Genetic Changes Associated with Immortalization
E. L. Duncan1 and R. R. Reddel1,2
1Children's Medical Research Institute, 214 Hawkesbury Rd., Westmead, Sydney, NSW 2145, Australia; fax: +61-2-9687-2120; E-mail: rreddel@mail.usyd.edu.au2To whom correspondence should be addressed.
Submitted July 28, 1997.
Human fibroblasts appear to have three terminal proliferative arrest (TPA) states that act as independent barriers to immortalization. The first of these TPA states is senescence, and several recent studies have shown that abrogation of p53 function permits temporary escape from senescence that ends in a poorly characterized form of arrest (referred to as p53-minus TPA) in which the pRB and p16INK4 genes appear to be involved. Abrogation of the function of both p53 and pRB (or p16INK4) results in continued proliferation until the cells enter crisis. Escape from crisis is always associated with the activation of a telomere maintenance mechanism. We also review evidence for the involvement of other genes in the immortalization process. Immortalization appears to be a complex process involving many genetic changes, not all of which are necessarily related to telomere maintenance.
KEY WORDS: terminal proliferation arrest, senescence, immortalization, telomeres, telomerase, alternative lengthening of telomeres (ALT), p53, pRB, p16INK4, p21sdi1/waf1/cip1, cGMP-dependent protein kinase II, hic-5, prohibitin, ING1, O6-methylguanine-DNA methyltransferase, DNA methyltransferase, mortalin.
One major difference between normal mammalian cells and many cancer cells is their proliferative potential. Normal mammalian cells proliferate for a limited number of population doublings (PDs) and then enter a nondividing state known as senescence, whereas many cancer cell populations appear to be able to proliferate indefinitely and are therefore referred to as immortal. There is compelling evidence that immortalization is a key aspect of the cancer cell phenotype. It has recently been shown that the activation of a telomere maintenance mechanism, either telomerase or some other unknown process, is closely associated with immortalization, and this observation has justifiably attracted great interest. Various immortalization studies, however, have shown that other genetic alterations are also required for immortalization. Some but probably not all of the genes involved may function as regulators of telomerase expression. Hence, the activation of telomere maintenance, whether through telomerase or through other mechanisms, is not the only genetic event responsible for the immortal phenotype. This review will discuss the current understanding of the other genetic events involved in the immortalization of human cells.
Human Cells Exhibit Multiple Terminal Proliferation Arrest States
Immortalization may be regarded as escape from the terminal proliferation arrest (TPA) state that eventually occurs in all normal somatic cells. Apart from the well-known phenomenon of terminal differentiation, the first viable TPA state to be discovered was senescence. The pioneering work of Hayflick and colleagues in the early 1960s revealed that normal human diploid fibroblasts proliferate only a limited number of times in vitro before they permanently cease cell division and become senescent [1]. Senescent cells can remain viable for long periods of time, indicating that the senescence process is markedly different from, and should not be confused with, any form of cell death such as apoptosis. Senescence has been observed in a variety of other human cell types, as well as in other vertebrate species, indicating that it is a general phenomenon (reviewed in [2]). The observation that the senescent phenotype is dominant over the immortal phenotype [3-6] has been interpreted by many investigators to mean that the limited lifespan of normal cells is a genetically programmed process and that immortal cells have lost the function of one or more putative senescence genes that are active in normal cells.
The second TPA state, referred to as crisis, was described in 1965 following studies of the effects of simian virus 40 (SV40) on human fibroblasts [7]. Normal human cells rarely, if ever, become immortalized spontaneously in vitro. However, they can be immortalized reliably, although at a very low frequency, following infection by DNA tumor viruses such as SV40 and papillomaviruses (reviewed in [8]). SV40 has been extensively studied, and is the most commonly used agent to induce immortalization of non-hematopoietic human cells. SV40-induced immortalization is a widely used in vitro model system for elucidating the mechanisms of limited lifespan, senescence, and immortalization (reviewed in [9]).
SV40-induced immortalization of human cells proceeds via two stages. The first stage is a temporary lifespan extension: following introduction of the SV40 early region DNA (encoding the large T antigen (LTAg) and other oncoproteins), normal human cells become morphologically transformed and their proliferative lifespan is increased by approximately 20-60 PD beyond the point at which their normal counterparts enter senescence. Usually these cells then enter crisis, during which no net population increase occurs. The second stage is the acquisition of an unlimited proliferative potential: some of the cells in crisis may resume proliferation and become immortal. Immortalization is a rare event, occurring at a frequency of between 10-5 [10] and 10-9 (E. Duncan et al., unpublished data). Similarly, human papilloma virus (HPV)-induced immortalization also proceeds via two stages [11] between the two tumor viruses.
These early studies thus revealed the existence of two TPA states, senescence which characterizes normal cells, and crisis which occurs in DNA tumor virus-transformed cells after a temporary escape from senescence. Crisis therefore appears to be the final TPA barrier that needs to be breached before cells can become immortal. The abbreviations M1 and M2 ("Mortality" stages 1 and 2) have been suggested for senescence and crisis, respectively [12], although as discussed above it must be kept in mind that these are viable arrest states that should not be confused with cell death.
Recent studies have shown the existence of at least one additional TPA state. Introduction of a dominant-negative mutant p53 gene permitted fibroblasts to proliferate for a limited number of PDs beyond the point at which their normal counterparts became senescent [13, 14]. Evidence that this effect was due to loss of wt p53 function and not due to a p53 gain-of-function mutant was provided by studies demonstrating that IIICF fibroblasts, obtained from an individual with Li--Fraumeni syndrome (LFS) and containing one wild-type (wt) and one null-mutant p53 allele, temporarily escaped from senescence and proliferated for an additional 32-35 PD when the wt allele was spontaneously deleted or mutated [15]. In most cases, these cells entered a proliferation arrest state from which they never emerged [15]. When the same heterozygous p53 wt/null-mutant IIICF cells were transformed with SV40 early region genes, the cells proliferated for an additional 60-70 PD beyond senescence before entering crisis [16] (Fig. 1). A previous study concluded that introduction of HPV-16 E6 (which binds p53 and induces its degradation) into normal human fibroblasts was not sufficient to bypass M1, but the data were consistent with a small increase in proliferative potential being induced (approximately 10 PD) [17]. The quantitative differences in the number of additional PDs achieved in these studies may reflect the completeness with which wt p53 was inactivated and/or the differing genetic backgrounds of the fibroblasts that were analyzed. These studies indicate that fibroblasts in which normal p53 function has been abrogated escape temporarily from senescence before entering a TPA state (referred to here as the "p53-minus TPA") that in terms of PD level is intermediate between senescence and crisis.
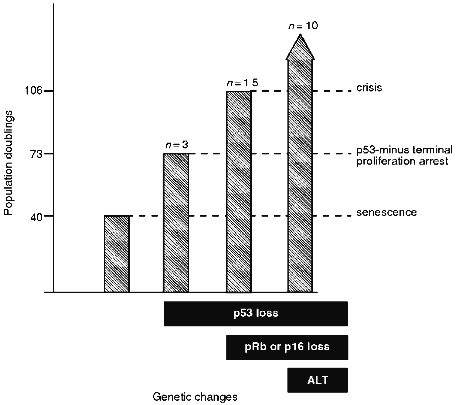
Thus, there are at least three TPA states (also referred to as lifespan checkpoints [18]) that provide a series of barriers against immortalization (Fig. 1). It is not yet clear whether there are additional arrest mechanisms in fibroblasts and whether the TPA mechanisms are different in cells of different lineages. The remainder of this review deals with the genes that are thought to be involved in these arrest states and the genetic changes required for breaching these proliferation barriers.Fig. 1. Proliferative capacity of IIICF Li--Fraumeni syndrome fibroblasts from a 36 year old female [15, 16, 62, 208]. The heterozygous p53 wt/null-mutant fibroblasts senesced at 40 population doublings (PDs) as would be expected for cells from an individual of this age. The genetic changes shown are as follows. Loss of functional p53 was due to spontaneous deletion or mutation of the wt allele or due to expression of a viral oncoprotein that inactivates p53. Loss of functional pRB was due to expression of a viral oncoprotein that inactivates pRB, and loss of p16INK4 was due to spontaneous deletion of both alleles of this gene. Immortalization was associated with activation of the ALT telomere maintenance mechanism in 10 of 10 cell lines derived from IIICF cells [124].
Genes Involved in Senescence
As outlined above, loss of wt p53 function is associated with escape from senescence. This observation suggests that p53 may be directly involved in the activation of senescence [19]. While it remains controversial as to whether the level of p53 protein changes as cells enter senescence [20-23], both the transcriptional activation and the DNA binding activities of the protein increase as cells near senescence [22, 24].
