The most dramatic evidence for the importance of these associations is chromosomal pairing in the fission yeast [75], where prophase chromosome movement apparently is directed by the telomeres.
A gene NDJ1 encodes a telomere associated protein required for meiotic chromosome segregation in S. cerevisiae [76]. This protein accumulates at the telomeres during meiotic prophase, and its absence results in high levels of failed meiotic chromosome segregation. The ndj1 mutant phenotype includes delays in the formation of synaptonemal complex axial elements, in synapsis and in the first meiotic division; loss of telomeric localization of Rap1p; reduced levels of sporulation and spore viability; and distributive segregation of linear heterologs. However, there is no effect of the absence of Ndj1p on the segregation of telomere-less ring chromosomes. This means that the mentioned protein is not required for meiotic chromosome separation per se, but rather that Ndj1p is essential to separate chromosomes that have telomeres.
As described below, associations between telomeres and other nuclear structures such as the nuclear envelope or nuclear matrix may be related to gene repression.
Telomere Position Effect
Genes positioned adjacent to or within the telomeric region are subject to variegated transcriptional repression. Telomeric position effect (TPE) in D. melanogaster is observed when P-transposable elements carrying a reporter gene are inserted into telomeric regions [77-82]. TPE has also been described in both fission and budding yeasts [83-85]. This repression resembles centromeric position effect variegation (PEV) in D. melanogaster [86-89]. TPE in yeast has been analyzed in detail because of its relationship and resemblance to mating-type gene silencing.
Cis-Acting Components of Silencers. Mating type in yeast is determined by genes present at the expressed MAT locus. Mating-type genes also exist at two other locations, HML and HMR, on the same chromosome near the left and right telomeres. At these sites, however, the mating-type genes are not expressed, even though all the signals for expression are present at these loci [90]. This position-dependent but gene-independent repression is known as silencing. It extends to other yeast genes placed at HML and HMR and results from formation of a specific chromatin structure that appears to be the yeast equivalent of metazoan heterochromatin. Silencing depends on the concerted action of cis-acting sequences and several trans-acting factors, including the products of the SIR genes.
Cis-acting sequences that flank HML and HMR, known as the E- and I-silencers are composed of consensus sequences for the DNA-binding proteins Rap1p, Abf1p, and proteins of the origin recognition complex ORC [91-94]. All four silencers contain a site that conforms to an 11-bp consensus found in all known autonomous replicated sequences (ARS). This core element, or ARS consensus sequence (ACS), is essential for the function of the origin of replication and is a binding site for a complex of six polypeptides of the ORC [95, 96]. A synthetic silencer consisting of a single binding site for ORC, Rap1p, and Abf1p is fully functional in silencing.
The arrangement of binding elements specific for silencers is not specific for telomeres. Telomeres in S. cerevisiae consist of a variable length terminal repeat C1-3A, about 150-450 bp in length, joined to a longer and more complex array of X and Y´ elements. The important feature of poly(C1-3A) tracts is the regular occurrence of Rap1p-binding sites [97]. Although short stretches of poly(C1-3A) tracts are not sufficient for repression when located at an internal chromosomal site [84], longer tracts do function as silencer elements [98]. Additional structural elements of telomere also play a role in TPE. Repression of a URA3 reporter gene extends farther from a native chromosome telomere containing a Y´ element than it does from a chromosome without this sequence [99]. The function of X elements in TPE is unclear.
The presence of the silent mating-type loci near the telomeres of chromosome III has led to the speculation that this location may contribute to the HM silencing [49]. Indeed, moving the silent mating-type loci away from the telomere decreases their silencing properties [100, 101]. Using a reporter gene flanked by complete mating-type silencers HML-E and HML-I at various chromosomal sites, Maillet and colleagues [64] demonstrated that proximity to telomeric repeat sequences is necessary for the repression of this gene. The reporter gene is not repressed when integrated >200 kb from a telomere. Repression is restored by either creation of a new telomere with 13 kb from the reporter or by the insertion of a 350 bp of telomeric tract, which is not sufficient to repress transcription on its own [98].
Silencers serve as organization sites for heterochromatin formation [100]. The overexpression of PPR1, the URA3 transcriptional activator, relieves silencing of URA3 inserted at a telomere [102]. Itcan be explained as a competition between the silencer promoting a condensation of the chromatin and an enhancer or activator promoting an open chromatin configuration.
One additional function of the silencers may be to help to localize loci to their specific subnuclear compartment. Rap1 protein is localized to the nuclear periphery [60], whereas ARS sequences are preferentially bound to the nuclear scaffold in yeast [103]. Genetic evidence suggests that a critical local concentration of Sir proteins is necessary to establish or maintain a repressed chromatin state even at an internal chromosomal location [65, 98, 104, 105].
Important Protein Components of Transcriptional Silencing. Rap1p plays an important role in transcriptional silencing at HM loci and at telomeres. At the same time Rap1p-binding sites are found within the promoters of a large number of yeast genes, where it appears to function as a transcriptional activator. The same was found for Abf1p, which appears to stimulate firing of the ARS1 replication origin. Genetic evidence for the involvement of Rap1p, Abf1p, and ORC proteins in silencing has been obtained by the isolation of mutations that disrupt silencing [91, 93-95, 106, 107]. Such mutations identify SIR-2, -3, -4, and RIF1, whose products interact with the Rap1p to form a multimeric complex [108, 109]. The fact that mutations in histones H3 and H4 suppress silencing suggests that the mechanism involves chromatin packaging [110-115].
