Immunochemical Characterization of Steroid Hydroxylases of Adrenocortical Mitochondria. 2. Localization of Antigenic Determinants in the Cytochrome P450scc Molecule (CYP11A1)
A. A. Chernogolov1,2 and S. A. Usanov1
1Institute of Bioorganic Chemistry, Academy of Sciences of Belarus, ul. Zhodinskaya 5/2, Minsk, 220141 Belarus; fax: (375-17) 263-7274; E-mail: usanov@ns.iboch.ac.by2To whom correspondence should be addressed.
Submitted July 4, 1997.
The antigenic structure of cytochrome P450scc was investigated by immunochemical identification of the peptides formed by chymotryptic cleavage of the protein and separated by reversed-phase and cation-exchange HPLCs. These procedures resulted in isolation and structural characterization of four homogeneous immunoreactive peptides which corresponded to the sequences Asn236-Tyr241, Leu266-Leu272, Ala381-Phe388, and Ser390-Trp400. These peptides contained several positively charged residues which were previously shown to participate in electrostatic interactions with adrenodoxin. Our data indicate that the positively charged residues of cytochrome P450scc are involved in formation of antigenic sites, and the antigenic determinants of the protein molecule coincide or overlap with the regions of polypeptide chain responsible for the interaction of the heme protein with adrenodoxin.
KEY WORDS: adrenal cortex, cytochrome P450scc, adrenodoxin, antigenic structure, antibodies, steroids.
Abbreviations: P450scc) cytochrome P450 catalyzing cholesterol side-chain cleavage reaction; AR) adrenodoxin reductase; AD) adrenodoxin; EIA) enzyme immunoassay.
The synthesis of pregnenolone from cholesterol in adrenocortical
mitochondria is the key reaction of corticosteroid biosynthesis that is
carried out with participation of cytochrome P450-dependent
monooxygenase systems consisting of adrenodoxin reductase
(AR)3, adrenodoxin (AD), and cytochrome P450scc (P450scc)
[1]. Investigations of intra- and intermolecular
interactions in the cholesterol cleavage-catalyzing enzyme complex have
established the process of monooxygenase catalysis proceeding through
several elementary stages (binding of oxygen and the substrate in the
active center, interaction of the heme protein with AD, intermolecular
electron transfer) involves various regions of the cytochrome P450scc
molecule [2-5]. A significant
role in cytochrome P450scc--AD interaction is due to the
carboxyl-containing residues of the ferredoxin molecule [6-10] which are suggested to
interact with lysine residues in the heme protein molecule. The
involvement of positively charged residues of the cytochrome P450scc
molecule in the interaction with AD was demonstrated by chemical
modification [11-13] and
site-directed mutagenesis [14].
Determination of the antigenic structure of proteins is an essential step in investigation of their molecular organization, functional activity, evolution, and classification. The structural representation of antigenic determinants and their correlation with the functions of given structural elements allows assessment of the relationship between the structure of the protein, its function, and its spatial organization. Immunochemical methods are a potent technological approach and have often been used to study the structural--functional relations in cytochrome P450 [15-17], its catalytic activity [18, 19], antigenic structure [20, 21], and membrane topology [22-24]. Investigation of the antigenic structures of cytochromes P450scc and P45011beta is a high-priority task, because the antibodies against heme proteins inhibit reactions of steroid hydroxylation in reconstituted system and may be used to control the activities of these very important enzymes [25].
To study the antigenic structure of cytochrome P450scc, we used an approach consisting of immunochemical identification of purified peptide products of chymotryptic cleavage of the heme protein by enzyme immunoassay (EIA) and dot immunoblotting. Our data confirmed that positively charged residues of cytochrome P450scc are involved in forming the antigenic structure of the heme protein, and the antigenic determinants of heme protein molecule coincide or overlap with the regions of the polypeptide chain responsible for the interaction of cytochrome P450scc with AD.
MATERIALS AND METHODS
Chemicals. Chemicals used in the present study were the following: alpha-chymotrypsin from bovine pancreas (45 units per mg), dansyl chloride and dansyl amino acid standards, gelatin, Tris, sodium cholate, 3,3´,5,5´-tetramethylbenzidine from Serva (Germany); Triton X-100 from Loba Chemie (Austria); guanidine hydrochloride from Merck (Germany); CNBr-activated Sepharose 4B and Sephadex G-25 (medium) from Pharmacia (Sweden); Bio-Gel P-4 and nitrocellulose membranes from Bio-Rad (USA); a reversed-phase carrier Ultrasphere-ODS (C18) and an HPLC carrier Ultrasil Cx from Altex (USA); reagents for automated Edman’s method from Fluka (Switzerland).
Isolation of Cytochrome P450scc and Preparation of Anti-P450scc Antibodies. Cytochrome P450scc was isolated as described previously [18, 26]. The protein was homogeneous by electrophoresis and immunochemical analysis and was characterized by spectrophotometrical index A393/A280 = 0.85-0.88. The concentration of cytochrome P450scc was determined from difference spectra of the reduced heme protein and its carbon monoxide complex [27] using molar absorption coefficient 91 mM-1·cm-1 at 450 nm.
To isolate antibodies, the antiserum against cytochrome P450scc obtained as described previously [17] was dialyzed against 10 mM sodium phosphate buffer (PB), pH 8.0, chromatographed on DEAE-cellulose [28], and applied to a column (1.5 × 10 cm) with protein A from Staphylococcus aureus immobilized on CNBr-activated Sepharose 4B (5 mg protein A per ml gel) according to the instructions of Pharmacia (Sweden). The column was washed subsequently with 50 mM PB (pH 7.4), with 50 mM PB containing 1 M NaCl, 1 mM EDTA, and 0.3% sodium cholate (pH 7.4), and once more with 50 mM PB (pH 7.4). The IgG fraction was eluted with 0.1 M glycine-HCl (pH 2.3) dialyzed against 5 mM PB containing 0.2 M NaCl (pH 7.0), and stored at -70°C.
