The Kinetics of Senescence in Retinal Pigmented Epithelial Cells: a Test for the Telomere Hypothesis of Ageing?
V. Rawes,1 D. Kipling,2 I. R. Kill,3 and R. G. A. Faragher1,4
1Ocular Research Group, Department of Pharmacy, University of Brighton, Brighton BN2 4GJ, UK; fax: +44-1273-679-333; E-mail: rgaf@brighton.ac.uk2Department of Pathology, College of Medicine, University of Wales, Heath Park, Cardiff CF4 4XN, UK; fax: +44-1222-743-524; E-mail: KiplingD@cardiff.ac.uk
3Department of Biological Sciences, The University, Dundee DD1 4HN, UK; E-mail: i.r.kill@dundee.ac.uk
4To whom correspondence should be addressed.
Submitted July 7, 1997.
Senescence, or replicative failure, has been reported for a wide variety of human cell types but has seldom been characterized in any detail. The senescence of human fibroblast cultures has been shown to be due to a steadily decreasing percentage of cells able to proliferate in standard media. This paper reports the serial subculture of a strain of adult retinal pigmented epithelial (RPE) cells until replicative failure after ~15 population doublings. Measurement of the growth fraction of the RPE cells at each passage using antibodies to the proliferation marker pKi67 demonstrated a rate of decline in the proliferating fraction of 3.66% per population doubling. Similar experiments carried out using a strain of human fibroblasts yielded a decline of approximately 0.88% per population doubling. Thus, individual RPE cells enter senescence significantly faster than control fibroblasts (p < 0.001). At growth arrest the RPE cells retained viability for extended periods but showed elevated endogenous autofluorescence, analogous to observations on post-mitotic human fibroblasts. Taken together these findings suggest that the process of senescence is a common feature of different cell lineages but that the specific rate can differ between them. The significance of these observations for the telomere hypothesis of senescence is discussed.
KEY WORDS: ageing, telomere, retinal pigmented epithelial cell, senescence, macular degeneration.
The classic work of Hayflick and Moorhead [1] demonstrated that the growth of human fibroblasts is characterized by a finite and predictable lifespan (reviewed in [2, 3]). For example, cultured human fibroblasts will typically undergo around 30-60 population doublings before finally ceasing cell division. This loss of growth potential is not cell death by either apoptosis or necrosis, nor is it terminal differentiation, but is rather a specialized form of viable cell cycle arrest that has been termed replicative senescence or the Hayflick limit. Such senescent cells are characterized by altered cell morphology and biochemical properties, reflecting, at least in part, an altered spectrum of gene expression. Their inability to divide in the face of mitogenic stimuli that cause proliferation of otherwise identical but pre-senescent cells, together with alterations in gene expression, mean that such senescent cells have the potential to have wide-ranging effects on organs and tissue systems where they arise, and in some instances may underlie or contribute to a variety of age-related degenerations and pathologies.
A large body of data is available regarding the behavior of cultured cells as they progress to senescence. Cloning studies, based on the analysis of colony size variation, demonstrated that the cells composing a fibroblast culture were reproductively heterogeneous. Some clones displayed reproductive potentials similar to that of the bulk culture whilst others demonstrated a severely restricted ability to proliferate [4]. The proportions of these two types of clone shifted in the direction of smaller colonies as the culture was passaged in vitro. Further analysis indicated that these clonal characteristics were not fixed since recloning of cells from a clone with a high reproductive potential yielded colonies with a range of proliferative abilities [5]. It was thus concluded that the decision to continue to divide or to enter senescence was to a large degree a stochastic (chance) process, with the chance of becoming senescent being more likely the more times the cell has been called on to divide. When analyzed at the level of a bulk culture this behavior leads to an essentially bimodal population of growing and senescent cells. The growing pool enters senescence at a finite rate which can be measured cytochemically by repeated determination of the fraction of a test culture which retains reactivity for markers of proliferation such as [3H]thymidine incorporation or pKi67 [6]. In fibroblasts this transition from division to arrest (the "cell kinetics") has been measured by increasingly sensitive techniques over the last twenty years [7, 8].