The molecular nature of the establishment of silencing in yeast is actively studied. Rap1p plays a central role in this process. Mutagenesis studies revealed that the carboxy-terminal 28 amino acids of Rap1p is critical in TPE, telomere length regulation and HML silencing [116]. Because Rap1p is able to either activate or repress transcription, depending on the context of its binding site, it must act together with other factors. To explain its action at the HM silencers and telomeres it was proposed that Rap1p recruits a complex of Sir proteins to the DNA via protein--protein interaction [61, 117, 118]. It was shown in a two-hybrid system that Sir3p and Sir4p interact with Rap1p. The Rap1p--Sir3p interaction is probably direct, and Sir3p and Sir4p themselves likely interact to form a protein complex [118]. One possible function of this interaction is the formation of an extended structure involved in propagating repressed chromatin from its site of initiation [119].
Establishment of silencing requires passage through S-phase [120], which suggest a role of ORC [94, 107]. Replication initiation at silencers, however, is not essential to the mechanism ofsilencing. Tethering a Gal4--Sir1 fusion protein directly to the silencer with a GAL4 DNA-binding domain bypasses the requirement both for ORC-binding sites in the silencer and for ORC function [121]. This confirms the suggestionthat ORC recruits Sir1p to the silencer [122].
On the other hand, some mutations in genes involved in replication affect silencing. A mutation in CDC7, which encodes a protein kinase required for replication initiation, restores silencing [123]. Similarly, mutations in the D. melanogaster gene encoding the proliferating cell nuclear antigen (PCNA), a processivity factor for replication, suppress heterochromatic gene inactivation [124].Silencing involves the assembly of a specialized repressive chromatin structure. The passage of a replication fork may be necessary for chromatin assembled in one state to be reassembled into another state. It is possible that other S-phase events, for example phosphorylation, are important for silencing.
Telomeric silencing requires several of the same proteins required to silence HMR and HML, but does not require Sir1p [125]. As mutations in ORC genes decrease telomeric silencing, ORC has a Sir1p-independent role in this function.The nature of this role is unclear; however, a few possible suggestions have been made. First, telomeres assemble into a large structure, often near the periphery of the nucleus [59]. Thus, an ORC bound to an ACS of one telomere can promote telomeric silencing by a mechanism akin to transvection in D. melanogaster. Alternatively, orc mutations may affect telomeric silencing by altering the timing of replication.
Telomeres themselves delay origin activation until late in S-phase [126]. Site-specific recombination was used to separate an origin of replication from the adjacent telomere in vivo, and it was shown that the signal for late activation of replication is established between mitosis and START-stage in the subsequent G1-phase. Once set, the signal persists through S-phase in the absence of the telomere. It is possible that this persistence is caused by chromatin structure. Alternatively, subnuclear localization of the telomeres at the nuclear periphery may be the primary cause of late replication. One difference between the telomere's effect on transcription and its effect on replication timing is the genomic distances involved. The origin can be affected over a distance of 27 kb from the telomere, whereas transcriptional silencing normally extends only about a tenth of that distance [99].
None of the Sir proteins appears to contain DNA-binding motifs, nor do mutations in these genes affect the binding of other proteins to silencer DNA. It now seems clear, that Sir3p and Sir4p proteins are directed to HM silencers and telomeres by interaction with Rap1p [118]. All four Sir proteins are necessary for silencing at HM, but only three (Sir-2, -3, -4) are required for telomeric silencing [125]. How the Sir proteins repress transcription once recruited to the silencer and what determines the size of the repressed domain are unknown. Deletion of the C-terminal part of Sir4p, which bears weak similarity to the mammalian nuclear lamins [127], results in loss of silencing at the HM loci and telomeres and promotes an increase in life span [128]. Much more is known about two other proteins Sir3p and Sir1p.
Overexpression of Sir3p protein has been shown to increase the efficacy and extent of spreading of silencing from a yeast telomere [99]. The probability of repression of the URA3 reporter gene decreases as a function of the distance of the promoter from the telomere, dropping approximately 10-fold for every kilobase. Thus, it can be argued that telomeric silencing is a cooperative process involving the assembly of a multiprotein complex, similar to that proposed for PEV in D. melanogaster [129, 130]. Increasing the dosage of Sir3p extends the silent telomeric domain. Therefore, Sir3p is a crucial component of the silent chromatin domain that initiates at the telomere and is assembled along the yeast chromosome. Because the SIR3 gene is located near a telomere, position-effect repression of the SIR locus would provide a feedback mechanism for control of position effect spreading in yeast. Thus, the spread of TPE in S. cerevisiae is modulated by numerous factors, including promoter distance from the telomere, promoter strength, transcriptional status of telomere-proximal genes, presence of Y´ elements, and intracellular concentration of the Sir3p.
While silencing at HM loci is extremely stable, at telomeres it is not [84]. This instability is seen as sectoring in yeast colonies or variegating in D. melanogaster. One obvious difference is the involvement of Sir1p in transcriptional repression at HM loci. Actually, loss of SIR1 function leads to variegated repression at HML [125, 131]. Support for the idea that Sir1p plays a role in establishing repression comes from the observation that silencer mutations, or rap1 mutations, can lead to phenotypes similar to sir1 mutations [132-134]. Additionally, targeting of Gal4--Sir1p hybrid protein to HMR can bypass the requirement of the HMR-E silencer for repression [135]. It suggests that Sir1p is sufficient to recruit the other Sir proteins to the HMR locus. This hybrid protein also affects telomeric silencing. When an artificial telomere with GAL4-binding sites adjacent to the C1-3A repeats was targeted by Gal4--Sir1p hybrid protein, the stability of URA3 repression was significantly increased.