Dot Immunoblotting. Peptide fractions (5 µl) were applied in the center of 1 × 1 cm squares laid out on a nitrocellulose membrane, dried, and washed in 50 mM Tris-HCl containing 0.2 mM NaCl (pH 7.4) followed with incubation of membranes in the same buffer containing 0.5% gelatin (for 15 h at 20°C) to block the nonspecific adsorption of immunoreactive agents on the nitrocellulose membrane. The membranes were transferred into a solution of antibodies (5 µg per ml) prepared in blocking buffer containing 0.05% Triton X-100, and incubated for 2 h at 30°C. Then the nitrocellulose membrane was washed with 50 mM Tris-HCl containing 0.2 M NaCl and 0.05% Triton X-100 (pH 7.4), and incubated under the same conditions with horseradish peroxidase-conjugated donkey IgG antibodies against rabbit IgG diluted 1:1000 (at the IgG/peroxidase ratio 1:2 and 12 µM peroxidase concentration in the original conjugate preparation). The membranes were washed free of conjugate and stained in 50 mM PB containing 0.8 mM o-dianisidine, 10 mM hydrogen peroxide, and 1.7% dimethylformamide (pH 6.1).
Enzyme Immunoassay. Peptide fractions were applied to polyvinyl chloride 96-well plates (Flow, USA), 10-20 µl per well. The plates were dried in air, washed with 20 mM PB (pH 7.6, 200 µl per well), and coated with a blocking buffer (0.5% gelatin prepared in the same buffer containing 0.2 M NaCl, 200 µl per well) followed by a 2-h incubation at 37°C. Then, antibodies were injected into the wells (0.2-0.5 µg in 100 µl blocking buffer containing 0.03% Tween 20 per well) and incubated for 1.5 h at 37°C. After washing with 20 mM PB containing 0.2 M NaCl and 0.03% Tween 20 (pH 7.6), the plates were coated with horseradish peroxidase-conjugated donkey IgG antibodies against rabbit IgG diluted 1:1500 with the blocking buffer containing 0.03% Tween 20 (100 µl per well), followed with a 1.5-h incubation at 37°C. Then the plates were washed first with the same buffer, then with 20 mM PB (pH 7.6), and coated with 50 mM PB containing 0.5 mM 3,3´,5,5´-tetramethylbenzidine and 30 mM hydrogen peroxide, pH 6.1 (100 µl per cell). After a 15-min incubation, 0.1 M PB (pH 7.8) was added (120 µl per well), and the wells were scanned photometrically on a Specol 211 (Carl Zeiss, Germany).
Hydrolysis of Cytochrome P450scc by Chymotrypsin and Separation of Immunoreactive Peptide Fractions. As the source of peptide fragments of cytochrome P450scc, we used a hydrolysate resulting from the treatment of the heme protein with chymotrypsin. Before the treatment with chymotrypsin, heme was removed as described in [29], and the resulting preparation of apo-cytochrome P450scc was carboxymethylated. Lyophilized cytochrome P450scc (1.5 µmoles) was dissolved in 40% water solution of acetone and acidified with 0.1 M HCl. After a 30-min incubation the protein was precipitated with 9-fold excess of acetone, the sediment was removed by centrifugation, washed with 80% water solution of acetone, and dissolved in 0.2 M Tris-HCl containing 8 M guanidine-HCl (pH 8.6). Apo-cytochrome P450scc was carboxymethylated according to [30]. After addition of 120 µl 2-mercaptoethanol, the protein solution was stirred for 30 min at 37°C. The reaction vessel was filled with argon, then 150 mg iodoacetamide was added, following with stirring in the dark for 1.5 h, then 2-mercaptoethanol was added to 1%, and 15 min later the preparation was intensively dialyzed in the dark against water. Carboxymethylated protein preparation (a water suspension) was denatured for 10 min in a boiling water bath. After the protein suspension was cooled to 37°C, NH4HCO3 was added to 0.2 M, and the pH of the medium was adjusted to 8.3, followed by addition of chymotrypsin to enzyme/substrate ratio 1:100 (w/w). Hydrolysis was performed under argon at 37°C. Two hours later, another portion of chymotrypsin, equal to the first, was added. After a 24-h incubation, the hydrolysate was lyophilized, mixed with 5 ml of water, and re-lyophilized.
The hydrolysate was dissolved in 20% CH3COOH and chromatographed on a column (0.9 × 115 cm) with Bio-Gel P-4 in the same solvent, resulting in 8 main fractions. The fraction corresponding to the void volume of the column was rejected, and the pooled fractions corresponding to individual peaks on the chromatogram were analyzed by EIA and dot immunoassay as described above for their reactivity with anti-P450scc antibodies. These fractions were separated on a Ultrasil Cx carrier with 0-0.15% triethylamine and 0-15% acetonitrile gradients at pH 4.6. Analysis of interaction with antibodies by EIA and dot immunoassay was performed in 5-15 µl aliquots.
Immunoreactive fractions were chromatographed on an Ultrasphere-ODS carrier with a 0-60% gradient of acetonitrile in 0.1% CF3COOH (pH 2.2), and once more subjected to immunochemical analysis. The fractions which positively reacted with antibodies were re-analyzed by HPLC on an Ultrasphere-ODS carrier with 0-80% gradient of acetonitrile in 0.1% CF3COOH, pH 2.2.
The N-terminus identification was performed by the dansyl method [31]. Amino acid sequencing of peptides was performed by the manual Edman method as modified by Hartley [32]. Amino acid analysis was performed according to [33] on a LKB-3201 amino acid analyzer (LKB, Sweden).