In parallel with studies conducted using fibroblasts, "senescence" has been reported for a wide range of other cell types [9]. However, there are no data for these other the cell types regarding the kinetics of the transition from growth to senescence that can be compared with the fibroblast data. In this study human retinal pigmented epithelial (RPE) cells are analyzed, an ocular cell type very different to human fibroblasts. These have previously been shown to undergo senescence, but no further observations have been reported. Here we describe the kinetics of transition of RPE cells to the senescent state and a parallel study of normal human fibroblasts. The implications of these new data to the telomere hypothesis [10, 11], first recognized as a potential mechanism for senescence in the classic theoretical work of Olovnikov, are discussed.
MATERIALS AND METHODS
Culture of Primary Human RPE Cells. The primary human retinal pigmented epithelial cell strain HRPE-8 (kind gift of Prof. J. Forrester, University of Aberdeen) was cultured in Eagle's minimal essential medium, supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 1 mM sodium pyruvate, 100 µg/ml streptomycin, and 10 IU/ml penicillin. Sodium pyruvate was purchased from Sigma (USA). All other reagents and tissue culture plastics were purchased from Gibco-BRL (USA). Cells were passaged every 4 to 5 days by trypsin-EDTA dispersion and seeded at an initial density of 6·103 cells per cm2.
Determination of Culture Growth Fraction. At each passage HRPE-8 cells were grown on sterile coverslips at 6·103 cells per cm2. After 3 days in culture the coverslips were washed in phosphate buffered saline and fixed in a 1:1 mixture of methanol--acetone. The fixed cells were then incubated overnight with 10 ml of antiserum to the proliferation-associated antigen pKi67 (DaKo, diluted 1:50) at 4°C in a humidified chamber. Excess anti-pKi67 was then removed by washing and the coverslip incubated overnight with a diluted solution of fluorescein-conjugated rabbit anti-mouse secondary antiserum (DaKo, diluted 1:30). The fixed cells were then washed, counterstained with DAPI, mounted, and viewed using an epifluorescence microscope. To assess the growth fraction of the culture 1000 total or 200 positive nuclei were scored in randomly selected fields.
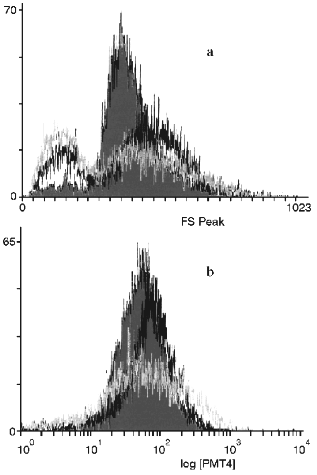
Determination of Cell Size and Lipofuscin. Early passage (2 PD), middle (9 PD), and late passage (13 PD) cells were harvested and 10,000 cells at each age were fixed in 4% (w/v) paraformaldehyde. These were then analyzed for cell size (using forward-angle light scatter) and autofluorescence (between 550-605 nm, after excitation at 488 nm). Fluorescence intensity was expressed on an arbitrary relative scale. The analyses was performed on a fluorescence activated cell sorter (FACS, Coulter Instruments, USA) linked to a personal computer to store and process the fluorescence profiles. Optimization of alignment and calibration was performed using standardized fluorescent microspheres.
RESULTS AND DISCUSSION
The proliferative fractions of both HRPE-8, a primary human retinal pigmented epithelial cell strain, and IBR.3, a normal adult dermal fibroblast strain, are shown in Fig. 1. Both strains began primary culture with a high proliferative fraction, as assessed using antisera to the pKi67 protein, a widely studied nuclear marker of proliferation active essentially throughout the cell cycle. In both cases the fraction of cycling cells declines continuously and gradually with culture and does not undergo an abrupt collapse. To obtain a numerical estimate of the rate of decline straight lines were fitted to the data by least squares regression analysis. Human fibroblasts gave a rate of change in growth fraction of 0.88 ± 0.13 percent of labelling index per population doubling (PLPD). By contrast the HRPE-8 cells entered senescence at a rate of 3.66 ± 0.24 PLPD. Statistical analysis based upon a modified Student's t-test for difference of slope showed this difference in rate to be highly significant (p < 0.001).