Direct interactions between Sir4p and Sir2p, Sir4p and Sir3p, Sir2p and Sir3p, Sir2p and Sir2p, and Sir4p and Sir4p were observed in vitro and in vivo [136]. Additionally, two other proteins are tightly associated with Sir2p--Sir3p--Sir4p complex [69]. One of these is Ubp3p, one of several yeast enzymes that deubiquitinate target proteins. Deletion of the UBP3 gene results in markedly increased silencing of genes inserted near a telomere or at one of the silent mating type loci. It indicates that Ubp3p is an inhibitor of silencing. Ubp3p may regulate silencing by controlling either the activity or the assembly of the Sir protein complex during the S-phase [69]. The regulation of silencing by ubiquitination appears to be an evolutionarily conserved process; mutations in the ubiquitin C-terminal hydrolase gene enhance position effect variegation in D. melanogaster [137].
Nucleosomes spanning the silent mating-type cassettes and other silenced regions are hypoacetylated, like those in inactive chromatin from metazoan cells [138], suggesting that higher-order chromatin structure reminiscent of heterochromatin is involved in the silencing in yeast [139]. In addition, TPE in yeast is accompanied by inaccessibility of the DNA to DAM methylase [140, 141]. Mutations in histones H3 and H4 [101, 113, 114] that remove potential acetylation sites in the amino-terminal domain cause silencing defects at HML, HMR, and telomeres. Sir2p also plays a role in promoting histone deacetylation in yeast [138]. It has long been known that histone gene dosage and inhibitors of histone deacetylases affect PEV in D. melanogaster [142-145]. Furthermore, deletion of NAT1 and ARD1, which encode subunits of an N-terminal acetyltransferase, causes a silencing defect in yeast [146]. While N-terminal acetylation is a modification distinct from the acetylation of lysine residues, it establishes the importance of acetylation in silencing.
The transcriptional regulator Rpd3p in yeast, which shares some homology with human histone deacetylase, affects the level of histone acetylation. Strains with rpd3 deletions exhibit decreased transcriptional activity near telomeres [147, 148]. In addition, enhancement of PEV was seen in D. melanogaster dRPD3 mutant strains [147]. It implies that the acetylation of chromatin components may regulate gene expression.
Heritability of the transcription state is not intrinsic to the silenced locus or the chromatin encompassing it [149]. Rather, it appears to be a property of the silencer. Repression in the absence of the silencer is erased within a single cell cycle. This indicates that silencers are required for inheritance of the repressed state. It is possible that epigenetic inheritance of repressive state observed in many cases of position-effect repression results from specific sequences. In addition to the silencer itself, silencer-binding proteins (Rap1p, Abf1p, and ORC), as well as Sir1p likely belong in the category of silencing components that serve to organize silencing. Mutation of several of these components yields a common phenotype: a partial silencing that exhibits epigenetic inheritance, yield mixed populations of repressed and derepressed cells, each of which gives rise at high frequency to progeny with the same transcriptional state as the parents. Thus, the silencers, their associated binding proteins, and Sir1p likely comprise a structure whose role is to direct formation of heterochromatin and ensure its persistence. So the silencers provide the genomic memory of the expression state. Once assembled, this complex may persist throughout the cell cycle and may be inherited efficiently in daughter cells. If the heterochromatin dissolves at some stage in the cell cycle, the silencer could ensure its efficient reassembly. Two observations favor this disassembly--reassembly model. First, silenced promoters appear to be more accessible to specific transcription factors close to or during mitosis [102]. Second, silenced domains that have been activated by dissolution of the heterochromatin over the region must pass through a specific stage in the cell cycle, S-phase or later, to regain the silenced state [120].
Rif1p and Rif2p interact with Rap1p. Mutations in RIF1 or RIF2 result in moderate telomere elongation and increased telomeric silencing [92, 150]. Simultaneous deletion of both genes results in a dramatic increase in telomere length, while overexpression of either decreases telomere length. While the function of these proteins in telomere length regulation requires the Rap1p carboxyl terminus, it is possible that Rif1p and Rif2p interact with each other in vivo [150]. Rif1p at one telomere may interact with Rif2p at another and may play a role in the clustering telomere [59, 60, 62, 151]. RIF1 may play a critical role in the balance between HM and telomeric silencing mediated by competition for the Sir proteins [152].
Telomere length may be regulated by a negative feedback mechanism that can sense the precise number of Rap1p carboxyl termini at the chromosome end. Marcand et al. [153] proposed a model in which Rap1p--Sir and Rap1p--Rif complexes are partitioned to opposite ends of the telomeric C1-3A repeats. Rap1p--Sir interactions are favored at the proximal end of telosome by cooperative interactions between the Sir proteins themselves and between Sir proteins and histones H3 and H4. The critical parameter controlling telomere length regulation appears to be the number of the Rif complexes. The shortening of telomeres caused by the overexpression of Rif proteins would thus result from an extension of the Rif complex along the telomere. The Rif1p--Rif2p complex is likely to act at the distal telomere end, and assembly may be promoted by interaction with telomere end-binding proteins. The Est1 [154], Cdc13 [155-157], and Stn1p [158] belong to the group of these proteins. Another candidate for Rif action is the Pif1p, a helicase that inhibits telomere elongation [159].
