REVIEW: Effect of Lysophosphatidylcholine on Transmembrane Signal Transduction
N. V. Prokazova1*, N. D. Zvezdina2, and A. A. Korotaeva1
1Institute of Experimental Cardiology, Cardiology Research Center, ul. 3-ya Cherepkovskaya 15a, Moscow, 121552 Russia; fax: (095) 415-29622Kol'tsov Institute of Developmental Biology, Russian Academy of Sciences, ul. Vavilova 26, Moscow, 117334 Russia; fax: (095) 135-8012
* To whom correspondence should be addressed.
Received July 14, 1997
Lysophosphatidylcholine (LPC), 1-acyl-sn-glycero-3-phosphocholine, is well known as an intermediate of metabolism of phosphatidylcholine (PC), the main phospholipid component in all eukaryotic and many prokaryotic cells. LPC is produced as a result of PC hydrolysis by several isoforms of phospholipase A2 (PLA2) and in the reaction mediated by lecithin-cholesterol acyltransferase that transfers the fatty acid residue from PC to cholesterol. LPC is classified as a second messengers that is produced by activation of cytosolic hormone-activated PLA2. It was shown that LPC inhibits transmembrane signaling via receptors, which in their active form are linked to G-proteins. There is a viewpoint that LPC abolishes formation of the complex between the receptor and G-protein. The effect of LPC on protein kinase C (PKC) activation is considered in this review. It was shown that low (less than 20 µM) and high (more than 30 µM) concentrations of LPC activated and inhibited PKC, respectively. The mechanism of LPC-induced activation of PKC still remains unclear. However, the studies of the effect of LPC on signal transduction through the PKC-mediated pathway showed that LPC probably plays an auxiliary role. It was suggested that LPC may prolong the effect of the direct activators of PKC (such as 1,2-diacylglycerol or phorbol esters). The physiological role of the elevation of LPC level in tissues is associated with its ability to enhance or even evoke cell proliferation, stimulate adhesion and differentiation of lymphoid cells, have mitogenic effect on macrophages, activate human T-lymphocytes, initiate monocyte chemotaxis, decrease myocardial sensitivity to cholinergic stimulation, impair contractility of arterial smooth muscle, and modulate aggregation of platelets.
KEY WORDS: lysophosphatidylcholine, phospholipase A2, G-proteins, protein kinase C
Abbreviations: LPC) lysophosphatidylcholine (1-acyl-sn-glycero-3-phosphocholine); DAG) 1,2-diacylglycerols; UFA) unsaturated fatty acids; PAF) platelet-activating factor; PKC) protein kinase C; PLA2, PLC) phospholipases A2 and C, respectively.
Studies of lipid second messengers in signal transduction continue as
new data on the biological activity of lipids of different classes
appear. LPC is a well-known intermediate of metabolism of
phosphatidylcholine, the main phospholipid component in all eukaryotic
and many prokaryotic cells. LPC is produced as a result of hydrolysis
of phosphatidylcholine with various isoforms of phospholipase
A2 or in reactions catalyzed by lecithin-cholesterol
acyltransferase, which transfers a fatty acid residue from
phosphatidylcholine to cholesterol and is the key enzyme in lipoprotein
metabolism in animal and human blood plasma. The real LPC content in
cells can hardly be measured because its quantitative determination
requires isolation of lipids from tissues, cells, and biological
liquids, during which this lipid may occur as an artifact. According to
published data, LPC content in tissues does not exceed 5 mole % of the
total phospholipids. The sole exception is animal and human blood
plasma, in which LPC content reaches 20% of the total phospholipids. In
the plasma LPC is present mainly in complexes with albumin and other
proteins. During recent decades many reports which describe a
significant elevation of LPC level in cells and tissues under different
diseases have been published.
During almost four decades the problem of the biological activity of LPC has attracted the attention of researchers in biological and medical chemistry. In the middle of 1960s the membrane-modulating properties of LPC were intensely studied. These properties are manifested at concentrations of LPC higher than 50 µM. The majority of scientists noted that the effects of LPC could be characterized as "everything or nothing", i.e., the effect was not observed up to a certain concentration, but after reaching the critical concentration of micelle formation it dramatically occurred. Based on these data, it was suggested that LPC is only a cytolytic lipid factor. For this reason, in the 1970s its biological functions were studied considerably less intensively. However, in this period we discovered a specific effect of LPC. It appeared that LPC at the concentration of 3 µM decreased the sensitivity of the myocardium of various animals to acetylcholine [1]. Later it was shown that LPC at low concentrations enhances or induces cell proliferation [2], stimulates adhesion and differentiation of lymphoid cells [2-5], has mitogenic effect on macrophages [6], activates T-lymphocytes [7], induces monocyte chemotaxis [8], modulates muscle contractility [9, 10] and platelet aggregation [11], etc. (see the table). With regard to these findings, in the 1990s another mechanism of action of this bifunctional lipid was proposed. It is clearly established to date that LPC is produced in cell membranes under the action of different external signals and modulates their effect. Although the mechanism of action of LPC is not completely understood, it is classified now with lipid second messengers.