Figure 2 shows the change in endogenous autofluorescence when early, middle and late passage HRPE-8 cells were flow sorted using a FACS. There was a tendency for the maximal levels of autofluorescence to increase on serial passage and for the range of autofluorescence to increase.Fig. 1. Decrease of the cyclic fraction of a strain of human retinal pigmented cells (HRPE-8) (1) and a strain of human diploid fibroblasts (IBR.3) (2). Decrease was measured by the proliferation marker pKi67.
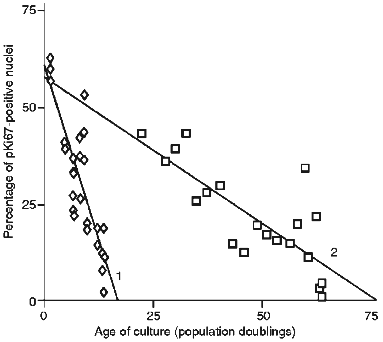
These results indicate that adult RPE cells in culture initially have a similar growth fraction to adult fibroblasts but lose this fraction approximately three times faster. This difference in rate of loss of the growth fraction appears to be responsible for the shorter lifespan (in PD) of HRPE-8 cells compared to IBR.3. Since RPE cells already represent a terminally differentiated cell population, loss of dividing cells through a differentiation or transdifferentiation process seems unlikely and would be an unsatisfactory explanation for the difference in decline rate observed [12].Fig. 2. Monoparametric histograms of the relative cell size (by frontal light scattering data) (a) and of endogenous autofluorescence of HRPE-8 cells (b) after 2 (grey), 9 (black), and 13 (white) population doublings.
In general, the rate of decline in a growth fraction is the cumulative result of a stochastic decision-making process by every individual cell within the population. The considerable difference in rates of decline seen between HRPE-8 and IBR.3 thus seems to indicate that individual RPE cells are far more likely to enter senescence if called on to divide than are fibroblasts. An extension of this argument is that any process or insult which triggers cell turnover in vivo would be predicted to shift the proportions of growing and senescent cells to a greater extent in the retinal layer than in a corresponding layer of dermal fibroblasts.
The cell hypothesis of ageing, first proposed by Hayflick, proposes that the accumulation of senescent cells is a principle cause of age-linked degenerative diseases [2, 3]. The hypothesis only applies to tissues which divide during adult life, rather than tissues composed of permanently non-dividing cells. Although RPE cells senescence has been suggested to be linked to age-related retinal disease [12, 13], the fact that individual cells cycle infrequently in the normal adult retina has tended to make the accumulation of a compromising number of senescent RPE cells appear unlikely. However, the kinetics of HRPE-8 cells now suggest that individual RPE cells readily enter senescence, making their appearance in vivo more plausible. Aged RPE cells in vivo show increased levels of lipofuscin and we have observed a similar increase in senescent RPE cells in vitro. It is thus possible that some in vivo aged RPE cells have become senescent, but histochemical analysis of tissue sections using a marker for senescent cells would be required to demonstrate this. The recent demonstration that senescent RPE cells show senescence-associated beta-galactosidase activity now renders this study possible.
The mechanism by which cells become senescent remains controversial [2, 3]. One of the most plausible hypotheses for human (as opposed to rodent) cellular senescence is provided by telomere shortening [14-16]. In the absence of any mechanism to produce compensatory de novo synthesis of telomeric DNA, human chromosomes are faced with what has been termed the "end replication problem" (reviewed by Kipling [17]). This results from the inability of all known DNA-dependent DNA polymerases to commence synthesis de novo. At the very termini a region at least as large as the priming RNA primer for lagging strand DNA synthesis will unavoidably be lost each cell division. In most immortal human cells there appears to be compensatory de novo synthesis of telomeric DNA by the enzyme telomerase and a stable maintenance of telomere length [15, 17-19].