Telomeric Position Effect in D. melanogaster. Telomeric silencing in D. melanogaster has not been studied as extensively as in yeast. TPE in D. melanogaster is observed when P-elements carrying a reporter gene are inserted into telomeric regions and variegate [77-82]. Little is known about cis- and trans-acting factors influencing the efficacy of silencing exerted on these transgenes. The variegating P[w] inserts studied to date have all inserted into subterminal minisatellites (H. Biessmann, personal communication; P. K. Geyer, personal communication; M. Pavlova and R. W. Levis, personal communication). Therefore, these minisatellites are good candidates for conferring transcriptional silencing and possibly heterochromatic structure within the subtelomeric region. In order to examine the subterminal minisatellite from chromosome 2L [160] as a potential cis-acting sequence for TPE, it has been placed upstream of the mini-white gene in a P-element construct. Several transgenic lines were generated in which the silencing activity of the subtelomeric minisatellite could be tested in nontelomeric locations. It has been shown that minisatellite represses reporter gene activity in ectopic chromosomal positions, and this repression depends on the orientation and length of the minisatellite (Kurenova et al., submitted). This repression is not responsive to changes in temperature, to an additional Y-chromosome or to trans-acting modifiers of centromeric PEV, suggesting that it is not related to classical PEV. However, it is suppressed by a mutation Su(z)2 a chromatin structural protein [161] that is necessary for TPE (L. Wallrath, personal communication).
The observed directionality of silencing is not without precedent; it has been shown for the silencers at the yeast mating type loci [100]. Both HMR-E and HML-E exhibit orientation-dependent silencing, and the particular organization of binding elements within the silencer is critical for its function. Different types and organizations of silencers have different potencies in establishing repression of an adjacent locus. TPE in D. melanogaster and yeast may have more in common with each other than D. melanogaster TPE has with PEV, which is induced by pericentric heterochromatin. However, long-term developmentally controlled maintenance of transcriptional silencing in D. melanogaster may share some common features with TPE. Zink and Paro [162] tested the functional role of a PRE (PcG Response Element) of the bithorax complex and found that the element behaves as an orientation-dependent silencer, capable of inducing mosaic expression of the neighboring genes. The PcG proteins are thought to induce heterochromatin-like structures at PRE sites which stably inactivate transcription [163]. It is possible that these, or an additional set of proteins, encoded by yet unidentified genes, is required for TPE. These proteins might form polymeric aggregates similar to the multimeric complexes found in the yeast that could interact with other nuclear structures, creating a local telomere-specific heterochromatic domain.
While the 2L subterminal minisatellite of D. melanogaster can repress the expression of the w gene expression in non-telomeric positions, it does not cause variegation (Kurenova et al., submitted). There are a number of possible reasons why these transgenes do not variegate. One is that the minisatellite may be responsible for nucleating heterochromatin, or localizing the telomere to a specific domain within the nucleus. Associations between telomeres and a nuclear structure such as the nuclear envelope or nuclear matrix correlate with repression in yeast [98]. Similarly telomeres of D. melanogaster are thought to attach to the nuclear envelope and interact with each other [48, 49]. Insertions of a P-element into the subtelomeric minisatellite may disrupt telomere function by destabilizing these structures or associations. The minisatellite in the transgene is left intact and does not cause variegation. Arguing against this model, however, is the fact that short minisatellite arrays cause less repression, without variegation.
A second possible explanation is that the minisatellite contains all structures necessary to cause transcriptional repression in all cells, thus preventing variegation. This explanation is in accord with data that different types and organizations of silencers have different potencies in establishing repression of an adjacent locus [100]. Thus, other components in cis may cause variegation by competing with the silencer. For example, overexpression the URA3 transcriptional activator relieves silencing of URA3 inserted at a telomere in yeast [102]. The same is true in the case of a PRE silencer in D. melanogaster [162]. Candidates for competitors of repression by the subtelomeric minisatellite in D. melanogaster are the telomeric HeT-A and TART elements. The 3´ non-coding region of a HeT-A element is located adjacent to the minisatellite at the tip of 2L [160]. This portion of HeT-A is not able to induce on its own variegated expression of an adjacent yellow gene [46, 47].The 3´ noncoding region of HeT-A element, however, contains a promoter [22]and may derepress silencing by competing with the silencer. This may explain the variegated expression of the genes inserted into telomere regions and the absence of variegation in subtelomeric minisatellite transgenes in ectopic positions.
Telomeric Transcriptional Silencing in a Natural Context. Telomeric silencing affects the yeast retrotransposon Ty5-1, which is located 1.8 kb from the left telomere of S. cerevisiae chromosome III. It is an inactive member of the Ty5 family of yeast retrotransposons, bearing a deletion that encompasses the integrase coding sequence. Ty5-1 expression is almost undetectable in wild-type strains, decreases in a SIR3 overexpression strain, but is enhanced in a sir3 mutant and in the histone H4 N-terminal deletion mutant [164]. The Ty5-1 expression pattern indicates that it is subject to telomeric silencing. The observation that Ty5 elements transpose preferentially near silenced areas like telomeres and HM loci suggests that this localization could be selected by Ty5 elements to turn off their own expression. Variegated and semi-stable silencing could be useful for some retrotransposons as a transcriptional and transpositional regulatory mechanism. Retrotransposon jumps may cause mutagenic damage. In a population of cells where retrotransposon expression is semi-stable, the mutagenic damage could be restricted to a small part of the population. This telomeric silencing could function as a safety net for the mobile element.
Thus, telomeres perform a number of cellular functions. Some of these, such as chromosome elongation and capping, are necessary in order to maintain linear chromosomes. The purpose of other telomere behaviors, such as nuclear localization and silencing, are less well understood. While most organisms have a simple repeat array at their termini, and this array is required by those organisms for both elongation and capping, some organisms, such as D. melanogaster, manage to survive without such a telomeric sequence. Thus, it is obvious that there are other structures and mechanisms that will perform these functions.
We are only beginning to understand other telomere behaviors. While the simple terminal repeat may be important for telomeric position effect in yeast, similar position effects are seen in D. melanogaster without this repeat. It remains to be seen whether Drosophila will be an exception in this case as well. Even if it is it will broaden our understanding of what a telomere is.