Although the trigger for this increased activity of p53 is unknown, it could be related to the telomeric shortening postulated in the telomere hypothesis of senescence [25-27]. This hypothesis is reviewed in detail in other articles in this issue, but suffice it to say here that it was proposed to account for the observation that the lifespan of cultured cells is determined by the number of PD, not by elapsed time [1], indicating that cells must possess a molecular "clock" that counts the number of times they have divided. Before the structure of chromosomes had been elucidated Olovnikov [25] hypothesized that, due to the inability of DNA polymerases to replicate chromosome ends, a portion of telomeric DNA would be lost with each cell division, and that when the telomeres shortened to a critical length the senescence process would be activated. DNA damage induces p53 expression and temporary growth arrest [28, 29], and it seems possible that shortened telomeres might be recognized as "DNA damage" resulting in p53 activation [13, 19, 30].
p53-mediated growth arrest following DNA damage is due in part to the activity of p21sdi1/waf1/cip1 (referred to hereafter as p21), a protein whose expression is induced by p53 [31] and which binds to, and inhibits the activity of, various cyclin--cyclin-dependent kinase (CDK) complexes thereby preventing phosphorylation events, including pRB phosphorylation, necessary for cell cycle progression [32, 33]. A role for p21 in senescence is supported by the observations that p21 expression is upregulated as cells approach senescence [34, 35] and that there is marked increase in complexes containing both p21 and E2F in senescent cells [36]. Thus, it is possible that in response to shortened telomeres, p53 activates p21 which then mediates the G1/S block in senescent cells.
However, while a high level of p21 may be involved in senescence, it will be important to determine whether this is sufficient for senescence of fibroblasts and whether it has the same role in cells of other lineages. p21 expression is high throughout the lifespan of human fetal adrenocortical cells, although it appeared to be even higher at senescence [37]. Interestingly, high levels of p21 expression are not necessarily maintained in cells after the onset of senescence [35]. p53 may activate other, as yet unidentified, proteins required for senescence since near-senescent fibroblasts continue proliferating following introduction of a dominant-negative p53 even though there is no decrease in p21 expression [38].
The expression of many genes has been documented to change as cells enter senescence (reviewed in [39, 40]). At present it is not known which of these are causal changes and which are secondary. However, the activities of a few genes do appear to directly effect the senescent phenotype. As already described, p21 activity appears to be involved in the p53 mediated senescence pathway. However, p21 expression can be induced independently from p53 activation [41, 42], one mechanism of which is through the basic helix-loop-helix (bHLH) transcription factors [43]. The bHLH transcription factors are inhibited by the products of the Id1 and Id2 genes [44], and the expression of these genes is downregulated in senescent cells [45]. Thus, the upregulation of p21 expression in senescent cells could alternatively be due to derepression of the bHLH transcription factors [46]. The finding that Id1 was able to complement a pRB-binding mutant of SV40 TAg to induce proliferation in senescent human fibroblasts supports a role for the bHLH transcription factors in maintaining the senescent phenotype [47].
Expression of the hydrogen peroxide-inducible gene hic-5 is upregulated as cells approach senescence and is frequently repressed in human tumor-derived cell lines [48]. Expression of exogenous hic-5 in immortal cells inhibited cell growth, and induced a senescent-like morphology concomitant with an increased level of p21 expression. However, induction of a senescent phenotype did not occur in all cells, and expression of anti-sense hic-5 did not have any effect on normal human fibroblasts [49]. Therefore, it is difficult at present to determine what role this gene plays in senescence.
The candidate tumor suppressor gene ING1, which has limited homology to retinoblastoma binding protein 2, is overexpressed in old compared to young human fibroblasts [50, 51]. While expression of an anti-sense ING1 construct in normal human fibroblasts appeared to increase their lifespan [51], it also protected cells from apoptosis [52]; thus it is as yet unknown if this gene is directly involved in senescence.
Another gene which has been implicated in the senescence process is cGMP-dependent protein kinase II (cGKII). This gene is expressed in normal cells and may be upregulated in senescent cells, although it is not clear if this has any biological significance [53]. However, cGKII expression was abolished following the introduction of SV40 T antigens into normal cells both prior to and after the cells become immortal. cGKII expression was also absent in non-virally immortalized human cell lines and tumor-derived cell lines. The lack of cGKII expression in these cell types does not appear to be through loss of p53 function since introduction of p53 into immortal cells did not induce expression of cGKII [53]. Fujii and colleagues [54] showed that cells immortalized following introduction of a temperature-sensitive TAg reverted to a mortal, senescent phenotype when grown at the non-permissive temperature; interestingly, it appeared that inhibitors of cGKII activity were able to prevent this reversion. This result suggests that induction of the senescent state requires cGKII.
Genes Involved in the p53-Minus TPA State
It is not certain which genes are responsible for proliferation arrest following the temporary escape from senescence occurring in the absence of wt p53 function, but prime candidates include those in the "RB pathway". The negative growth regulator pRB plays a role in cell cycle control throughout the lifespan of normal cells. During the G1-phase of the cell cycle pRB is underphosphorylated. In this state it binds to members of the E2F family of transcription factors preventing expression of proteins required during S-phase. pRB is phosphorylated by the cyclin D/CDK4 and cyclin D/CDK6 complexes as cells approach the G1/S boundary; this phosphorylation dissociates pRB from E2F and allows progression through S-phase (reviewed in [55, 56]). The cell cycle inhibitor p16INK4 binds to CDK4 and CDK6 in competition with cyclin D, thereby inhibiting the kinase activity of these CDKs and, hence, the phosphorylation of pRB [57-61].
A number of genetic alterations in IIICF cells that had lost wt p53 function allowed them to avoid arresting at the PD level characteristic of the p53-minus TPA state (Fig. 1). These included spontaneous loss of both p16INK4 alleles, or transfection with a mutant SV40 early region plasmid encoding a LTAg that binds pRB (but not p53) [15, 16, 62]. Thus, the RB pathway is likely to play a major role in p53-minus TPA.
In addition to inducing a TPA state in the absence of p53, it is also possible that the RB pathway normally cooperates with wt p53 in inducing senescence. pRB remains hypophosphorylated in senescent cells; therefore, the G1/S block in senescent cells may be due to this constitutively active pRB [63, 64]. This hypophosphorylation may be maintained by the elevated p16INK4 expression observed in senescent cells [35, 65, 66]. The trigger for this elevated p16INK4 expression is not yet clear. A negative feedback loop between pRB and p16INK4 has been postulated, whereby inactivation of pRB leads to increased expression of p16INK4 [57, 67, 68], but since pRB is constitutively active in senescent cells there must be some other mechanism operating to maintain a high p16INK4 expression level. p53 does not appear to be involved in this since, unlike p21 expression, p16INK4 expression is independent of p53 [66, 69]. Hara and co-workers [65] observed that p16INK4 mRNA and protein were very stable and that inactivation of pRB by SV40 LTAg did not increase the p16INK4 transcription rate enough to account for its increased mRNA level in SV40-transformed cells. They postulated that p16INK4 may simply accumulate with each population doubling, leading to a high level in old cells.
One consequence of constitutively active pRB in senescent cells is transcriptional repression of the proto-oncogene c-fos [70], whose protein product is thought to be essential for fibroblast proliferation [71]. However, c-fos repression in senescent cells is not the sole cause of their failure to proliferate, since the introduction of c-fos into senescent cells did not induce DNA synthesis [70] and induction of c-fos expression by phorbol 12-myristate 13-acetate did not increase DNA synthesis in senescent fibroblasts [72].
A question that has yet to be answered definitively is whether inactivation of the RB pathway alone is sufficient to permit normal human fibroblasts to escape from senescence. A mutant SV40 TAg, that binds pRB but not p53, was unable to induce lifespan extension in normal fibroblasts [16, 73], but was able to do so in the context of spontaneous loss of the wt p53 allele in IIICF LFS cells [16] (Fig. 1). Another study found that antisense oligomer-induced inhibition of pRB synthesis increased the proliferation rate of human fibroblasts but did not permit their escape from senescence [74]. A seemingly contradictory result was that normal human fibroblasts incubated with an antisense pRB oligomer exhibited an extended lifespan of about 10 PD beyond the point at which the controls senesced, but in this study an antisense p53 oligomer had no effect whereas the combination of antisense pRB and antisense p53 oligomers resulted in an extended lifespan of about 20 PD [75]. Furthermore, introduction of the gene for the HPV-16 E7 protein (which binds to pRB) into normal human fibroblasts induced a small extension of lifespan [17]. Thus, whether pRB inactivation may result in temporary escape from senescence requires further analysis. It is possible that inactivation of pRB in the presence of wt p53 results in increased cell death which counterbalances any effect it might have in permitting an increase in proliferation potential. There also seem to be major cell lineage differences in the response to pRB inactivation [76].