In most primary human cells telomerase is absent, and telomere shortening with proliferation in vitro has been reported [15, 18, 20, 21]. This shortening is presumed to reflect losses from a failure in end-replication. An analogous decline in telomere length with donor age has also been reported for a variety of human tissues [18, 22]. Since the loss of telomeric DNA is linked to passage through S-phase it forms an attractive mechanism by which cells can count divisions [23].
Although telomere length reduction could form the basis of a clock [14], there is as yet little direct evidence that it does so. Some of the strongest evidence in support of a telomere clock, but still circumstantial, is that telomere length among unrelated fibroblast cultures is a good predictor of proliferative potential [24]. Intervention tests of the role of telomere shortening in senescence are currently not possible since not all subunits of the enzyme have been cloned. It is thus pertinent to ask if the cell kinetics of primary cultures can be used to construct a test of the telomere hypothesis.
One immediate kinetic problem for the telomere hypothesis is the bimodal nature of primary cultures which yield senescent cells at very low PDs. On the surface this behavior contrasts markedly with the kinetics predicted for a telomere clock [25], in which the small amount of telomeric sequence lost each S-phase would seem unlikely to produce senescent daughter cells with very short telomeres early in a culture's lifespan. Indeed, it would require considerable telomere shortening (from a start of 12-20 kbp) before such potentially growth-arresting short telomeres arose (at ~5 kbp). Thus a simple telomere-driven clock would be predicted to yield cultures in which the growth fraction was stably high for the first portion of the lifespan and then dropped markedly.
One possible explanation for this discrepancy is the nature of the starting biological material used in such experiments. If an explant is being cultured, as opposed to a clone, it will naturally consist of cells that have undergone different numbers of prior divisions in vivo. This heterogeneity in divisional history provides one route to "smear out" the kinetics and turn a telomere-style precipitous collapse into the more gradual decline that is observed.
An additional factor that will provide heterogeneity comes from the fact that end replication losses are limited to the lagging strand of the telomere, and thus the amount of sequence loss depends on whether the maternal T- or C-strand of the telomere is inherited by the daughter cell. Thus an initial telomere of defined length will intrinsically eventually yield a population of cells which differ among themselves in the length of that telomere [25]. Together, this aspect of end replication losses and the heterogeneity of the biological sample used in mass culture experiments may be sufficient to produce the gradual decline in the cycling fraction that is usually seen. This is supported by studies on the senescence of clonal cell strains, which show a much more abrupt late-stage drop in cycling fraction [26, 27] that the telomere hypothesis would predict.
Thus, a gradual decline in growth fraction is, with reasonable assumptions, consistent with a telomere-driven clock. It is far harder to explain differences in the rate of decline of this fraction between different cell types, such as that described in this report between IBR.3 and HRPE-8. Such a change in rate is unlikely to be caused by differences in the size of the starting telomere (or the length detected as "critically short"), because this would be expected to affect only the remaining lifespan and not the rate at which the growth fraction declined. This is incompatible with the observation that IBR.3 and HRPE-8 cultures start with similar proliferative indices but lose them at different rates. For the telomere hypothesis to hold in RPE cells as well as fibroblasts, the amount of telomeric sequence lost must be greater in HRPE-8 than in IBR.3. However, it is not immediately obvious how two cell types, both completely lacking telomerase activity, could have different rates of telomere loss. One possibility is lineage-type variation in the position of placement of the most terminal RNA primer, perhaps reflecting variation in RNA primase activity. Another is a difference in the rate of C-strand degradation. A pathway to degrade the C-strand, that complementary to the strand synthesized by telomerase, has been suggested by work in budding yeast and can be affected by mutation in the yeast CDC13 gene [27, 28]. Recently data have been reported consistent with a pathway of C-strand degradation in mammalian cells [29]. Modulation of this pathway opens the way to differences in rates of telomere loss above the basal level predicted by a straightforward loss because of the end replication problem. Whatever the underlying mechanism that might modulate telomere loss rates in telomerase-negative somatic cells, the very existence of markedly different rates of cell senescence in RPE cells and fibroblasts provides the basis of a critical test of the telomere hypothesis of cell senescence.