LITERATURE CITED
1.Wellinger, R. J., Ethier, K., Labrecque, P., and
Zakian, V. A. (1996) Cell, 85, 423-433.
2.Greider, C. W., and Blackburn, E. H. (1996) Sci.
Amer., 274, 92-97.
3.Greider, C. W., and Blackburn, E. H. (1989)
Nature, 337, 331-337.
4.Autexier, C., and Greider, C. W. (1996) Trends
Biochem. Sci., 21, 387-391.
5.Yu, G. L., Bradley, J. D., Attardi, L. D., and
Blackburn, E. H. (1990) Nature, 344, 126-132.
6.McEachern, M. J., and Blackburn, E. H. (1995)
Nature, 376, 403-409.
7.Singer, M. S., and Gottschling, D. E. (1994)
Science, 266, 404-409.
8.Feng, J., Funk, W. D., Wang, S. S., Weinrich, S.
L., Avilion, A. A., Chiu, C. P., Adams, R. R., Chang, E., Allsopp, R.
C., and Yu, J. (1995) Science, 269, 1236-1241.
9.Collins, K., Kobayashi, R., and Greider, C. W.
(1995) Cell, 81, 677-686.
10.Harrington, L., Hull, C., Crittenden, J., and
Greider, C. (1995) J. Biol. Chem., 270, 8893-8901.
11.Cohn, M., and Blackburn, E. H. (1995)
Science, 269, 396-400.
12.Collins, K., and Greider, C. W. (1993) Gene
Develop., 7, 1364-1376.
13.Melek, M., Greene, E. C., and Skippen, D. E.
(1996) Mol. Cell Biol., 16, 3437-3445.
14.Harley, C. B. (1991) Mutat. Res.,
256, 271-282.
15.Shay, J. W., and Wright, W. E. (1996) Trends
Genet., 12, 129-131.
16.Bryan, T. M., Englezou, A., Gupta, J., Bacchetti,
S., and Reddel, R. R. (1995) EMBO J., 14, 4240-4248.
17.Kim, N. W., Piatyszek, M. A., Prowse, K. R.,
Harley, C. B., West, M. D., Ho, P. L. C., Coviello, G. M., Wright, W.
E., Weinrich, S. L., and Shay, J. W. (1994) Science,
266, 2011-2015.
18.Rogan, E. M., Bryan, T. M., Hukku, B., Maclean,
K., Chang, A. C., Moy, E. L., Englezou, A., Warneford, S. G., Dalla, P.
L., and Reddel, R. R. (1995) Mol. Cell Biol., 15,
4745-4753.
19.Murnane, J. P., Sabatier, L., Marder, B. A., and
Morgan, W. F. (1994) EMBO J., 13, 4953-4962.
20.Biessmann, H., and Mason, J. M. (1997)
Chromosoma, in press.
21.Mason, J. M., and Biessmann, H. (1995) Trends
Genet., 11, 58-62.
22.Danilevskaya, O. N., Arkhipova, I. R., Traverse,
K. L., and Pardue, M. L. (1997) Cell, 88, 647-655.
23.Muller, H. J. (1938) The Collecting Net,
8, 182-195.
24.McClintock, B. (1939) Proc. Natl. Acad. Sci.
USA, 25, 405-416.
25.McClintock, B. (1941) Genetics,
26, 234-282.
26.Wang, S., Lapitan, N. L. V., and Tsuchiya, T.
(1992) Genome, 35, 975-980.
27.Werner, J. E., Kota, R. S., Gill, B. S., and
Endo, T. R. (1992) Genome, 35, 844-848.
28.Biessmann, H., and Mason, J. M. (1994)
Chromosoma, 103, 154-161.
29.Melek, M., and Shippen, D. E. (1996)
Bioessays, 18, 301-308.
30.Wang, S. S., and Zakian, V. A. (1990)
Nature, 345, 456-458.
31.Yao, M.-C., Yao, C.-H., and Monks, B. (1990)
Cell, 63, 763-772.
32.Farr, C., Fantes, J., Goodfellow, P. N., and
Cooke, H. (1991) Proc. Natl. Acad. Sci. USA, 88,
7006-7010.
33.Hanish, J. P., Yanowitz, J. L., and De Lange, T.
(1994) Proc. Natl. Acad. Sci. USA, 91, 8861-8865.
34.Biessmann, H., and Mason, J. M. (1988) EMBO
J., 7, 1081-1086.
35.Flint, J., Craddock, C. F., Villegas, A.,
Bentley, D. P., Williams, H. J., Galanello, R., Cao, A., Wood, W. G.,
Ayyub, H., and Higgs, D. R. (1994) Am. J. Hum. Genet.,
55, 505-512.
36.Sandell, L. L., and Zakian, V. A. (1993)
Cell, 75, 729-739.
37.Morin, G. B. (1991) Nature, 353,
454-456.
38.Lee, M. S., and Blackburn, E. H. (1993) Mol.
Cell Biol., 13, 6586-6599.
39.Kramer, K. M., and Haber, J. E. (1993) Gene
Develop., 7, 2345-2356.
40.Harrington, L. A., and Greider, C. W. (1991)
Nature, 353, 451-454.
41.Klobutcher, L. A., Swanton, M. T., Donini, P.,
and Prescott, D. M. (1981) Proc. Natl. Acad. Sci. USA,
78, 3015-3019.
42.Surosky, R. T., and Tye, B. K. (1985) Proc.
Natl. Acad. Sci. USA, 82, 2106-2110.