Regardless of the precise roles the p53 and pRB pathways play in inducing TPA states, their importance is supported by the well-known observation that these pathways are frequently disrupted in tumor derived cell lines and primary tumors. p53 is the most commonly mutated gene in human cancer [77-81] and a mutant p53 gene is inherited in many LFS cancer kindreds [82, 83]. Wild-type pRB expression is lost in many cancers [84-90] and is the familial gene responsible for retinoblastoma [91]. Loss of p16INK4 expression has been observed in human tumor-derived cell lines as well as in primary tumor samples [92-98], and it was identified as a familial melanoma gene [99, 100]. Inactivation or loss of p16INK4 appears to be functionally equivalent to inactivation or loss of pRB: for example, all immortal cell lines examined to date, including in vitro immortalized and tumor derived cell lines, and primary tumors, have inactive pRB or inactive p16, but not both [62, 67, 95, 101-108]. Overexpression of cyclin D1, and/or overexpression of cdk4, is associated with human cancer and is presumably equivalent to loss of p16INK4 or pRB expression [109-119].
Genes Involved in the Crisis TPA State
The additive effect of loss of function in the p53 and pRB pathways is to permit cells to proliferate until the final barrier of crisis is reached. As already noted, escape from senescence and proliferation until crisis was first documented in studies of SV40-transformed human fibroblasts [7] and many subsequent studies have shown that this effect is dependent on the ability of SV40 LTAg to bind to p53 and pRB (reviewed in [9]). The existence of the crisis TPA state in such cells clearly indicates that functional inactivation of p53 and pRB is insufficient for immortalization. Escape from crisis occurs at a low frequency, presumably as a result of deletions, mutations, or epigenetic alterations of the genes that would otherwise ensure that crisis occurs. The identity of these genes is essentially unknown, but some interesting candidates are emerging, primarily from analyses of cells that have escaped from crisis. These analyses are summarized below and include complementation via somatic cell hybridization, microcell mediated chromosome transfer, and gene expression studies. But first, some comments on telomeres are needed to place these other genetic changes in context.
A number of studies have shown that cells escaping from senescence continue to undergo telomere shortening until they enter the p53-minus TPA state [15] or crisis [120-122]. This raises the possibility that crisis may be triggered in response to telomere shortening, which is consistent with the observation that cells escaping from crisis have stabilized telomere lengths [15, 120-124]. In most cases this is associated with expression of telomerase, a specialized reverse transcriptase that maintains telomere length [125]. Activity of this enzyme has been detected in the great majority of tumor derived cell lines and primary tumor samples [126-129]. In some cases, telomere stabilization occurs due to one or more alternative mechanisms, ALT (alternative lengthening of telomeres) (reviewed in [130] and elsewhere in this issue). ALT cell lines are characterized by the combination of no detectable telomerase activity and the presence of telomeres of very heterogeneous length, ranging from very short to much longer than normal [124]. To date, no exceptions have been found to the rule that immortalization of cells in vitro is associated with activation of a telomere maintenance mechanism. This provides much stronger evidence, albeit circumstantial, that shortened telomeres have a major role in crisis than is currently available for their proposed role in senescence. It follows from this that the genes responsible for the crisis TPA state are likely to include those involved in responding to critically shortened telomeres, and that the genetic changes involved in escape from crisis may include loss of function of the putative repressors of telomere maintenance processes.
It is not yet clear how telomerase expression is controlled, especially since telomerase is a multisubunit enzyme encoded by multiple genes not all of which have been identified and cloned. The telomerase RNA component was the first mammalian telomerase subunit to be identified [131, 132]. The human telomerase RNA gene, hTR, is expressed in both mortal and immortal human cells [133], making it unlikely that upregulation of hTR is the major means of controlling telomerase activity, although increased hTR gene dosage or expression may contribute to increased telomerase activity in cancer cells [134]. A protein component of mammalian telomerase, TP1/TLP1, was recently identified [135, 136]; however, it is not yet known if and how expression of this protein changes during the immortalization process. TP1/TLP1 is homologous to the Tetrahymena 80 kD telomerase subunit; a mammalian homolog for the Tetrahymena 95 kD telomerase subunit remains to be identified. Telomerase proteins with reverse transcriptase motifs and bearing no homology to the 80 and 95 kD proteins identified in Tetrahymena have been identified in the ciliate Euplotes aediculatus and in yeast [137] raising the possibility that there is more than one type of telomerase and/or that the subunit structure of telomerase is more complex than previously realized.
Immortalization may not simply be associated with switching of telomerase activity from off to on. Some normal somatic cells, despite exhibiting shortening telomeres when passaged in vitro, already have a low level of telomerase activity [129, 138-142]. In addition, some virally transformed cells in the pre-immortal extended lifespan phase have detectable telomerase activity [143]. There may be a threshold level of telomerase activity which is required for telomere maintenance [144] and which can be attained via dysregulation of the mechanisms involved in the temporal control of telomerase activity during development [145] or those involved in the regulation of telomerase activity following growth and/or differentiation signals [139, 144, 146-157].
There is preliminary evidence indicating that at least some of the genetic changes required for escape from crisis involve repressors of telomerase or ALT expression. Somatic cell hybridization studies have shown that the senescent phenotype that is the mortal phenotype is dominant over the immortal phenotype [3-6], so it seems likely that immortal cells have lost the function of one or more putative senescence genes that are active in normal cells. It may thus be expected that the expression of a telomere maintenance mechanism in immortalized cells will be found to be due to loss of one or more telomerase gene repressors present in normal cells rather than due to an activation event [26]. This prediction is consistent with the observation that hybrids between normal and immortal cells have only 1% of the telomerase activity of the immortal parent cells [158].
Cell hybridization studies between different immortal cell lines initially identified four complementation groups for immortalization [159], and further studies raise the possibility that there may be more (E. Moy et al., unpublished data), suggesting that there may be at least four genes involved in repressing the mechanisms of telomere maintenance. Interestingly, telomerase-positive immortal cells have been assigned to all four immortalization complementation groups, while ALT-positive cell lines have been assigned to two of these groups [105]. Thus, if each complementation group represents a mutation in one TPA gene then, in at least two cases, mutation of the same TPA gene can lead to derepression of either telomerase or ALT. This suggests that the same pathways may be involved in derepression of both telomere maintenance mechanisms. An alternative view is that the complementation groups are independent of either telomere maintenance mechanism. According to this view, immortalization requires derepression of a telomere maintenance mechanism and the loss of a TPA gene corresponding to the complementation group.
Many Human Chromosomes Encode Putative TPA Genes
Microcell-mediated monochromosome transfer experiments have shown that when various normal human chromosomes are transferred into immortal human cell lines the cells proliferate for a number of PDs before entering an uncharacterized TPA state. The chromosomes that are capable of doing this presumably contain one or more genes involved in TPA and include 1q [160], 2 [161], 3 [162], 4 [163], 6q [164], 7 [165], 11 [166], 18 [167], and X [168, 169]. The involvement of chromosome 6q in immortalization was determined from the observation that this region of the genome is frequently lost following immortalization of SV40 transformed and spontaneously immortalized human cells [170-172]; DNA polymorphism analysis localized the region involved to 6q26-6q27 [173]. Chromosome 17q24-q25 causes growth arrest in human breast cancer cells [174], alteration of chromosome 18 is associated with immortalization of human T-lymphocytes [175], and amplification of chromosome 20q13.2 is associated with immortalization of human HPV-16 E7 transformed uroepithelial cells [176].
Three chromosomes have been identified as inducing TPA in immortal cells from a particular complementation group. Chromosome 4 induces TPA in cells assigned to complementation group B but not in cells assigned to groups A, C, or D [163]. Similarly, chromosome 1 specifically induces TPA in cells assigned to group C [160], and chromosome 7 induces TPA in cells assigned to group D [177]. In addition, chromosome 6 induces TPA in a cell line SVHF39 [164] that has been assigned to group A [159]. The finding that more chromosomes carry putative TPA genes than there are complementation groups could indicate that there are more than four complementation groups (E. Moy et al., unpublished data), or that inactivation of multiple pathways is involved in breaching the barriers to unlimited proliferation [178].
Some Genes Have an Altered Expression Pattern Following Immortalization
One gene which may be involved in immortalization is prohibitin, which was originally identified through its ability to inhibit DNA synthesis in human diploid fibroblasts [179, 180]. Prohibitin appears to be a ubiquitous protein localized to the inner mitochondrial membrane [181] whose expression is maximal at G1/S [182, 183]. Prohibitin mRNA levels are lower in senescent fibroblasts compared to young fibroblasts [182]. Prohibitin protein exists in more than one form, presumably due to post-translational modification. One of these forms comprises 10% of the total prohibitin and is absent from senescent cells [183]. Immortal cells appear to have an increased copy number of the prohibitin gene, and a corresponding increased level of one prohibitin mRNA isoform and protein [184, 185]. These observations appear somewhat paradoxical given that prohibitin appears to be a growth suppressor. The protein isoform missing in senescent cells might be the inactive form of prohibitin, so that senescent cells have constitutively active prohibitin [183]. When injected into immortal cell lines, prohibitin mRNA inhibits DNA synthesis only in cell lines assigned to complementation group B [184, 185], suggesting that this gene is specifically involved in one mechanism of human cell immortalization. Two alleles have been identified for prohibitin, one of which has an additional EcoR1 site. The normal cells examined were heterozygous for the two alleles, but cells assigned to complementation group B were homozygous for the allele containing the additional EcoR1 site [184, 185]. In addition, the sequence of the 3´ untranslated region of the mRNA isoform overexpressed in immortal cells was found to be wild-type in all cells except those from complementation group B. This 3´ untranslated region was shown to be responsible for the growth inhibitory activity of prohibitin mRNA [185]. These results suggest that mutation and loss of heterozygosity of the prohibitin gene are involved in the immortalization of cell lines assigned to group B, and that cell lines assigned to the other complementation groups are insensitive to the negative effect of prohibitin mRNA. However, prohibitin is unlikely to be the primary TPA gene for complementation group B since microcell mediated chromosome transfer experiments identified chromosome 4 as being associated with complementation group B [163]. Prohibitin gene is located on chromosome 17 and is thus more likely to be involved downstream in the complementation group B immortalization process [181].
The DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) is expressed in normal human cells; however, expression is frequently lost in immortal cell lines. In SV40-transformed cells, loss of MGMT expression occurs only after escape from crisis and is associated with altered methylation of the gene [186]. It is not clear whether MGMT is directly involved in the immortalization process; it may be that MGMT- cells in a population of MGMT+ cells have a higher rate of DNA mutation due to absence of the gene, and hence are more likely to become immortal. Alternatively, since the MGMT gene is located at the telomere of chromosome 10, its changed expression pattern may reflect a change in telomere organization during immortalization [186].
Immortalization of cells is associated with resistance to the potent protein kinase inhibitor staurosporine [187]. Interestingly, virally-transformed cells exhibiting extended lifespan are as sensitive to staurosporine as normal cells; these cells only acquire staurosporine resistance following escape from crisis [188]. This suggests that genetic events resulting in altered protein kinase activity are associated with immortalization.
The antiproliferative protein mortalin was identified as being present in normal, but absent in immortal, mouse cells [189]. In normal human cells, mortalin is distributed throughout the cytoplasm. However, immortal human cells exhibit one of four characteristic patterns: anti-mortalin antibody staining shows either a granular juxtanuclear pattern, a gradient from the nucleus to the cell membrane, a juxtanuclear arch pattern, or a fibrous perinuclear pattern [190, 191]. Interestingly, the mortalin staining pattern in different human cells from various origins correlates with their complementation group for immortalization, suggesting that different alterations in mortalin are involved in different mechanisms of immortalization [191].
The activities of Orpheus, a putative transcriptional repressor, and Pluto, a putative transcriptional activator, have been documented to change during in vitro cellular aging and following immortalization [192]. These transcription factors appear to be involved in type I collagenase regulation. It is not yet clear how significant a role these putative transcription factors play in senescence and immortalization.
The expression of several cytoskeletal genes, including genes of talin, vinculin, vimentin, alpha-actinin, tropomyosin 1, lamin A and lamin C, and/or the localization of their protein products have been reported to change following immortalization [193]. These authors speculated that, since the intermediate filaments may be involved in telomere binding, alterations in these proteins may play a significant role in telomere maintenance during immortalization.
DNA Methylation May Play a Role in Senescence and Immortalization
DNA can be methylated at CpG by the enzyme DNA (cytosine-5-)methyltransferase (DNA MTase) and methylation of promoter sequences usually results in reduced gene expression [194]. In cultured fibroblasts, the level of 5-methylcytosine (5mC) and of DNA MTase activity decreased with increased PDs [195, 196]. Interestingly, the rate of 5mC decline was found to be directly proportional to a species lifespan of the cultivated cell population: mouse cells had the highest rate of decline and the shortest lifespan, human cells had the lowest rate of decline and the longest lifespan, and hamster cells had an intermediate lifespan and rate of decline. Furthermore, treatment of young cells with demethylating agents reduced their lifespan [197]. Following immortalization, 5mC levels appear to be stabilized [195]. Some immortal cells have increased expression of DNA MTase [198, 199]. Aberrant DNA methylation may play a role in immortalization by repressing the expression of genes required for senescence [200].
The dynamics of 5mC level and telomere length during cellular lifespan and following immortalization are very similar. Progressive alteration of methylation has been proposed as a possible counting mechanism for the number of PDs occurring during development [201]. However, there is as yet no evidence that decreasing 5mC content acts as the counting mechanism that determines the onset of senescence [202]. Unlike telomere shortening, which continues in cells which have escaped from senescence, 5mC levels appear to stabilize in SV40-transformed cells prior to their becoming immortal [203]. In addition, DNA MTase activity in pre- and post-immortal SV40-transformed cells is the same as that in young cells [196]. Thus, while decreasing 5mC levels may be involved in triggering senescence and while 5mC maintenance may be necessary for the immortal phenotype, it appears that activation of the 5mC maintenance mechanism is not sufficient for immortalization.
The Requirement for Multiple Genetic Changes in Immortalized Cells
When cells transduced with the SV40 genes have escaped from crisis and become immortalized, they have presumably undergone the genetic or epigenetic changes required for overcoming the crisis TPA state. A number of laboratories have addressed the question of whether the continued proliferation of such cells is dependent on continued expression of the viral oncoproteins that permitted the initial escape from the earlier TPA state, senescence. In two laboratories, immortal human cell lines were generated using a temperature sensitive mutant of SV40 LTAg [204, 205]. These mutant LTAgs were functional at 35°C, but non-functional at 39°C. The immortal cells grew normally at 35°C, but when incubated at 39°C they exhibited defective colony formation, rapid cessation of growth, reduced number of cells in S-phase, and a progressive loss of viability. In a similar experiment, immortal human cells were generated following transfection of normal fibroblasts with a plasmid encoding SV40 LTAg under the control of the mouse mammary tumor virus promoter. In this system, LTAg was only expressed when the cells were grown in dexamethasone. When the immortal cells were grown in the absence of dexamethasone they entered G1 arrest [12] and acquired a senescent phenotype as assayed by both morphological and biochemical markers [206]. Introduction of an antisense LTAg gene into SV40-transformed immortal human fibroblasts caused >70% reduction in LTAg expression and partial reversion of the cells to the senescent phenotype [207]. These experiments all appear to indicate that the continued expression of SV40 LTAg, the protein that is able to inactivate the senescence TPA mechanism, is essential even after the crisis TPA mechanism has been inactivated.
The simplest interpretation of these data is that each of the TPA mechanisms is independently capable of acting as an effective barrier to unlimited proliferation. An alternative explanation might be that because of the number of genetic changes required, the probability of breaching the crisis TPA mechanism is normally so low that it is unlikely to occur in the absence of prior genetic changes that, first, permit increased genomic instability and, second, expand the target cell population. In the process of accumulating the required genetic changes, the amount of collateral damage may be sufficiently large as to preclude proliferation if normal p53 function is experimentally restored. When the appropriate experimental tools become available, it will therefore be very interesting to determine whether telomerase or an alternative telomere maintenance mechanism can be activated in the absence of other genetic damage, and if so whether any other genetic changes are essential for immortalization.
A number of genetic changes within proliferating cells are associated with escape from the permanent arrest states that act as barriers to immortalization. The best characterized of these changes are the inactivation of p53 and the pRB pathway and the activation of a telomere maintenance mechanism. Other genetic changes have been shown to accompany escape from senescence or escape from crisis, and chromosome transfer studies suggest that there are other important genetic changes that have not yet been characterized. Some of these changes may be required for derepression of one or more of the telomere maintenance mechanisms, and some may be a consequence of telomeric alterations, but for others there is no obvious connection with the requirement for telomere maintenance. Determining the relevance of each of these changes and the causal relationships among them continues to be a major challenge in the study of immortalization.
Work in the authors' laboratory was supported by the Carcinogenesis Fellowship of the New South Wales Cancer Council and the National Health and Medical Research Council of Australia project grants.
LITERATURE CITED
1. Hayflick, L., and Moorhead, P. S. (1961) Exp.
Cell Res., 25, 585-621.
2. Cristofalo, V. J., and Pignolo, R. J. (1993)
Physiol. Rev., 73, 617-638.
3. Bunn, C. L., and Tarrant, G. M. (1980) Exp.
Cell Res., 127, 385-396.
4. Muggleton-Harris, A. L., and DeSimone, D. W.
(1980) Somatic Cell Genet., 6, 689-698.
5. Pereira-Smith, O. M., and Smith, J. R. (1981)
Somatic Cell Genet., 7, 411-421.
6. Pereira-Smith, O. M., and Smith, J. R. (1983)
Science, 221, 964-966.
7. Girardi, A. J., Jensen, F. C., and Koprowski, H.
(1965) J. Cell. Comp. Physiol., 65, 69-84.
8. Linder, S., and Marshall, H. (1990) Exp. Cell
Res., 191, 1-7.
9. Bryan, T. M., and Reddel, R. R. (1994) Crit.
Rev. Oncogenesis, 5, 331-357.