Regulation of transmembrane signaling by LPC
| Organ or cell | Effect | Mechanism | References |
|---|---|---|---|
| Mouse and human monocytes | chemotaxis | PKC activation | [8, 41] |
| Human T-lymphocytes | activation | PKC activation | [7] |
| HL-60 cells (transformed human leukocytes) | differentiation | PKC activation | [5] |
| Human arterial endothelial cells | induction of adhesion of human monocytes, | inhibition of degradation or processing of mRNA | [2] |
| human and pig leukocytes or TPH-1 cells | PKC activation and enhancement of expression of ICAM6 and VCAM | [3, 42, 4] | |
| Mouse macrophages | mitogenic effect | PKC activation | [6] |
| Mouse macrophage foam cells | cholesterol excretion | activation of apolipoprotein apoE secretion | [46] |
| Rat glial cells | modulation of ionic currents | unknown | [43] |
| Human platelets | inhibition of aggregation and receptor-dependent Ca2+ regulation | disruption of transmembrane signaling | [11] |
| Human platelets and endothelial cells | enhancement of P-selectin expression | PKC activation | [44] |
| Rabbit cardiomyocytes | modification and synchronization of Na-channel gating | G-protein-dependent mechanism, formation of Na-channel dimers and trimers | [33] |
| Pig coronary artery | suppression of endothelium-dependent hyperpolarization | PKC activation | [10] |
| Rabbit aorta | suppression of endothelium-dependent relaxation | PKC activation | [9, 37] |
| Pig mesenteric artery | suppression of endothelium-dependent relaxation | inhibition of phosphoinositide hydrolysis | [38] |
| Rat myocardium | adenylate cyclase activation, Ca2+ influx through "slow" Ca-channels, and accelerated Na+/Ca2+ exchange | PKC activation, inhibition of Na,K-ATPase, modulation of Na-channel gating | [45] |
| Frog myocardium | decrease of sensitivity to acetylcholine | cooperativity in binding of the ligand to receptor | [1, 34] |
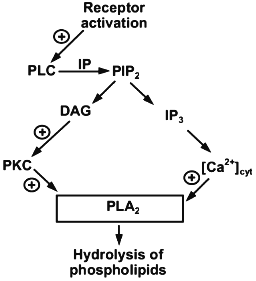
LPC is a second messenger resulting from the activation of hormone-activated cytosolic phospholipase A2 (Fig. 1). This enzyme significantly differs from the secreted form of phospholipase A2 and enzymes found in the pancreas and snake or bee venom. The molecular weight of cytosolic phospholipase A2 is 6 times higher than the secreted form. In addition, it is activated by Ca2+ concentrations which are an order lower than those required for the activation of the secreted form. Cytosolic phospholipase A2 is activated by hormones and agonists via G-protein-linked receptors and by growth factors or elevation of the concentration of intracellular Ca2+ [12, 13]. Activation is associated with translocation of the enzyme from the cytoplasm to membrane and results in the production of three types of second messengers, i.e., arachidonic acid (the precursor of prostaglandins), other unsaturated fatty acids, and LPC [13].
Hormonal activation of cytosolic phospholipase A2 is not the sole reason for the appearance of LPC in the cell membrane. It may also be derived from lipoproteins which are the main form of lipids. In this case, LPC is produced either in the reaction catalyzed by lecithin-cholesterol acyltransferase or as a result of oxidative processes due to activation of endogenous phospholipase A2 characteristic of apolipoprotein B [14]. During the interaction of lipoproteins with cells, LPC is probably actively incorporated into the cell membrane, because it was shown that the addition of LPC to the incubation medium entails its binding to cultured mammal cells. Exogenous LPC is detected in the cell membrane 30 sec after its addition to the incubation medium. Maximal incorporation of LPC into the membrane is observed 15 min after its addition to the incubation medium; LPC is maintained in the membranes for several hours. The bound LPC is metabolized to phosphatidylcholine via metabolic cell structures responsible for metabolizing endogenous LPC [15].Fig. 1. Activation of cytosolic phospholipase A2 by hormones and agonists (according to [12]).
LPC probably does not have its own binding sites and receptors on the plasma membrane. However, even 1-5 µM of LPC causes specific biological effects. LPC markedly modulates the effect of hormones and agonists, the signal from which is transmitted through G-proteins and PKC activation. Peculiarities of the mechanism of LPC action as a second messenger considerably depends on the type of cells, the animal species, and the mechanism of transduction of the signal from a particular agonist or hormone.