43.Murray, A. W., Claus, T. E., and Szostak, J. W.
(1988) Mol. Cell Biol., 8, 4642-4650.
44.Zhong, Z., Shiue, L., Kaplan, S., and De Lange,
T. (1992) Mol. Cell Biol., 12, 4834-4843.
45.Mason, J. M., Strobel, E., and Green, M. M.
(1984) Proc. Natl. Acad. Sci. USA, 81, 6090-6094.
46.Biessmann, H., Mason, J. M., Ferry, K., d'Hulst,
M., Valgeirsdottir, K., Traverse, K. L., and Pardue, M. L. (1990)
Cell, 61, 663-673.
47.Biessmann, H., Carter, S. B., and Mason, J. M.
(1990) Proc. Natl. Acad. Sci. USA, 87, 1758-1761.
48.Dernburg, A. F., Sedat, J. W., Zacheus Cande, W.,
and Bass, H. W. (1995) in Telomeres (Blackburn, E. H., and
Greider, C. W., eds.) Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY, pp. 295-338.
49.Gilson, E., Laroche, T., and Gasser, S. M. (1993)
Trends Cell Biol., 3, 128-134.
50.Rabl, C. (1885) Morphol. Jahrb.,
10, 214-330.
51.Hochstrasser, M., Mathog, D., Gruenbaum, Y.,
Saumweber, H., and Sedat, J. W. (1986) J. Cell Biol.,
102, 112-123.
52.Mathog, D., Hochstrasser, M., Gruenbaum, Y.,
Saumweber, H., and Sedat, J. (1984) Nature, 308,
414-421.
53.Marshall, W. F., Dernburg, A. F., Harmon, B.,
Agard, D. A., and Sedat, J. W. (1996) Mol. Biol. Cell,
7, 825-842.
54.Dernburg, A. F., Broman, K. W., Fung, J. C.,
Marshall, W. F., Philips, J., Agard, D. A., and Sedat, J. W. (1996)
Cell, 85, 745-759.
55.Johansen, K. M., Johansen, J., Baek, K. H., and
Jin, Y. (1996) J. Cell. Biochem., 63, 268-279.
56.Hiraoka, Y., Agard, D. A., and Sedat, J. W.
(1990) J. Cell. Biol., 111, 2815-2828.
57.Chung, H. M., Shea, C., Fields, S., Taub, R. N.,
Van der Ploeg, L. H., and Tse, D. B. (1990) EMBO J., 9,
2611-2619.
58.Shaw, P., Highett, M., and Rawlins, D. (1992)
J. Microscopy, 166, 87-97.
59.Palladino, F., Laroche, T., Gilson, E., Axelrod,
A., Pillus, L., and Gasser, S. M. (1993) Cell, 75,
543-555.
60.Klein, F., Laroche, T., Cardenas, M. E., Hofmann,
J. F.-X., Schweizer, D., and Gasser, S. M. (1992) J. Cell Biol.,
117, 935-948.
61.Cockell, M., Palladino, F., Laroche, T., Kyrion,
G., Liu, C., Lustig, A. J., and Gasser, S. M. (1995) J. Cell.
Biol., 129, 909-924.
62.Gotta, M., and Gasser, S. M. (1996)
Experientia, 52, 1136-1147.
63.Funabiki, H., Hagan, I., Uzawa, S., and Yanagida,
M. (1993) J. Cell Biol., 121, 961-976.
64.Maillet, L., Boscheron, C., Gotta, M., Marcand,
S., Gilson, E., and Gasser, S. M. (1996) Gene Develop.,
10, 1796-1811.
65.Konkel, L. M. C., Enomoto, S., Chamberlain, E.
M., McCunezierath, P., Iyadurai, S. J. P., and Berman, J. (1995)
Proc. Natl. Acad. Sci. USA, 92, 5558-5562.
66.Hawley, R. S. (1997) Science, 275,
1441-1443.
67.Kirk, K. E., Harmon, B. P., Reichardt, I. K.,
Sedat, J. W., and Blackburn, E. H. (1997) Science, 275,
1478-1481.
68.Cenci, G., Rawson, R. B., Belloni, G.,
Castrillon, D. H., Tudor, M., Petrucci, R., Goldberg, M. L., Wasserman,
S. A., and Gatti, M. (1997) Gene Develop., 11,
863-75.
69.Moazed, D., and Johnson, D. (1996) Cell,
86, 667-677.
70.Henderson, E. (1995) in Telomeres
(Blackburn, E. H., and Greider, C. W., eds.) Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, NY, pp. 11-34.
71.Fang, G., and Cech, T. R. (1995) in
Telomeres (Blackburn, E. H., and Greider, C. W., eds.) Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 69-106.
72.Chong, L., Van Steensel, B., Broccoli, D.,
Erdjument, B. H., Hanish, J., Tempst, P., and De Lange, T. (1995)
Science, 270, 1663-1667.
73.Marshall, I. C. B., and Wilson, K. L. (1997)
Trends Cell Biol., 7, 69-74.
74.Sundquist, W. I. (1993) Curr. Biol.,
3, 893-895.
75.Chikashige, Y., Ding, D. Q., Funabiki, H.,
Haraguchi, T., Mashiko, S., Yanagida, M., and Hiraoka, Y. (1994)
Science, 264, 270-273.
76.Conrad, M. N., Dominguez, A. M., and Dresser, M.
E. (1997) Science, 276, 1252-1255.
77.Gehring, W. J., Klemenz, R., Weber, U., and
Kloter, U. (1984) EMBO J., 3, 2077-2085.
78.Hazelrigg, T., Levis, R. W., and Rubin, G. M.
(1984) Cell, 36, 469-481.