Dr. D. Kipling is supported by the Lister Institute of Preventive Medicine.
LITERATURE CITED
1.Hayflick, L., and Moorhead, P. S. (1961) Exp.
Cell Res., 25, 585-621.
2.Campisi, J. (1996) Cell, 84,
497-500.
3.Campisi, J. (1997) Eur. J. Cancer,
33, 703-709.
4.Smith, J. R., and Hayflick, L. (1974) J. Cell
Biol., 63, 48-53.
5.Smith, J. R., and Whitney, R. G. (1980)
Science, 207, 82-84.
6.Cristofalo, V. J., and Scharf, B. B. (1972) Exp.
Cell Res., 76, 419-427.
7.Ponten, J., Stein, W. D., and Shall, S. (1983)
J. Cell Physiol., 117, 342-352.
8.Faragher, R. G. A., Kill, I. R., Hunter, J. A. A.,
Pope, F. M., Tannock, T. C., and Shall, S. (1993) Proc. Natl. Acad.
Sci. USA, 90, 12030-12034.
9.Stanulis-Prager, B. (1987) Mech. Ageing
Dev., 38, 1-37.
10.Olovnikov, A. M. (1971) Dokl. Akad. Nauk
SSSR, 201, 1496-1499.
11.Olovnikov, A. M. (1973) J. Theor. Biol.,
41, 181-190.
12.Eldred, G. E. (1993) Progr. Retinal Res.,
12, 101-121.
13.Silvestri, G. (1997) Mol. Med. Today,
3, 84-91.
14.Harley, C. B. (1991) Mut. Res.,
256, 271-282.
15.Harley, C. B., and Villeponteau, B. (1995)
Curr. Opin. Genet. Dev., 5, 249-255.
16.Autexier, C., and Greider, C. W. (1996) Trends
Biochem. Sci., 21, 387-391.
17.Kipling, D. (1995) The Telomere, Oxford
University Press, Oxford.
18.De Lange, T. (1995) in Telomeres
(Blackburn, E. H., and Greider, C. W., eds.) Cold Spring Harbor Press,
New York, pp. 265-293.
19.Counter, C. M., Avilion, A. A., LeFeuvre, C. E.,
Stewart, N. G., Greider, C. W., Harley, C. B. and Bacchetti, S. (1992)
EMBO J., 11, 1921-1929.
20.Harley, C. B., Futcher, A. B. and Greider, C. W.
(1990) Nature, 345, 458-460.
21.Henderson, S., Allsopp, R., Spector, D., Wang,
S.-S., and Harley, C. (1996) J. Cell Biol., 134,
1-12.
22.Hastie, N. D., Dempster, M., Dunlop, M. G.,
Thompson, A. M., Green, D. K., and Allshire, R. C. (1990)
Nature, 346, 866-868.
23.Olovnikov, A. M. (1996) Exp. Gerontol.,
31, 443-448.
24.Allsopp, R. C., Vaziri, H., Patterson, C.,
Goldstein, S., Younglai, E. V., Futcher, A. B., Greider, C. W., and
Harley, C. B. (1992) Proc. Natl. Acad. Sci. USA, 89,
10114-10118.
25.Levy, M. Z., Allsopp, R. C., Futcher, A. B.,
Greider, C. W., and Harley, C. B. (1992) J. Mol. Biol.,
225, 951-960.
26.Bond, J., Haughton, M., Blaydes, J., Gire, V.,
Wynford-Thomas, D., and Wyllie, F. (1996) Oncogene, 13,
2097-2104.
27.Wynford-Thomas, D. (1997) Eur. J. Cancer,
33, 716-726.
28.Lin, J.-J., and Zakian, V. A. (1996) Proc.
Natl. Acad. Sci. USA, 93, 13760-13765.