Computer analysis of secondary structure elements and antigenic sites in the cytochrome P450scc molecule and its graphic representation were performed with the program package PC Gene, version 6.60 (Intelligenetics Inc., Switzerland) with kind permission of Dr. Rita Bernhardt (Universitat des Saarlandes, Germany).
RESULTS
Preparation and Separation of Peptide Fragments of Cytochrome P450scc. As the source of peptide fragments of cytochrome P450scc, we used a hydrolysate obtained by the treatment of the heme protein with chymotrypsin. The chymotrypsin-catalyzed hydrolysis was selected because it results in several peptides which are soluble in aqueous buffer solutions, long enough (5-6 amino acids) to form antigenic determinants, and cover most of the putative AD-binding sites.
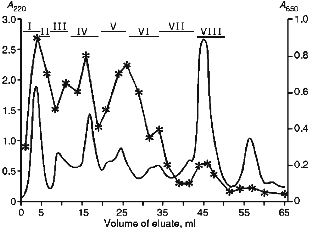
Prior to chymotryptic cleavage, cytochrome P450scc was treated with acidified acetone to remove its prosthetic group, protoporphyrin IX, and then carboxymethylated in the presence of 8 M guanidine hydrochloride to block the cysteine residues. Because carboxymethylated cytochrome P450scc is not soluble in aqueous buffer solutions, it was dialyzed against water to obtain a "fine" suspension of carboxymethylated cytochrome P450scc, and just before the hydrolysis, the protein preparation was denatured by incubation for 10 min at 98°C. The "fine" suspension of the protein provided a practically complete solution of the suspension after 24-h chymotryptic cleavage (at the final ratio enzyme/substrate 1:50). After hydrolysis, the preparation was lyophilized to remove NH4HCO3. The first step in separation of cytochrome P450scc hydrolysate was its chromatography on a Bio-Gel P-4 in 20% acetic acid. After chromatography was performed, 100-µl aliquots of each eluate fraction were lyophilized and used for EIA and immunoblotting. The eluate fractions resulting from chromatography were pooled in 8 main fractions (Fig. 1).
As follows from the data in Fig. 1, on the whole, the intensity of color in EIA correlates with peak fractions on the chromatogram, with maximal immunoreactivity observed in the free volume (fraction I), where high-molecular-weight products of hydrolysis are eluted (over 4000 daltons for Bio-Gel P-4). However, this fraction was not further separated, because it was impossible to localize individual antigenic sites in large peptide fragments of cytochrome P450scc resulting from the hydrolysis. No immunoreactive peptides were found in fraction VIII which contained low-molecular-weight peptide fragments and salts. Therefore, 6 immunoreactive fractions (II-VII) were selected for further separation.Fig. 1. Chromatography of chymotryptic digest of cytochrome P450scc on a Bio-Gel P-4 column (0.9 × 115 cm) with in 20% CH3COOH (elution rate, 2.5 ml/h). The broken line represents the data of enzyme immunoassay (the right scale is the absorption of peroxidase reaction products at 650 nm). The numbered sections above the chromatogram show the pooled fractions.
These fractions were successively subjected to HPLC on a cation-exchange carrier Ultrasil Cx and on a reverse-phase carrier Ultrasphere ODS (C18). The reversed-phase HPLC of the immunoreactive fractions required prior removal of the salts used in gradient elution of the peptides from the cation-exchanger. Only desalted samples could be used for further structural characterization. Second, at this step each of the fractions II, IV, V, and VI, detectable as a single peak after cation-exchange chromatography, was separated into individual immunoreactive fractions. The separations for fractions II-VIII are characterized below.
Fraction II. Chromatography of this fraction on a Ultrasil Cx resulted in 7 fractions. As shown by EIA and immunoblotting, fractions II-1 and II-6 (the last number indicates the order of elution of the fraction from the column in 0-15% acetonitrile and 0-0.15% triethylamine gradient) reacted with anti-P450scc antibodies.
Chromatography of fraction II-1 on a Ultrasphere ODS column resulted in 4 fractions, and only fractions II-1-1 and II-1-4 (the last number indicates the order elution of the fraction from the column with 0-60% gradient of acetonitrile in 0.1% CF3COOH) reacted with the antibodies. Six fractions were separated from fraction II-6, of which fractions II-6-5 and II-6-6 were immunoreactive.
Fraction III. Cation-exchange chromatography of this fraction resulted in 21 fractions. Only fraction III-19 characterized by a strong positive charge was immunoreactive. Chromatography on an Ultrasphere ODS column resulted in a single peptide peak (III-19-1) which reacted with the antibodies in EIA and immunoblotting.
Fraction IV. Cation-exchange chromatography of this fraction resulted in 11 fractions, of which IV-1 and IV-6 were immunoreactive and subjected to reverse-phase chromatography. Of the resulting fractions (4 for IV-1 and 6 for IV-6), only fractions IV-1-4 and IV-6-5 reacted with anti-P450scc antibodies.
Fraction V. Chromatography of this fraction on Ultrasil Cx resulted in 11 fractions, of which only fraction V-1 reacted with anti-P450scc antibodies. The latter was separated on a reverse-phase carrier into two subfractions, of which only one (V-1-2) reacted with the antibodies.
Fraction VI. Chromatography of this fraction on Ultrasil Cx resulted in 12 fractions, of which only fraction VI-1 reacted with the antibodies in EIA and immunoblotting. The latter was subjected to reversed-phase chromatography that resulted in 3 subfractions, of which only one (VI-1-1) reacted with the anti-P450scc antibodies.
Fraction VII. Chromatography of this fraction on Ultrasil Cx resulted in 11 fractions. The only immunoreactive fraction VII-1 was subjected to chromatography on Ultrasphere ODS. The immunoreactive peptide material was eluted in the first of three peak fractions (VII-1-1).