79.Karpen, G. H., and Spradling, A. C. (1992)
Genetics, 132, 737-753.
80.Levis, R. W., Ganesan, R., Houtchens, K., Tolar,
L. A., and Sheen, F. M. (1993) Cell, 75, 1083-1093.
81.Tower, J., Karpen, G. H., Craig, N., and
Spradling, A. C. (1993) Genetics, 133, 347-359.
82.Wallrath, L. L., and Elgin, S. C. R. (1995)
Gene Develop., 9, 1263-1277.
83.Allshire, R. C., Javerzat, J.-P., Redhead, N. J.,
and Cranston, G. (1994) Cell, 76, 157-169.
84.Gottschling, D. E., Aparicio, O. M., Billington,
B. L., and Zakian, V. A. (1990) Cell, 63, 751-762.
85.Nimmo, E. R., Cranston, G., and Allshire, R. C.
(1994) EMBO J., 13, 3801-3811.
86.Henikoff, S. (1992) Curr. Opin. Genet.
Dev., 2, 907-912.
87.Moehrle, A., and Paro, R. (1994) Dev.
Genet., 15, 478-484.
88.Muller, H. J. (1941) Cold Spring Harbor Symp.
Quant. Biol., 9, 151-165.
89.Spofford, J. B. (1976) in The Genetics and
Biology of Drosophila (Ashburner, M., and Novitski, E., eds.)
Academic Press, London, pp. 469-481.
90.Laurenson, P., and Rine, J. (1992) Microbiol
Rev., 56, 543-560.
91.Kurtz, S., and Shore, D. (1991) Gene
Develop., 5, 616-628.
92.Kyrion, G., Liu, K., Liu, C., and Lustig, A. J.
(1993) Gene Develop., 7, 1146-1159.
93.Loo, S., Laurenson, P., Foss, M., Dillin, A., and
Rine, J. (1995) Genetics, 141, 889-902.
94.Loo, S., Fox, C. A., Rine, J., Kobayashi, R.,
Stillman, B., and Bell, S. (1995) Mol. Biol. Cell, 6,
741-756.
95.Bell, S. P., Kobayashi, R., and Stillman, B.
(1993) Science, 262, 1844-1849.
96.Newlon, C. S., and Theis, J. F. (1993) Curr.
Opin. Genet. Dev., 3, 752-758.
97.Conrad, M. N., Wright, J. H., Wolf, A. J., and
Zakian, V. A. (1990) Cell, 63, 739-750.
98.Stavenhagen, J. B., and Zakian, V. A. (1994)
Gene Develop., 8, 1411-1422.
99.Renauld, H., Aparicio, O. M., Zierath, P. D.,
Billington, B. L., Chhablani, S. K., and Gottschling, D. E. (1993)
Gene Develop., 7, 1133-1145.
100.Shei, G. J., and Broach, J. R. (1995) Mol.
Cell Biol., 15, 3496-3506.
101.Thompson, J. S., Johnson, L. M., and Grunstein,
M. (1994) Mol. Cell Biol., 14, 446-455.
102.Aparicio, O. M., and Gottschling, D. E. (1994)
Gene Develop., 8, 1133-1146.
103.Amati, B. B., and Gasser, S. M. (1988)
Cell, 54, 967-978.
104.Lustig, A. J., Liu, C., Zhang, C., and Hanish,
J. P. (1996) Mol. Cell Biol., 16, 2483-2495.
105.Marcand, S., Buck, S. W., Moretti, P., Gilson,
E., and Shore, D. (1996) Gene Develop., 10,
1297-1309.
106.Foss, M., McNally, F. J., Laurenson, P., and
Rine, J. (1993) Science, 262, 1838-1844.
107.Fox, C. A., Loo, S., Dillin, A., and Rine, J.
(1995) Gene Develop., 9, 911-924.
108.Sandell, L. L., and Zakian, V. A. (1992)
Trends Cell Biol., 2, 10-14.
109.Tartof, K. D. (1994) Bioessays,
16, 713-714.
110.Hecht, A., Laroche, T., Strahlbolsinger, S.,
Gasser, S. M., and Grunstein, M. (1995) Cell, 80,
583-592.
111.Johnson, L. M., Kayne, P. S., Kahn, E. S., and
Grunstein, M. (1990) Proc. Natl. Acad. Sci. USA, 87,
6286-6290.
112.Kayne, P. S., Kim, U. J., Han, M., Mullen, J.
R., Yoshizaki, F., and Grunstein, M. (1988) Cell, 55,
27-39.
113.Megee, P. C., Morgan, B. A., Mittman, B. A.,
and Smith, M. M. (1990) Science, 247, 841-815.
114.Megee, P. C., Morgan, B. A., and Smith, M. M.
(1995) Gene Develop., 9, 1716-1727.
115.Park, E. C., and Szostak, J. W. (1990) Mol.
Cell. Biol., 10, 4932-4934.
116.Liu, C., Mao, X. D., and Lustig, A. J. (1994)
Genetics, 138, 1025-1040.
117.Hecht, A., Strahl, B. S., and Grunstein, M.
(1996) Nature, 383, 92-96.
118.Moretti, P., Freeman, K., Coodly, L., and
Shore, D. (1994) Gene Develop., 8, 2257-2269.
119.Shore, D. (1995) Curr. Biol., 5,
822-825.
120.Miller, A. M., and Nasmyth, K. A. (1984)
Nature, 312, 247-251.
121.Fox, A. C., Ehrenhofer-Murray, A. E., Loo, S.,
and Rine, J. (1997) Science, 276, 1547-1551.
122.Triolo, T., and Sternglanz, R. (1996)
Nature, 381, 251-253.