10. Shay, J. W., Van Der Haegen, B. A., Ying, Y.,
and Wright, W. E. (1993) Exp. Cell Res., 209, 45-52.
11. De Silva, R., Whitaker, N. J., Rogan, E. M., and
Reddel, R. R. (1994) Exp. Cell Res., 213, 418-427.
12. Wright, W. E., Pereira-Smith, O. M., and Shay,
J. W. (1989) Mol. Cell. Biol., 9, 3088-3092.
13. Bond, J. A., Wyllie, F. S., and Wynford-Thomas,
D. (1994) Oncogene, 9, 1885-1889.
14. Fushimi, K., Iijima, M., Gao, C., Kondo, T.,
Tsuji, T., Hashimoto, T., Mihara, K., and Namba, M. (1997) Int. J.
Cancer, 70, 135-140.
15. Rogan, E. M., Bryan, T. M., Hukku, B., Maclean,
K., Chang, A. C.-M., Moy, E. L., Englezou, A., Warneford, S. G.,
Dalla-Pozza, L., and Reddel, R. R. (1995) Mol. Cell. Biol.,
15, 4745-4753.
16. Maclean, K., Rogan, E. M., Whitaker, N. J.,
Chang, A. C.-M., Rowe, P. B., Dalla-Pozza, L., Symonds, G., and Reddel,
R. R. (1994) Oncogene, 9, 719-725.
17. Shay, J. W., Wright, W. E., Brasiskyte, D., and
Van Der Haegen, B. A. (1993) Oncogene, 8, 1407-1413.
18. Wynford-Thomas, D. (1997) Eur. J. Cancer
(A), 33A, 716-726.
19. Wynford-Thomas, D. (1996) J. Pathol.,
180, 118-121.
20. Kulju, K. S., and Lehman, J. M. (1995) Exp.
Cell Res., 217, 336-345.
21. Afshari, C. A., Vojta, P. J., Annab, L. A.,
Futreal, P. A., Willard, T. B., and Barrett, J. C. (1993) Exp. Cell
Res., 209, 231-237.
22. Atadja, P., Wong, H., Garkavtsev, I., Veillette,
C., and Riabowol, K. (1995) Proc. Natl. Acad. Sci. USA,
92, 8348-8352.
23. Vaziri, H., and Benchimol, S. (1996) Exp.
Gerontol., 31, 295-301.
24. Bond, J., Haughton, M., Blaydes, J., Gire, V.,
Wynford-Thomas, D., and Wyllie, F. (1996) Oncogene, 13,
2097-2104.
25. Olovnikov, A. M. (1971) Dokl. Akad. Nauk
SSSR, 201, 1496-1499.
26. Harley, C. B. (1991) Mutat. Res.,
256, 271-282.
27. Harley, C. B., Vaziri, H., Counter, C. M., and
Allsopp, R. C. (1992) Exp. Gerontol., 27, 375-382.
28. Kastan, M. B., Onyekwere, O., Sidransky, D.,
Vogelstein, B., and Craig, R. W. (1991) Cancer Res., 51,
6304-6311.
29. Lane, D. P. (1992) Nature, 358,
15-16.
30. De Lange, T. (1994) Proc. Natl. Acad. Sci.
USA, 91, 2882-2885.
31. El-Deiry, W. S., Tokino, T., Velculescu, V. E.,
Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E.,
Kinzler, K. W., and Vogelstein, B. (1993) Cell, 75,
817-825.
32. Harper, J. W., Adami, G. R., Wei, N., Keyomarsi,
K., and Elledge, S. J. (1993) Cell, 75, 805-816.
33. Dulic, V., Kaufmann, W. K., Wilson, S. J.,
Tlsty, T. D., Lees, E., Harper, J. W., Elledge, S. J., and Reed, S. I.
(1994) Cell, 76, 1013-1023.
34. Noda, A., Ning, Y., Venable, S. F.,
Pereira-Smith, O. M., and Smith, J. R. (1994) Exp. Cell Res.,
211, 90-98.
35. Alcorta, D. A., Xiong, Y., Phelps, D., Hannon,
G., Beach, D., and Barrett, J. C. (1996) Proc. Natl. Acad. Sci.
USA, 93, 13742-13747.
36. Afshari, C. A., Nichols, M. A., Xiong, Y., and
Mudryj, M. (1996) Cell Growth Differ., 7, 979-988.
37. Yang, L. Q., Didenko, V. V., Noda, A., Bilyeu,
T. A., Darlington, G. J., Smith, J. R., and Hornsby, P. J. (1995)
Exp. Cell Res., 221, 126-131.
38. Bond, J. A., Blaydes, J. P., Rowson, J.,
Haughton, M. F., Smith, J. R., Wynford-Thomas, D., and Wyllie, F. S.
(1995) Cancer Res., 55, 2404-2409.
39. Cristofalo, V. J., Pignolo, R. J., Cianciarulo,
F. L., DiPaolo, B. R., and Rotenberg, M. O. (1992) Exp.
Gerontol., 27, 429-432.
40. Meyyappan, M., Atadja, P. W., and Riabowol, K.
T. (1996) Biol. Signals, 5, 130-138.
41. Johnson, M., Dimitrov, D., Vojta, P. J.,
Barrett, J. C., Noda, A., Pereira-Smith, O. M., and Smith, J. R. (1994)
Mol. Carcinog., 11, 59-64.
42. Tahara, H., Sato, E., Noda, A., and Ide, T.
(1995) Oncogene, 10, 835-840.
43. Halevy, O., Novitch, B. G., Spicer, D. B.,
Skapek, S. X., Rhee, J., Hannon, G. J., Beach, D., and Lassar, A. B.
(1995) Science, 267, 1018-1021.
44. Sun, X.-H., Copeland, N. G., Jenkins, N. A., and
Baltimore, D. (1991) Mol. Cell. Biol., 11,
5603-5611.
45. Hara, E., Yamaguchi, T., Nojima, H., Ide, T.,
Campisi, J., Okayama, H., and Oda, K. (1994) J. Biol. Chem.,
269, 2139-2145.
46. Campisi, J., Dimri, G. P., Nehlin, J. O.,
Testori, A., and Yoshimoto, K. (1996) Exp. Gerontol.,
31, 7-12.
47. Hara, E., Uzman, J. A., Dimri, G. P., Nehlin, J.
O., Testori, A., and Campisi, J. (1996) Dev. Genet., 18,
161-172.
48. Shibanuma, M., Mashimo, J., Kuroki, T., and
Nose, K. (1994) J. Biol. Chem., 269, 26767-26774.
49. Shibanuma, M., Mochizuki, E., Maniwa, R.,
Mashimo, J.-I., Nishiya, N., Imai, S.-I., Takano, T., Oshimura, M., and
Nose, K. (1997) Mol. Cell. Biol., 17, 1224-1235.
50. Garkavtsev, I., Kazarov, A., Gudkov, A., and
Riabowol, K. (1996) Nature Genet., 14, 415-420.
51. Garkavtsev, I., and Riabowol, K. (1997) Mol.
Cell. Biol., 17, 2014-2019.
52. Helbing, C. C., Veillette, C., Riabowol, K.,
Johnston, R. N., and Garkavtsev, I. (1997) Cancer Res.,
57, 1255-1258.
53. Fujii, M., Ogata, T., Takahashi, E., Yamada, K.,
Nakabayashi, K., Oishi, M., and Ayusawa, D. (1995) FEBS Lett.,
375, 263-267.
54. Fujii, M., Ide, T., Wadhwa, R., Tahara, H.,
Kaul, S. C., Mitsui, Y., Ogata, T., Oishi, M., and Ayusawa, D. (1995)
Oncogene, 11, 627-634.
55. Nevins, J. R. (1992) Science,
258, 424-429.
56. Dyson, N. (1994) J. Cell Sci.,
S18, 81-87.
57. Serrano, M., Hannon, G. J., and Beach, D. (1993)
Nature, 366, 704-707.
58. Tam, S. W., Shay, J. W., and Pagano, M. (1994)
Cancer Res., 54, 5816-5820.
59. Schulze, A., Zerfass, K., Spitkovsky, D.,
Henglein, B., and Jansen-Dürr, P. (1994) Oncogene,
9, 3475-3482.
60. Della Ragione, F., Russo, G. L., Oliva, A.,
Mercurio, C., Mastropietro, S., Della Pietra, V., and Zappia, V. (1996)
J. Biol. Chem., 271, 15942-15949.
61. Serrano, M., Gómez-Lahoz, E., DePinho, R.
A., Beach, D., and Bar-sagi, D. (1995) Science, 267,
249-252.
62. Noble, J. R., Rogan, E. M., Neumann, A. A.,
Maclean, K., Bryan, T. M., and Reddel, R. R. (1996) Oncogene,
13, 1259-1268.
63. Stein, G. H., Beeson, M., and Gordon, L. (1990)
Science, 249, 666-669.
64. Futreal, P. A., and Barrett, J. C. (1991)
Oncogene, 6, 1109-1113.