Structural Characterization of Immunoreactive Fractions. After separation of cytochrome P450scc hydrolysate 10 peptide fractions which reacted with anti-P450 antibodies in EIA and immunoblotting were selected. As mentioned above, at each step of peptide separation, only the fractions containing peptides reacting with anti-P450scc antibodies in EIA and dot immunoblotting were selected for further structural characterization and immunochemical analysis (Fig. 2). EIA and dot immunoblotting were a helpful combination for reliable identification of immunoreactive products of hydrolysis because a number of peptide fractions show a weak reaction with antibodies if examined by only one of these methods; this was probably due to the fact that peptides of different amino acid composition are differently adsorbed on plastic plates and nitrocellulose membranes.
Analysis of the fractions for their homogeneity and structural identification of peptides were performed by standard methods of structural chemistry--N-terminus identification, amino acid analysis, and peptide sequencing by the Edman method. Unfortunately, the yield of immunoreactive peptides was low (Table 1), probably because of the multisite character of chymotrypsin action and the cleavage of bonds within antigenic domains. In theory, a cytochrome P450scc molecule has 137 chymotrypsin-sensitive peptide bonds, and their hydrolysis may result in up to 45 free amino acids and 52 oligopeptides (of 2-4 amino acid residues). However, organization of an immunogenic site requires at least 5 amino acids due to the peculiarities of immune response which involves the interaction of the antigenic site with a molecule of major compatibility complex and T-lymphocyte receptor [34].Fig. 2. Identification of immunoreactive peptide fractions of chymotryptic digest of cytochrome P450scc with dot immunoblotting (A7, II-6-6 (P4); C8, III-19-1 (P3); D6, IV-1-4; D10, IV-6-5 (P2); F10, V-1-1 (P1); I1, P450scc) and enzyme immunoassay (A12, P450scc; E4, II-1-4; G6, II-6-6 (P4); G10, III-19-1 (P3)). Only immunoreactive fractions are given.
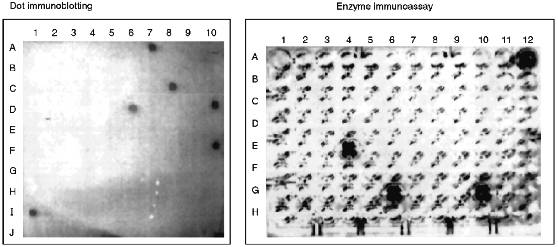
TABLE 1. Characterization of Immunoreactive
Peptide Products of Chymotryptic Cleavage of Cytochrome P450scc
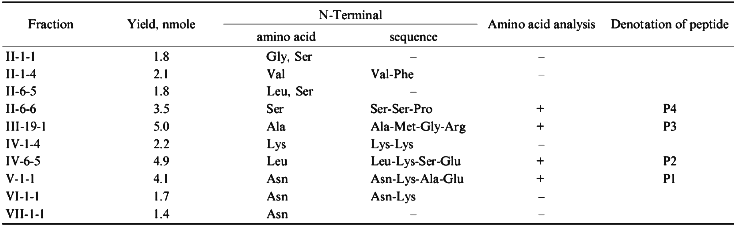
Because fractions II-1-1, II-1-4, II-6-5, IV-1-4, VI-1-1, and VII-1-1 were resulted from reverse-phase chromatography with minimal yield (1.4-2.1 nmoles peptide), it was impossible to perform their complete structural characterization including amino acid analysis. For these fractions, only the N-terminal amino acid was identified, and for fractions II-1-4, II-6-5, and IV-1-4, also the second amino acid in N-terminus was identified. However, comparison of these data with those available on the structural characterization of peptides resulting from chymotryptic cleavage of cytochrome P450scc [35] allowed us to identify several of the fractions.
By N-terminus identification, fractions II-1-1 and II-6-5 were not homogeneous, and their further purification was impossible because of the small amount of peptide material. Because of that, these fraction were excluded from further analysis.
Fraction II-1-4 was homogeneous, according to N-terminus identification and was isolated in an amount sufficient for identification of the second terminal amino acid. Based on the data on the primary structure of cytochrome P450scc [36, 37], we suggest that this peptide may correspond to the only sequence beginning with Val470. In the study cited above [35] a peptide containing the sequence Val-Phe-Arg-Pro-Phe was characterized. Though the amount of peptide material in this fraction was not enough to perform amino acid analysis, the available data suggest that this peptide corresponds to the sequence Val470-Phe474, i.e., it is located in the C-terminal region of cytochrome P450scc.
The N-terminal amino acid in fraction II-6-6 is Ser. The second and the third terminal amino acids are identified as Ser and Pro, respectively. In a cytochrome P450scc molecule, only the region beginning with Ser390 contains this sequence. There were isolated two peptides, one of which corresponded to the sequence Ser-Ser-Pro-Asp-Lys-Phe-Asp-Pro-Thr-Arg-Trp, and the second one is the same peptide shortened by the cleavage of the Asp-Pro bond and thus including the sequence Ser-Ser-Pro-Asp-Lys-Phe-Asp [35]. Therefore, it was established that the immunoreactive site is located within the region Ser390-Trp400. To identify the C-terminal border of this site, fraction II-6-6 was subjected to amino acid analysis (Table 2). Based on this analysis, it was concluded that this immunoreactive site is indeed represented by the sequence Ser390-Trp400.