123.Axelrod, A., and Rine, J. (1991) Mol. Cell
Biol., 11, 1080-1091.
124.Henderson, D. S., Banga, S. S., Grigliatti, T.
A., and Boyd, J. B. (1994) EMBO J., 13, 1450-1459.
125.Aparicio, O. M., Billington, B. L., and
Gottschling, D. E. (1991) Cell, 66, 1279-1287.
126.Raghuraman, M. K., Brewer, B. J., and Fangman,
W. L. (1997) Science, 276, 806-809.
127.Diffley, J. F. X., and Stillman, B. (1989)
Nature, 342, 24.
128.Kennedy, B. K., Austriaco, N. R., Zhang, J. S.,
and Guarente, L. (1995) Cell, 80, 485-496.
129.Locke, J., Kotarski, M. A., and Tartof, K. D.
(1988) Genetics, 120, 181-198.
130.Tartof, E. D., Bishop, C., Jones, M., Hobbs, C.
A., and Locke, J. (1989) Dev. Genet., 10, 162-176.
131.Pillus, L., and Rine, J. (1989) Cell,
59, 637-647.
132.Mahoney, D. J., Marquardt, R., Shei, G. J.,
Rose, A. B., and Broach, J. R. (1991) Gene Develop., 5,
605-615.
133.Sussel, L., and Shore, D. (1991) Proc. Natl.
Acad. Sci. USA, 88, 7749-7753.
134.Sussel, L., Vannier, D., and Shore, D. (1993)
Mol. Cell. Biol., 13, 3919-3928.
135.Chien, C. T., Buck, S., Sternglanz, R., and
Shore, D. (1993) Cell, 75, 531-541.
136.Moazed, D., Kistler, A., Axelrod, A., Rine, J.,
and Johnson, A. D. (1997) Proc. Natl. Acad. Sci. USA,
94, 2186-2191.
137.Henchoz, S., De Rubertis, F., Pauli, D., and
Spierer, P. (1996) Mol. Cell Biol., 16, 5717-5725.
138.Braunstein, M., Rose, A. B., Holmes, S. G.,
Allis, C. D., and Broach, J. R. (1993) Gene Develop., 7,
592-604.
139.Gottschling, D. E. (1992) Proc. Natl. Acad.
Sci. USA, 89, 4062-4065.
140.Loo, S., and Rine, J. (1994) Science,
264, 1768-1771.
141.Singh, J., and Klar, A. J. (1992) Gene
Develop., 6, 186-196.
142.Khesin, R. B., and Leibovitch, B. A. (1978)
Mol. Gen. Genet., 162, 323-328.
143.Moore, G. D., Procunier, J. D., Cross, D. P.,
and Grigliatti, T. A. (1979) Nature, 282, 312-314.
144.Mottus, R., Reeves, R., and Grigliatti, T. A.
(1980) Mol. Gen. Genet., 178, 465-469.
145.Reuter, G., Werner, W., and Hoffmann, H. J.
(1982) Chromosoma, 85, 539-551.
146.Mullen, J. R., Kayne, P. S., Moerschell, R. P.,
Tsunasawa, S., Gribskov, M., Colavito, S. M., Grunstein, M., Sherman,
F., and Sternglanz, R. (1989) EMBO J., 8, 2067-2075.
147.De Rubertis, F., Kadosh, D., Henchoz, S.,
Pauli, D., Reuter, G., Struhl, K., and Spierer, P. (1996)
Nature, 384, 589-591.
148.Rundlett, S. E., Carmen, A. A., Kobayashi, R.,
Bavykin, S., Turner, B. M., and Grunstein, M. (1996) Proc. Natl.
Acad. Sci. USA, 93, 14503-14508.
149.Holmes, S. C., and Broach, J. R. (1996) Gene
Develop., 10, 1021-1032.
150.Wotton, D., and Shore, D. (1997) Gene
Develop., 11, 748-760.
151.Palladino, F., Laroche, T., Gilson, E., Pillus,
L., and Gasser, S. M. (1993) Cold Spring Harb. Symp. Quant.
Biol., 58, 733-746.
152.Buck, S. W., and Shore, D. (1995) Gene
Develop., 9, 370-384.
153.Marcand, S., Gilson, E., and Shore, D. (1997)
Science, 275, 986-990.
154.Virta, P. V., Morris, D. K., and Lundblad, V.
(1996) Gene Develop., 10, 3094-3104.
155.Lin, J. J., and Zakian, V. A. (1996) Proc.
Natl. Acad. Sci. USA, 93, 13760-13765.
156.Nugent, C. I., Hughes, T. R., Lue, N. F., and
Lundblad, V. (1996) Science, 274, 249-252.
157.Garvik, B., Carson, M., and Hartwell, L. (1995)
Mol. Cell Biol., 15, 6128-6138.
158.Grandin, N., Reed, S. I., and Charbonneau, M.
(1997) Gene Develop., 11, 512-527.
159.Schulz, V. P., and Zakian, V. A. (1994)
Cell, 76, 145-155.
160.Walter, M. F., Jang, C., Kasravi, B., Donath,
J., Mechler, B. M., Mason, J. M., and Biessmann, H. (1995)
Chromosoma, 104, 229-241.
161.Wu, C. T., and Howe, M. (1995) Genetics,
140, 139-181.
162.Zink, D., and Paro, R. (1995) EMBO J.,
14, 5660-5671.
163.Pirrotta, V. (1995) Curr. Opin. Genet.
Dev., 5, 466-472.
164.Vega, P. M., Venditti, S., and Di Mauro, E.
(1997) Nat. Genet., 15, 232-233.