65. Hara, E., Smith, R., Parry, D., Tahara, H.,
Steven, S., and Peters, G. (1996) Mol. Cell. Biol., 16,
859-867.
66. Reznikoff, C. A., Yeager, T. R., Belair, C. D.,
Savelieva, E., Puthenveettil, J. A., and Stadler, W. M. (1996)
Cancer Res., 56, 2886-2890.
67. Li, Y., Nichols, M. A., Shay, J. W., and Xiong,
Y. (1994) Cancer Res., 54, 6078-6082.
68. Khleif, S. N., DeGregori, J., Yee, C. L.,
Otterson, G. A., Kaye, F. J., Nevins, J. R., and Howley, P. M. (1996)
Proc. Natl. Acad. Sci. USA, 93, 4350-4354.
69. Musgrove, E. A., Lilischkis, R., Cornish, A. L.,
Lee, C. S. L., Setlur, V., Seshadri, R., and Sutherland, R. L. (1995)
Int. J. Cancer, 63, 584-591.
70. Campisi, J. (1992) Exp. Gerontol.,
27, 397-401.
71. Cohen, D. R., and Curran, T. (1989)
Oncogenesis, 1, 65-88.
72. De Tata, V., Ptasznik, A., and Cristofalo, V. J.
(1993) Exp. Cell Res., 205, 261-269.
73. Lin, J.-Y., and Simmons, D. T. (1991) J.
Virol., 65, 6447-6453.
74. Strauss, M., Hering, S., Lieber, A., Herrmann,
G., Griffin, B. E., and Arnold, W. (1992) Oncogene, 7,
769-773.
75. Hara, E., Tsurui, H., Shinozaki, A., Nakada, S.,
and Oda, K. (1991) Biochem. Biophys. Res. Commun., 179,
528-534.
76. Foster, S. A., and Galloway, D. A. (1996)
Oncogene, 12, 1773-1779.
77. Cheng, J., and Haas, M. (1990) Mol. Cell.
Biol., 10, 5502-5509.
78. Levine, A. J., Momand, J., and Finlay, C. A.
(1991) Nature, 351, 453-456.
79. Hollstein, M., Sidransky, D., Vogelstein, B.,
and Harris, C. C. (1991) Science, 253, 49-53.
80. Caron de Fromentel, C., and Soussi, T. (1992)
Genes Chrom. Cancer, 4, 1-15.
81. Greenblatt, M. S., Bennett, W. P., Hollstein,
M., and Harris, C. C. (1994) Cancer Res., 54,
4855-4878.
82. Malkin, D., Li, F. P., Strong, L. C., Fraumeni,
J. F., Jr., Nelson, C. E., Kim, D. H., Kassel, J., Gryka, M. A.,
Bischoff, F. Z., Tainsky, M. A., and Friend, S. H. (1990)
Science, 250, 1233-1238.
83. Srivastava, S., Zou, Z., Pirollo, K., Blattner,
W., and Chang, E. H. (1990) Nature, 348, 747-749.
84. Friend, S. H., Horowitz, J. M., Gerber, M. R.,
Wang, X.-F., Bogenmann, E., Li, F. P., and Weinberg, R. A. (1987)
Proc. Natl. Acad. Sci. USA, 84, 9059-9063.
85. Harbour, J. W., Lai, S.-L., Whang-Peng, J.,
Gazdar, A. F., Minna, J. D., and Kaye, F. J. (1988) Science,
241, 353-357.
86. T´Ang, A., Varley, J. M., Chakraborty, S.,
Murphree, A. L., and Fung, Y.-K. T. (1988) Science, 242,
263-266.
87. Bookstein, R., Rio, P., Madreperla, S. A., Hong,
F., Allred, C., Grizzle, W. E., and Lee, W.-H. (1990) Proc. Natl.
Acad. Sci. USA, 87, 7762-7766.
88. Cheng, J., Scully, P., Shew, J.-Y., Lee, W.-H.,
Vila, V., and Haas, M. (1990) Blood, 75, 730-735.
89. Horowitz, J. M., Park, S.-H., Bogenmann, E.,
Cheng, J.-C., Yandell, D. W., Kaye, F. J., Minna, J. D., Dryja, T. P.,
and Weinberg, R. A. (1990) Proc. Natl. Acad. Sci. USA,
87, 2775-2779.
90. Bookstein, R., and Lee, W.-H. (1991) Crit.
Rev. Oncogenesis, 2, 211-227.
91. Weinberg, R. A. (1991) Science,
254, 1138-1146.
92. Kamb, A., Gruis, N. A., Weaver-Feldhaus, J.,
Liu, Q., Harchman, K., Tavtigian, S. V., Stockert, E., Day, R. S., III,
Johnson, B. E., and Skolnick, M. H. (1994) Science, 264,
436-440.
93. Mori, T., Miura, K., Aoki, T., Nishihira, T.,
Mori, S., and Nakamura, Y. (1994) Cancer Res., 54,
3396-3397.
94. Nobori, T., Miura, K., Wu, D. J., Lois, A.,
Takabayashi, K., and Carson, D. A. (1994) Nature, 368,
753-756.
95. Okamoto, A., Demetrick, D. J., Spillare, E. A.,
Hagiwara, K., Hussain, S. P., Bennett, W. P., Forrester, K., Gerwin,
B., Serrano, M., Beach, D. H., and Harris, C. C. (1994) Proc. Natl.
Acad. Sci. USA, 91, 11045-11049.
96. Merlo, A., Herman, J. G., Mao, L., Lee, D. J.,
Gabrielson, E., Burger, P. C., Baylin, S. B., and Sidransky, D. (1995)
Nature Med., 1, 686-692.
97. Siebert, R., Willers, C. P., Schramm, A., Fossa,
A., Dresen, I. M. G., Uppenkamp, M., Nowrousian, M. R., Seeber, S., and
Opalka, B. (1995) Br. J. Haematol., 91, 350-354.
98. Woloschak, M., Yu, A., Xiao, J., and Post, K. D.
(1996) Cancer Res., 56, 2493-2496.
99. Kamb, A., Shattuck-Eidens, D., Eeles, R., Liu,
Q., Gruis, N. A., Ding, W., Hussey, C., Tran, T., Miki, Y.,
Weaver-Feldhaus, J., McClure, M., Aitken, J. F., Anderson, D. E.,
Bergman, W., Frants, R., Goldgar, D. E., Green, A., MacLennan, R.,
Martin, N. G., Meyer, L. J., Youl, P., Zone, J. J., Skolnick, M. H.,
and Cannon-Albright, L. A. (1994) Nature Genet., 8,
22-26.
100. Gruis, N. A., Van der Velden, P. A.,
Sandkuijl, L. A., Prins, D. E., Weaver-Feldhaus, J., Kamb, A., Bergman,
W., and Frants, R. R. (1995) Nature Genet., 10,
351-353.
101. Otterson, G. A., Kratzke, R. A., Coxon, A.,
Kim, Y. W., and Kaye, F. J. (1994) Oncogene, 9,
3375-3378.
102. Aagaard, L., Lukas, J., Bartkova, J.,
Kjerulff, A.-A., Strauss, M., and Bartek, J. (1995) Int. J.
Cancer, 61, 115-120.
103. Parry, D., Bates, S., Mann, D. J., and Peters,
G. (1995) EMBO J., 14, 503-511.
104. Shapiro, G. I., Edwards, C. D., Kobzik, L.,
Godleski, J., Richards, W., Sugarbaker, D. J., and Rollins, B. J.
(1995) Cancer Res., 55, 505-509.
105. Whitaker, N. J., Bryan, T. M., Bonnefin, P.,
Chang, A. C.-M., Musgrove, E. A., Braithwaite, A. W., and Reddel, R. R.
(1995) Oncogene, 11, 971-976.
106. Yeager, T., Stadler, W., Belair, C.,
Puthenveettil, J., Olopade, O., and Reznikoff, C. (1995) Cancer
Res., 55, 493-497.
107. Sakaguchi, M., Fujii, Y., Hirabayashi, H.,
Yoon, H. E., Komoto, Y., Oue, T., Kusafuka, T., Okada, A., and Matsuda,
H. (1996) Int. J. Cancer, 65, 442-445.
108. Ueki, K., Ono, Y., Hensen, J. W., Efird, J.
T., Von Deimling, A., and Louis, D. N. (1996) Cancer Res.,
56, 150-153.
109. Lammie, G. A., and Peters, G. (1991) Cancer
Cells, 3, 413-420.
110. Schuuring, E., Verhoeven, E., Van Tinteren,
H., Peterse, J. L., Nunnink, B., Thunnissen, F. B. J. M., Devilee, P.,
Cornelisse, C. J., Van de Vijver, M. J., Mooi, W. J., and Michalides,
R. J. A. M. (1992) Cancer Res., 52, 5229-5234.
111. Buckley, M. F., Sweeney, K. J. E., Hamilton,
J. A., Sini, R. L., Manning, D. L., Nicholson, R. I., DeFazio, A.,
Watts, C. K. W., Musgrove, E. A., and Sutherland, R. L. (1993)
Oncogene, 8, 2127-2133.