TABLE 2. Amino Acid Analysis of
Immunoreactive Peptides of Cytochrome P450scc
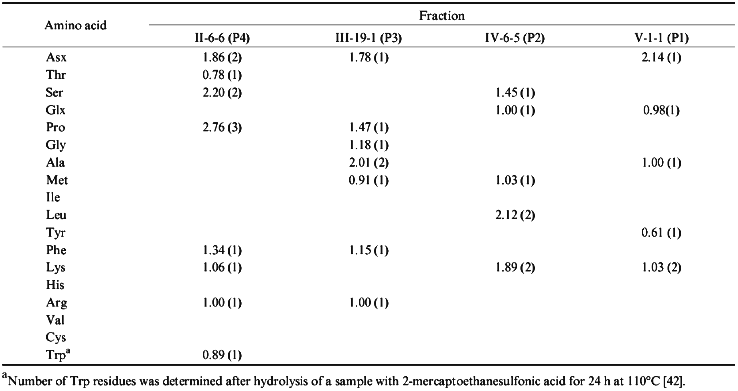
Fraction III-19-1 was obtained with the highest yield and by the data of structural analysis was an individual peptide containing one of the two peptides of the chymotryptic digest of cytochrome P450scc beginning with Ala, i.e., Ala-Met-Gly-Arg-Asn. According to the primary structure, the N-terminal amino acid in this peptide is Ala381. Amino acid analysis of this fraction has shown that in addition to 5 amino acids present in the peptide described in [35], fraction III-19-1 also contains Pro and Phe, and the amount of Ala in it is twice higher than that in the mentioned peptide [35]. These data suggest that the isolated immunoreactive site corresponds to the sequence Ala381-Met-Gly-Arg-Asp-Pro-Ala-Phe388.
Fraction IV-1-1 contains peptides with two lysine residues in the N-terminus. In the polypeptide chain of cytochrome P450scc, there are two regions with lysine residues located one after another, Lys103-Lys104 and Lys109-Lys110. These sites are N-terminal for the peptides of chymotryptic digest of cytochrome P450scc described previously, Lys-Lys-Ser-Gly-Thr-Trp and Lys-Lys-Asp-Arg-Val-Val-Leu, respectively [35]. The limited amount of peptide material did not allow us to identify the third terminal amino acid and perform the amino acid analysis for the fraction; however, the data available indicate that the region containing Lys103-Trp108 and/or Lys109-Leu114 may form the antigenic site.
According to the data of N-terminus identification, fraction IV-6-5 is homogeneous. Nine peptides with N-terminal Leu were described [35]; however, neither of them had Lys in the second position. The identifications of the third and the forth amino acid residues in the peptide from this fraction suggested that the isolated immunoreactive peptide contains the sequence Lys-Ser-Glu-Lys-Met described in the above-cited study, identical to the region Lys267-Met271 of cytochrome P450scc. The amino acid analysis confirmed this suggestion. In addition, it has been established that this peptide fraction is enriched with leucine and the peptide bond was cleaved not at Met271, but at Leu272, and thus this peptide corresponds to the sequence Leu266-Lys-Ser-Glu-Lys-Met-Leu272.
Fraction V-1-1 contains peptides with the Asn-Lys-Ala-Glu N-terminal amino acid sequence. The peptide Asn-Lys-Ala-Glu-Lys-Tyr was described in [35]. Amino acid analysis of this fraction confirmed the indicated composition of this peptide, thus providing the identification of the sequence Asn236-Tyr241 as an antigenic site of the cytochrome P450scc molecule.
Fraction VI-1-1 also contains peptides with the Asn as the N-terminal amino acid. The second terminal amino acid is Lys. This fraction was not further analyzed because of the limited amount of peptide material. However, because the polypeptide chain of cytochrome P450scc has only one region with this sequence, Asn236-Lys237, we suggest that the peptide material of this fraction is of the same composition as that of fraction V-1-1. Thus, the peptide corresponding to the cytochrome P450scc sequence Asn236-Tyr241 is eluted from Bio-Gel P-4 in the neighboring fractions V and VI.
Fraction VII-1-1 contains peptides with Asn as the N-terminal amino acid. This fraction was not subjected to further structural characterization because of the low yield of the peptides.
To summarize, we conclude that after separation of chymotryptic digest of cytochrome P450scc, of 10 immunoreactive fractions resulting only 4 were subjected to a complete structural analysis (Tables 1 and 2), providing a reliable identification of the immunoreactive sites in the polypeptide chain of cytochrome P450scc. According to localization of these sites in the cytochrome P450scc molecule, they were denoted as P1, P2, P3, and P4, which corresponded to the sequences Asn236-Tyr241, Leu266-Leu272, Ala381-Phe388, and Ser390-Trp400 (Fig. 3).
Fig. 3. Antigenic sites in the polypeptide chain of cytochrome P450scc including putative sites in the regions Lys103-Leu114 and Val470-Phe474 (underlined). Immunoreactive peptides P1, P2, P3, and P4 are given in bold font. For comparison, the figure shows the lysine residues (K*) involved in the interaction of the heme protein--AD [11] and the peptides of cytochrome P450scc isolated from a covalent complex AD--P450scc [38] (italicized).
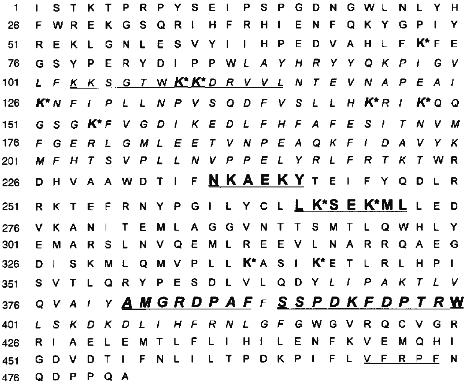
DISCUSSION
The data represented in our previous paper [25] indicate that the efficiency of hydrolysis of cholesterol in the reconstituted system is sharply decreased with addition of anti-P450scc antibodies, and the antibodies exert their effects at the level of interaction of cytochrome P450scc with AD. As shown previously by chemical modification of lysine residues with succinic anhydride [11, 13] and a structural analysis of the complex AD--P450scc covalently linked with bifunctional reagents [38], the ferredoxin-binding domain is formed by a number of regions of polypeptide chain located in the N- and C-terminal sequences of cytochrome P450scc including the regions Gly47-Thr107 and Leu368-Gly416. These data were prerequisite to searching for a correlation between the antigenic determinants and the functionally active domains of the heme protein molecule.
Comparison of the localized antigenic determinants with the sites in the cytochrome P450scc molecule responsible for the interaction with AD suggests that the antigenic determinants of heme proteins at least partly coincide or overlap with these regions; this agrees with the data of spectrophotometric titration of cytochrome P450scc in the presence of the antibodies. It is noteworthy that three of four immunoreactive sites are localized in fragment F3, which has been shown to be exposed mainly to the matrix [20]. It is known that antigenic determinants may be formed in the exposed regions of protein molecules which are mainly in beta-turns and in the regions of irregular secondary structure [39] which are organized with participation of proline and asparagine related to residues "hindering" formation of alpha-helix structures [40, 41].
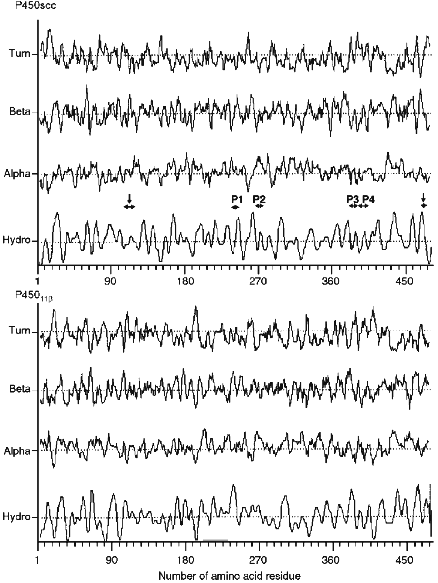
The calculation performed on the secondary structure elements indicates that unlike microsomal heme proteins, mitochondrial cytochromes P450 have no long alpha-helix regions in the N-terminal sequence, and the hydrophobic and hydrophilic regions are distributed rather evenly in the amino acid sequence (Fig. 4). Comparative analysis shows that the identified and localized in the present study antigenic sites are located in the beta-turn regions of the molecule (peptides P3 and P4, putative antigenic site around Lys103-Trp108 and/or Lys109-Leu114), beta-pleated sheet (Val470-Phe474), or short alpha-helix regions (peptides P1 and P2). This indicates the involvement of these regions in the interaction with AD. A sufficiently high concentration of positively charged amino acid residues in the indicated regions (pI for the peptides are: 9.4 for Asn236-Tyr241, 9.9 for Leu266-Leu272, 6.6 for Ala381-Phe388, and 6.6 for Ser390-Trp400) confirms the data indicating a substantial contribution of Lys and Arg residues of cytochrome P450scc in its interaction with AD.
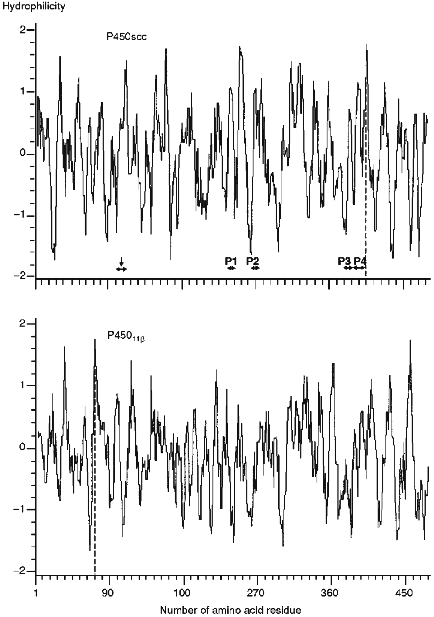
The calculation of the hydrophilic profile for cytochrome P450scc molecule (Fig. 5) indicates that the antigenic determinants may be located in N- and C-terminal sequences of the heme protein, and the longest hydrophilic regions are localized in the central part of the molecule (within 220-260 amino acid residues), and also in N-terminal and central parts of fragment F2 (within 260-290, 310-360, and 385-420 amino acid residues). Comparison of hydrophilic profiles for cytochromes P450scc and P45011beta demonstrates the presence of large homologous regions in the central and N-terminal parts of the two heme proteins. Because the inhibiting effect of anti-P45011beta antibodies on the hydroxylation of 11beta-deoxycorticosterone depends on the amount of the AD added, we suggest that the interaction with AD involves the positively charged amino acids of cytochrome P45011beta located in these homologous regions possessing antigenic properties.Fig. 4. Elements of secondary structure of cytochromes P450scc and P45011beta determined according to Novotny and Auffray [42]. Top to bottom, the probability curves for formations of beta-turn, beta-pleated sheet, and alpha-helix (calculated using the parameters from [43]), and also the hydrophobic profile (by the scale from [44]). Borders of the structural elements corresponding to immunoreactive peptides P1, P2, P3, and P4, and to putative antigenic sites Lys103-Leu114 and Val470-Phe474 (shown by vertical arrows) are marked with the symbol ***<-->.
The peptides P1, P3, and P4 identified as immunoreactive sites of cytochrome P450scc are located in the predicted antigenic domains of the heme protein molecule. It is noteworthy that peptides P3 and P4 are intermediate products in chymotryptic cleavage of cytochrome P450scc. Both peptides contain an unstable Asp-Pro bond. This may also be a reason for the low yield of immunoreactive peptides.Fig. 5. Hydrophilic profiles (probabilities of formation of antigenic determinants) for cytochromes P450scc and P45011beta calculated according to Hop [39]. The minimal design-basis element was the hexapeptide. For the denotations, see Fig. 4.