112. Gillett, C., Fantl, V., Smith, R., Fisher, C.,
Bartek, J., Dickson, C., Barnes, D., and Peters, G. (1994) Cancer
Res., 54, 1812-1817.
113. Hinds, P. W., Dowdy, S. F., Eaton, E. N.,
Arnold, A., and Weinberg, R. A. (1994) Proc. Natl. Acad. Sci.
USA, 91, 709-713.
114. Jares, P., Fernández, P. L., Campo, E.,
Nadal, A., Bosch, F., Aiza, G., Nayach, I., Traserra, J., and Cardesa,
A. (1994) Cancer Res., 54, 4813-4817.
115. Bartkova, J., Lukas, J., Müller, H.,
Strauss, M., Gusterson, B., and Bartek, J. (1995) Cancer Res.,
55, 949-956.
116. Michalides, R., van Veelen, N., Hart, A.,
Loftus, B., Wientjens, E., and Balm, A. (1995) Cancer Res.,
55, 975-978.
117. Courjal, F., Louason, G., Speiser, P.,
Katsaros, D., Zeillinger, R., and Theillet, C. (1996) Int. J.
Cancer, 69, 247-253.
118. Saxena, A., Robertson, J. T., and Ali, I. U.
(1996) Oncogene, 13, 661-664.
119. Zhang, T., Nanney, L. B., Luongo, C., Lamps,
L., Heppner, K. J., DuBois, R. N., and Beauchamp, R. D. (1997)
Cancer Res., 57, 169-175.
120. Counter, C. M., Avilion, A. A., LeFeuvre, C.
E., Stewart, N. G., Greider, C. W., Harley, C. B., and Bacchetti, S.
(1992) EMBO J., 11, 1921-1929.
121. Counter, C. M., Botelho, F. M., Wang, P.,
Harley, C. B., and Bacchetti, S. (1994) J. Virol., 68,
3410-3414.
122. Klingelhutz, A. J., Barber, S. A., Smith, P.
P., Dyer, K., and McDougall, J. K. (1994) Mol. Cell. Biol.,
14, 961-969.
123. Morin, G. B. (1989) Cell, 59,
521-529.
124. Bryan, T. M., Englezou, A., Gupta, J.,
Bacchetti, S., and Reddel, R. R. (1995) EMBO J., 14,
4240-4248.
125. Greider, C. W., and Blackburn, E. H. (1985)
Cell, 43, 405-413.
126. Counter, C. M., Hirte, H. W., Bacchetti, S.,
and Harley, C. B. (1994) Proc. Natl. Acad. Sci. USA, 91,
2900-2904.
127. Nilsson, P., Mehle, C., Remes, K., and Roos,
G. (1994) Oncogene, 9, 3043-3048.
128. Kim, N. W., Piatyszek, M. A., Prowse, K. R.,
Harley, C. B., West, M. D., Ho, P. L. C., Coviello, G. M., Wright, W.
E., Weinrich, S. L., and Shay, J. W. (1994) Science,
266, 2011-2015.
129. Counter, C. M., Gupta, J., Harley, C. B.,
Leber, B., and Bacchetti, S. (1995) Blood, 85,
2315-2320.
130. Bryan, T. M., and Reddel, R. R. (1997) Eur.
J. Cancer, 33A, 767-773.
131. Blasco, M. A., Funk, W., Villeponteau, B., and
Greider, C. W. (1995) Science, 269, 1267-1270.
132. Feng, J., Funk, W. D., Wang, S.-S., Weinrich,
S. L., Avilion, A. A., Chiu, C.-P., Adams, R. R., Chang, E., Allsopp,
R. C., Yu, J. H., Le, S. Y., West, M. D., Harley, C. B., Andrews, W.
H., Greider, C. W., and Villeponteau, B. (1995) Science,
269, 1236-1241.
133. Avilion, A. A., Piatyszek, M. A., Gupta, J.,
Shay, J. W., Bacchetti, S., and Greider, C. W. (1996) Cancer
Res., 56, 645-650.
134. Soder, A. I., Hoare, S. F., Muir, S., Going,
J. J., Parkinson, E. K., and Keith, W. N. (1997) Oncogene,
14, 1013-1021.
135. Harrington, L., McPhail, T., Mar, V., Zhou,
W., Oulton, R., Bass, M. B., Arruda, I., and Robinson, M. O. (1997)
Science, 275, 973-977.
136. Nakayama, J., Saito, M., Nakamura, H.,
Matsuura, A., and Ishikawa, F. (1997) Cell, 88,
1-20.
137. Lingner, J., Hughes, T. R., Shevchenko, A.,
Mann, M., Lundblad, V., and Cech, T. R. (1997) Science,
276, 561-567.
138. Broccoli, D., Young, J. W., and de Lange, T.
(1995) Proc. Natl. Acad. Sci. USA, 92, 9082-9086.
139. Hiyama, K., Hirai, Y., Kyoizumi, S., Akiyama,
M., Hiyama, E., Piatyszek, M. A., Shay, J. W., Ishioka, S., and
Yamakido, M. (1995) J. Immunol., 155, 3711-3715.
140. Härle-Bachor, C., and Boukamp, P. (1996)
Proc. Natl. Acad. Sci. USA, 93, 6476-6481.
141. Yasumoto, S., Kunimura, C., Kikuchi, K.,
Tahara, H., Ohji, H., Yamamoto, H., Ide, T., and Utakoji, T. (1996)
Oncogene, 13, 433-439.
142. Hsiao, R., Sharma, H. W., Ramakrishnan, S.,
Keith, E., and Narayanan, R. (1997) Anticancer Res., 17,
827-832.
143. Klingelhutz, A. J., Foster, S. A., and
McDougall, J. K. (1996) Nature, 380, 79-82.
144. Weng, N.-P., Levine, B. L., June, C. H., and
Hodes, R. J. (1996) J. Exp. Med., 183, 2471-2479.
145. Wright, W. E., Piatyszek, M. A., Rainey, W.
E., Byrd, W., and Shay, J. W. (1996) Dev. Genet., 18,
173-179.
146. Sharma, H. W., Sokoloski, J. A., Perez, J. R.,
Maltese, J. Y., Sartorelli, A. C., Stein, C. A., Nichols, G., Khaled,
Z., Telang, N. T., and Narayanan, R. (1995) Proc. Natl. Acad. Sci.
USA, 92, 12343-12346.
147. Albanell, J., Han, W., Mellado, B.,
Gunawardane, R., Scher, H. I., Dmitrovsky, E., and Moore, M. A. S.
(1996) Cancer Res., 56, 1503-1508.
148. Bestilny, L. J., Brown, C. B., Miura, Y.,
Robertson, L. D., and Riabowol, K. T. (1996) Cancer Res.,
56, 3796-3802.
149. Buchkovich, K. J., and Greider, C. W. (1996)
Mol. Biol. Cell, 7, 1443-1454.
150. Holt, S. E., Wright, W. E., and Shay, J. W.
(1996) Mol. Cell. Biol., 16, 2932-2939.
151. Igarashi, H., and Sakaguchi, N. (1996)
Biochem. Biophys. Res. Commun., 219, 649-655.
152. Kruk, P. A., Balajee, A. S., Rao, K. S., and
Bohr, V. A. (1996) Biochem. Biophys. Res. Commun., 224,
487-492.
153. Morrison, S. J., Prowse, K. R., Ho, P., and
Weissman, I. L. (1996) Immunity, 5, 207-216.
154. Norrback, K.-F., Dahlenborg, K., Carlsson, R.,
and Roos, G. (1996) Blood, 88, 222-229.
155. Savoysky, E., Yoshida, K., Ohtomo, T.,
Yamaguchi, Y., Akamatsu, K.-I., Yamazaki, T., Yoshida, S., and
Tsuchiya, M. (1996) Biochem. Biophys. Res. Commun., 226,
329-334.
156. Zhang, W., Piatyszek, M. A., Kobayashi, T.,
Estey, E., Andreeff, M., Deisseroth, A. B., Wright, W. E., and Shay, J.
W. (1996) Clin. Cancer Res., 2, 799-803.
157. Ramirez, R. D., Wright, W. E., Shay, J. W.,
and Taylor, R. S. (1997) J. Invest. Dermatol., 108,
113-117.
158. Wright, W. E., Brasiskyte, D., Piatyszek, M.
A., and Shay, J. W. (1996) EMBO J., 15, 1734-1741.
159. Pereira-Smith, O. M., and Smith, J. R. (1988)
Proc. Natl. Acad. Sci. USA, 85, 6042-6046.
160. Hensler, P. J., Annab, L. A., Barrett, J. C.,
and Pereira-Smith, O. M. (1994) Mol. Cell. Biol., 14,
2291-2297.
161. Uejima, H., Mitsuya, K., Kugoh, H., Horikawa,
I., and Oshimura, M. (1995) Genes Chrom. Cancer, 14,
120-127.