Because in our study, due to a low yield of peptide material, only four immunoreactive fractions have been characterized, it is necessary to continue the investigation of antigenic structure of cytochrome P450scc. As shown earlier, one of the antigenic sites of cytochrome P450scc (fraction IV-1-1) is located within the region Lys103-Leu114 containing the modifiable by succinic anhydride Lys103 and Lys104 [11]. It should be noted that the region Val470-Phe474 (fraction II-1-4) is also immunogenic. This is an interesting fact because it indicates the presence of antigenic determinants in the C-terminal sequence of fragment F2, for as we showed earlier, anti-F2 antibodies bind both with submitochondrial particles (SMP, formed during ultrasonic treatment of mitochondria with subsequent sedimentation at 105,000g for 1 h) and mitoplasts, while anti-F3 antibodies were shown to bind only with SMP, i.e., localization of the antigenic sites within the region Trp400-Ala481 would confirm that the C-terminal sequence of F2 contain regions is exposed to the cytosol. An indication of this suggestion is the fact that the antibodies raised against synthetic C-terminal peptide of rat cytochrome P450scc react with the antigen both in solution and in sections of adrenocortical tissue [45].
Investigation of the antigenic structure of cytochrome P450scc is a continuation of the structural--functional study of the heme protein based on immunochemical approaches started in our previous paper [25]. Comparison of the data of immunochemical analysis and those obtained using chemical modification and examination of covalent complexes suggests that the central part of the cytochrome P450scc molecule and fragment F3 are the most probable regions to form the AD-binding site of cytochrome P450scc. This suggestion is supported by data indicating that phosphorylation of cytochrome P450scc by protein kinase C may occur at Thr253, and AR and cytochrome b5 which form strong complexes with cytochrome P450scc protects the heme protein from phosphorylation [46]. Chemical modification of the free Cys264 located just near the antibody-binding site P2, decreases the affinity of the heme protein to AD and results in inhibition of cholesterol-hydrolyzing activity in the reconstituted system [47]. However, a detailed mapping of the AD-binding site requires data accounting for the spatial configuration of the cytochrome P450scc molecule.
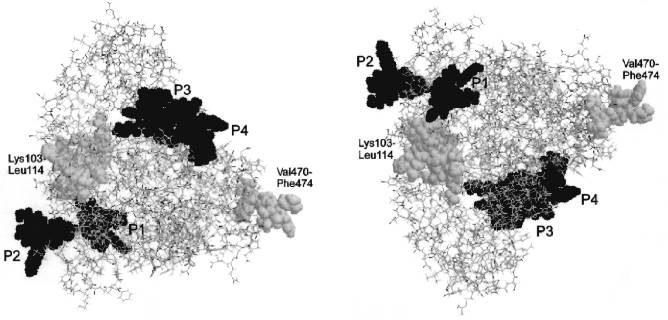
The published theoretical model for the tertiary structure of cytochrome P450scc molecule [48] based on the analysis and matching of the regions in the P450scc molecule homologous to those of cytochrome P450cam, whose crystal structure is known [49], indicates that the domains corresponding to the localized immunoreactive peptides are located in a peripheral part of the cytochrome P450scc molecule, within the region homologous to the alpha-helix G in cytochrome P450cam (peptide P1), in the loop between the helices G and I (peptide P2), and in a surface region Met382-Lys405 (peptides P3 and P4) (Fig. 6). In addition, according to this model, the identified immunoreactive region in the N-terminal sequence including Lys103-Trp108 and/or Lys109-Leu114, and also in the C-terminal region of cytochrome P450scc with putative immunoreactive peptide Val470-Phe474, are also exposed on the surface of protein globule.
The authors express their gratitude to Dr. T. B. Adamovich, a senior scientist of the Institute of Bioorganic Chemistry, Academy of Sciences of Belarus, for help in structural analysis of the peptide fractions.Fig. 6. Tertiary structure of cytochromes P450scc and P45011beta reconstructed according to Vijayakumar and Salerno [48]. Either image is transformed into the other one by a 180°-turn about the horizontal axis. The antigenic sites corresponding to immunoreactive peptides P1, P2, P3, and P4 are given in black, and the putative antigenic sites Lys103-Leu114 and Val470-Phe474 are given in gray color.
LITERATURE CITED
1.Usanov, S. A., Chashchin, V. L., and Akhrem, A. A.
(1990) in Molecular Mechanisms of Adrenal Steroidogenesis and
Aspects of Regulations and Application (Ruckpaul, K., and Rein, H.,
eds.) Frontiers in Biotransformation, Vol. 3, Akademie-Verlag,
Berlin, pp. 1-57.
2.Usanov, S. A., Pikuleva, I. A., Chashchin, V. L.,
and Akhrem, A. A. (1984) Biochim. Biophys. Acta, 790,
259-267.
3.Tsubaki, M., Iwamoto, Y., Hiwatashi, A., and
Ichikawa, Y. (1989) Biochemistry, 28, 6899-6907.
4.Tulls, J., Geren, L., and Millet, F. (1989) J.
Biol. Chem., 264, 16421-16425.
5.Tsujita, M., and Ichikawa, Y. (1993) Biochim.
Biophys. Acta, 1161, 124-130.
6.Akhrem, A. A., Shkumatov, V. M., and Chashchin, V.
L. (1977) Bioorg. Khim., 3, 1064-1069.
7.Geren, L. M., O’Brien, P., Stonenhuerner, L.,
and Millet, F. (1984) J. Biol. Chem., 259,
2155-2160.
8.Coghlan, V. M., and Vickery, L. E. (1991) J.
Biol. Chem., 266, 18606-18612.
9.Coghlan, V. M., and Vickery, L. E. (1992) J.
Biol. Chem., 267, 8932-8935.
10.Palin, M. F., Sygush, J., and LeHoux, J. G.
(1994) in Cytochrome P450. Biochemistry, Biophysics, and Molecular
Biology (Lechner, M. C., ed.) John Libbey Eurotext, Paris, pp.
721-724.
11.Adamovich, T. B., Pikuleva, I. A., Chashchin, V.
L., and Usanov, S. A. (1991) Biochim. Biophys. Acta, 996,
247-253.