162. Ohmura, H., Tahara, H., Suzuki, M., Ide, T.,
Shimizu, M., Yoshida, M. A., Tahara, E., Shay, J. W., Barrett, J. C.,
and Oshimura, M. (1995) Jpn. J. Cancer Res., 86,
899-904.
163. Ning, Y., and Pereira-Smith, O. M. (1991)
Mutat. Res., 256, 303-310.
164. Sandhu, A. K., Hubbard, K., Kaur, G. P., Jha,
K. K., Ozer, H. L., and Athwal, R. S. (1994) Proc. Natl. Acad. Sci.
USA, 91, 5498-5502.
165. Ogata, T., Ayusawa, D., Namba, M., Takahashi,
E., Oshimura, M., and Oishi, M. (1993) Mol. Cell. Biol.,
13, 6036-6043.
166. Koi, M., Johnson, L. A., Kalikin, L. M.,
Little, P. F. R., Nakamura, Y., and Feinberg, A. P. (1993)
Science, 260, 361-364.
167. Sasaki, M., Honda, T., Yamada, H., Wake, N.,
Barrett, J. C., and Oshimura, M. (1994) Cancer Res., 54,
6090-6093.
168. Klein, C. B., Conway, K., Wang, X. W., Bhamra,
R. K., Lin, X., Cohen, M. D., Annab, L., Barrett, J. C., and Costa, M.
(1991) Science, 251, 796-799.
169. Wang, X. W., Lin, X., Klein, C. B., Bhamra, R.
K., Lee, Y.-W., and Costa, M. (1992) Carcinogenesis, 13,
555-561.
170. Hubbard-Smith, K., Patsalis, P., Pardinas, J.
R., Jha, K. K., Henderson, A. S., and Ozer, H. L. (1992) Mol. Cell.
Biol., 12, 2273-2281.
171. Ray, F. A., and Kraemer, P. M. (1992)
Cancer Genet. Cytogenet., 59, 39-44.
172. Rice, R. H., Steinmann, K. E., DeGraffenried,
L. A., Qin, Q., Taylor, N., and Schlegel, R. (1993) Mol. Biol.
Cell, 4, 185-194.
173. Ozer, H. L., Banga, S. S., Dasgupta, T.,
Houghton, J., Hubbard, K., Jha, K. K., Kim, S.-H., Lenahan, M., Pang,
Z., Pardinas, J. R., and Patsalis, P. C. (1996) Exp. Gerontol.,
31, 303-310.
174. Plummer, S. J., Adams, L., Simmons, J. A., and
Casey, G. (1997) Oncogene, 14, 2339-2345.
175. Kaltoft, K., Pedersen, C. B., Hansen, B. H.,
and Thestrup-Pedersen, K. (1995) Cancer Genet. Cytogenet.,
81, 13-16.
176. Savelieva, E., Belair, C. D., Newton, M. A.,
DeVries, S., Gray, J. W., Waldman, F., and Reznikoff, C. A. (1997)
Oncogene, 14, 551-560.
177. Ogata, T., Oshimura, M., Namba, M., Fujii, M.,
Oishi, M., and Ayusawa, D. (1995) Jpn. J. Cancer Res.,
86, 35-40.
178. Vojta, P. J., and Barrett, J. C. (1995)
Biochim. Biophys. Acta, 1242, 29-41.
179. McClung, J. K., Danner, D. B., Stewart, D. A.,
Smith, J. R., Schneider, E. L., Lumpkin, C. K., Dell´Orco, R. T.,
and Nuell, M. J. (1989) Biochem. Biophys. Res. Commun.,
164, 1316-1322.
180. Nuell, M. J., Stewart, D. A., Walker, L.,
Friedman, V., Wood, C. M., Owens, G. A., Smith, J. R., Schneider, E.
L., Dell´Orco, R., Lumpkin, C. K., Danner, D. B., and McClung, J.
K. (1991) Mol. Cell. Biol., 11, 1372-1381.
181. Dell´Orco, R. T., McClung, J. K., Jupe,
E. R., and Liu, X.-T. (1996) Exp. Gerontol., 31,
245-252.
182. McClung, J. K., King, R. L., Walker, L. S.,
Danner, D. B., Nuell, M. J., Stewart, C. A., and Dell´Orco, R. T.
(1992) Exp. Gerontol., 27, 413-417.
183. Liu, X.-T., Stewart, C. A., King, R. L.,
Danner, D. A., Dell´Orco, R. T., and McClung, J. K. (1994)
Biochem. Biophys. Res. Commun., 201, 409-414.
184. Jupe, E. R., Liu, X.-T., Kiehlbauch, J. L.,
McClung, J. K., and Dell´Orco, R. T. (1995) Exp. Cell
Res., 218, 577-580.
185. Jupe, E. R., Liu, X.-T., Kiehlbauch, J. L.,
McClung, J. K., and Dell´Orco, R. T. (1996) Exp. Cell
Res., 224, 128-135.
186. Harris, L. C., Von Wronski, M. A., Venable, C.
C., Remack, J. S., Howell, S. R., and Brent, T. P. (1996)
Carcinogenesis, 17, 219-224.
187. Gadbois, D. M., Crissman, H. A., Tobey, R. A.,
and Bradbury, E. M. (1992) Proc. Natl. Acad. Sci. USA,
89, 8626-8630.
188. Chang, T., Khalsa, O., Wang, H., Lee, M.-E.,
and Schlegel, R. (1996) Cell Growth Differ., 7,
361-372.
189. Wadhwa, R., Kaul, S. C., Ikawa, Y., and
Sugimoto, Y. (1993) J. Biol. Chem., 268, 6615-6621.
190. Wadhwa, R., Kaul, S. C., Mitsui, Y., and
Sugimoto, Y. (1993) Exp. Cell Res., 207, 1-7.
191. Wadhwa, R., Pereira-Smith, O. M., Reddel, R.
R., Sugimoto, Y., Mitsui, Y., and Kaul, S. C. (1995) Exp. Cell
Res., 216, 101-106.
192. Imai, S.-I., Fujino, T., Nishibayashi, S.,
Manabe, T., and Takano, T. (1994) Mol. Cell. Biol., 14,
7182-7194.
193. Kaneko, S., Satoh, Y., Ikemura, K., Konishi,
T., Ohji, T., Karasaki, Y., Higashi, K., and Gotoh, S. (1995) Cell
Struct. Funct., 20, 107-115.
194. Holliday, R., Ho, T., and Paulin, R. (1996) in
Epigenetic Mechanisms of Gene Regulation (Russo, V. E. A,
Martienssen, R. A., and Riggs, A. D., eds.) Cold Spring Harbor
Laboratory Press, New York, pp. 47-59.
195. Wilson, V. L., and Jones, P. A. (1983)
Science, 220, 1055-1057.
196. Vertino, P. M., Issa, J.-P., Pereira-Smith, O.
M., and Baylin, S. B. (1994) Cell Growth Differ., 5,
1395-1402.
197. Honda, S., and Matsuo, M. (1987) Cell Biol.
Int. Rep., 11, 141.
198. El-Deiry, W. S., Nelkin, B. D., Celano, P.,
Yen, R.-W. C., Falco, J. P., Hamilton, S. R., and Baylin, S. B. (1991)
Proc. Natl. Acad. Sci. USA, 88, 3470-3474.
199. Issa, J.-P. J., Vertino, P. M., Wu, J.,
Sazawal, S., Celano, P., Nelkin, B. D., Hamilton, S. R., and Baylin, S.
B. (1993) J. Natl. Cancer Inst., 85, 1235-1240.
200. Antequera, F., Boyes, J., and Bird, A. (1990)
Cell, 62, 503-514.
201. Holliday, R., and Pugh, J. E. (1975)
Science, 187, 226-232.
202. Howard, B. H. (1996) Exp. Gerontol.,
31, 281-293.
203. Matsumura, T., Hunter, J. L., Farooq, M., and
Holliday, R. (1989) Exp. Cell Res., 184, 148-157.
204. Radna, R. L., Caton, Y., Jha, K. K., Kaplan,
P., Li, G., Traganos, F., and Ozer, H. L. (1989) Mol. Cell.
Biol., 9, 3093-3096.
205. Tsuyama, N., Miura, M., Kitahira, M.,
Ishibashi, S., and Ide, T. (1991) Cell Struct. Funct.,
16, 55-62.
206. Shay, J. W., West, M. D., and Wright, W. E.
(1992) Exp. Gerontol., 27, 477-492.
207. Tanaka, Y., Tang, X., Hou, D.-X., Gao, H.,
Kitabayashi, I., Gachelin, G., and Yokoyama, K. (1992) Cell Struct.
Funct., 17, 351-362.
208. Warneford, S. G., Witton, L. J., Townsend, M.
L., Rowe, P. B., Reddel, R. R., Dalla-Pozza, L., and Symonds, G. (1992)
Cell Growth Differ., 3, 839-846.