12.Lapko, A. G., Smettan, G., Ruckpaul, K., and
Usanov, S. A. (1991) Bioorg. Khim., 17, 921-932.
13.Adamovich, T. B., Pikuleva, I. A., Usanov, S. A.,
and Chashchin, V. L. (1989) Biokhimiya, 54,
1206-1215.
14.Wada, A., and Waterman, M. R. (1992) J. Biol.
Chem., 267, 22877-22882.
15.Usanov, S. A., Turko, I. V., Akhrem, A. A., and
Chashchin, V. L. (1984) Biokhimiya, 49, 1810-1818.
16.Usanov, S. A., Chernogolov, A. A., and Chashchin,
V. L. (1989) FEBS Lett., 255, 125-128.
17.Efimov, A. V., Chernogolov, A. A., Petrashin, A.
I., Kiril’chik, E. P., Usanov, S. A., and Chashchin, V. L. (1990)
Biomed. Sci., 1, 171-177.
18.Usanov, S. A., Chernogolov, A. A., Akhrem, A. A.,
and Chashchin, V. L. (1987) Biokhimiya, 52, 110-122.
19.Sugano, S., Ohnishi, T., Hatae, N., Ishimura, K.,
Fujita, H., Yamano, T., and Okamoto, M. (1985) J. Steroid
Biochem., 23, 1013-1021.
20.Usanov, S. A., Chernogolov, A. A., and Chashchin,
V. L. (1990) FEBS Lett., 275, 33-35.
21.Edwards, R. J., Murray, B. P., and Boobis, A. R.
(1991) Meth. Enzymol., 206, 220-223.
22.De Lemos-Chiarandini, C., Frey, A. B., Sabatini,
D. D., and Kreibich, G. (1987) J. Cell. Biol., 104,
209-219.
23.Usanov, S. A., Chernogolov, A. A., Petrashin, A.
I., Akhrem, A. A., and Chashchin, V. L. (1987) Biol. Membr.
(Moscow), 4, 1102-1116.
24.Black, M. D., Martin, S. T., and Smith, C. A.
(1994) Biochemistry, 33, 6945-6951.
25.Chernogolov, A. A., and Usanov, S. A. (1997)
Biochemistry (Moscow), 62, 1609-1619 (Russ.).
26.Usanov, S. A., Pikuleva, I. A., Chashchin, V. L.,
and Akhrem, A. A. (1984) Bioorg. Khim., 10, 32-45.
27.Omura, T., and Sato, R. (1964) J. Biol.
Chem., 239, 2370-2378.
28.Phillips, A. P., Martin, K. L., and Horton, W. H.
(1984) J. Immunol. Meth., 74, 385-393.
29.Strittmatter, P. (1960) J. Biol. Chem.,
235, 2492-2497.
30.Crestfield, A. M., Moore, S., and Stein, V. H.
(1963) J. Biol. Chem., 238, 622-627.
31.Gray, W. R., and Hartley, B. S. (1963)
Biochem. J., 89, 235.
32.Bruton, C. J., and Hartly, B. C. (1970) J.
Mol. Biol., 52, 165-178.
33.Pacman, D. H., Stein, W. H., and Moore, S. (1958)
Anal. Biochem., 30, 1190-1205.
34.Reddehase, M. J., Rothbard, J. B., and
Koszinovski, U. H. (1989) Nature, 337, 651-653.
35.Chashchin, V. L., Lapko, V. N., Adamovich, T. B.,
Lapko, A. G., Kuprina, N. S., and Akhrem, A. A. (1982) Bioorg.
Khim., 8, 1307-1320.
36.Chashchin, V. L., Lapko, V. N., Adamovich, T. B.,
Lapko, A. G., Kuprina, N. S., Kirillova, N. M., Berikbaeva, T. M.,
Akhrem, A. A., and Zolotarev, A. I. (1985) Bioorg. Khim.,
11, 1048-1067.
37.Morohashi, K., Yoshioka, H., Gotoh, O., Okada,
K., Yamamoto, K., Miyata, T., Sogawa, K., Fujii-Kuriuama, Y., and
Omura, T. (1984) Proc. Natl. Acad. Sci. USA, 81,
4647-4651.
38.Turko, I. V., Adamovich, T. B., Kirillova, N. M.,
Usanov, S. A., and Chashchin, V. L. (1989) Biochim. Biophys.
Acta, 996, 37-42.
39.Hop, T. P. (1988) J. Immunol. Meth.,
88, 1-18.
40.Degli Esposti, D., Chelli, A., Crimi, M., and
Lenaz, G. (1989) Ital. J. Biochem., 38, 1-22.
41.Penke, B., Ferenczi, R., and Coracs, K. (1974)
Anal. Biochem., 60, 45-50.
42.Novotny, J., and Auffray, C. (1984) Nucleic
Acids Res., 12, 243-255.
43.Chou, P. Y., and Fasman, G. D. (1974)
Biochemistry, 13, 211-222.
44.Rose, G. D., and Roy, S. (1980) Proc. Natl.
Acad. Sci. USA, 77, 4643-4647.
45.Roby, K. F., Larsen, D., Deb, S., and Soares, M.
(1991) Mol. Cell Endocrinol., 79, 13-20.
46.Lobanov, N. A., Hansey, K., Usanov, S. A., and
Azzi, A. (1993) Biochemistry (Moscow), 58, 1529-1537
(Russ.).
47.Chernogolov, A. A., Usanov, S. A., and Shvarts,
D. (1996) Biochemistry (Moscow), 61, 2103-2115
(Russ.).
48.Vijayakumar, S., and Salerno, J. C. (1992)
Biochim. Biophys. Acta, 1160, 281-286.
49.Poulos, T. L., Finzel, B. C., and Howard, A. J.
(1987) J. Mol. Biol., 195, 687-700.