MODIFICATION OF G-PROTEIN-DEPENDENT SIGNALING
Flavahan studied the mechanism whereby LPC affects relaxation of the aorta induced by effectors with different mechanisms of action [16]. The motility of aorta smooth muscle tissues is regulated by factors secreted by endothelial cells, which cover the vessel on the side of the lumen. In particular, endothelium-dependent relaxation is mediated by the releasing endothelium-derived relaxation factor-NO (EDRF-NO), in response to the effect of serotonin and other agonists on specific receptors on endothelial cells, whose signals are realized via G-proteins. It was suggested that LPC at concentrations of 1-10 µM disrupt transduction of transmembrane signal that depends on Gi-protein, which is specifically inhibited by pertussis toxin (Fig. 2).
Gi-Proteins are present in vascular endothelial cells. Their activation either with receptor-dependent signals or due to the direct effect of fluoride ions evokes the release of endothelium-derived relaxation factor-NO. When studying the mechanism of LPC action on the vascular relaxation system, it was shown that 10 µM LPC inhibited endothelium-dependent relaxation caused by activation of serotonin and alpha2-adrenergic receptors, whose active form is linked to Gi-protein. Conversely, LPC had no effect on endothelium-dependent relaxation induced by bradykinin or ADP, whose receptors in active form are linked to pertussis toxin-insensitive G-proteins. In addition, LPC did not suppress vessel relaxation induced by direct stimulation of NO production. Moreover, LPC did not affect the ability of vascular smooth muscle to respond the endothelium-derived relaxation factors-NO themselves and did not suppress the ability of endothelium to synthesize and secrete these factors [16].Fig. 2. Hypothetical mechanism of effect of LPC on transmembrane signaling in endothelial cells (according to [16]). alpha2-AR, 5-HTR, B2R, and P2yR designate membrane alpha2-adrenergic, serotonin, bradykinin, and ADP/ATP receptors; Gi and Gq are G-proteins.
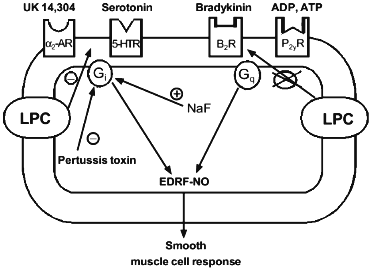
The following facts are also indicative of a specific effect of LPC. Serotonin receptors in endothelial cells are associated with at least two different intracellular processes, of which only one is mediated by Gi-proteins that are specifically inhibited by pertussis toxin. To establish which of the two processes is affected by LPC, the authors of [16] performed a comparative study of inhibitory effects of LPC and pertussis toxin. The latter suppressed serotonin-induced endothelium-dependent relaxation only partially. LPC suppressed serotonin-induced endothelium-dependent relaxation in the control artery and did not affect the residual endothelium-dependent relaxation observed after inactivation of Gi-protein with pertussis toxin. Based on these data, Flavahan suggested that the LPC-induced suppression of the response of endothelial cells to serotonin is due to inhibition of the Gi-protein. LPC suppressed the serotonin effect less effectively than the pertussis toxin. These data indicate that LPC probably disrupts the Gi-protein-dependent pathway of signal transduction; however, it does not make transduction of hormonal signals completely impossible [16].
It was found that LPC did not suppress relaxation of the aorta of experimental animals induced by fluoride ions, which directly activate G-proteins. After treatment with LPC, the response to fluoride ions could be inhibited by pertussis toxin (Fig. 2). Therefore, LPC does not change the nature of G-proteins activated with fluoride ion. Flavahan holds to the idea that LPC prevents formation of the complex between Gi-protein and the receptor [16]. He corroborated this hypothesis by the finding that sensitivity of Gi-protein-activated serotonin and alpha2-adrenergic receptors to LPC is different. For instance, LPC much more efficiently inhibited the response to serotonin than that to UK-14,304 (an activator of alpha2-adrenergic receptors). Receptors for different hormones differ in their ability to be activated by G-proteins and thus may differ in their sensitivity to auxiliary factors that affect the formation of an active complex. Actually, adrenergic activation by UK-14,304 causes more intensive Gi-protein-mediated response than serotonin-induced activation. This may reflect more effective complexation of alpha2-adrenoceptor to Gi-protein. On the contrary, this may cause a higher resistance of the alpha2-adrenergic response to the inhibitory effect of LPC [16].
We observed an inhibitory effect of LPC on the hormone-induced increase in the concentration of intracellular Ca2+ in platelets. This phenomenon is probably realized via impairing the receptor or Ca-channel and G-protein [11]. This assumption emerges from the pharmacological studies performed on platelets loaded with the fluorescent probe Fura-2. We found that a 2-min incubation of such platelets with 1-10 µM LPC prior to the treatment with the platelet-activating factor (PAF) almost completely suppressed the PAF-induced increase in Ca2+ concentration. Half-maximal inhibition was observed at 2-4 µM LPC.
The inhibitory effect of LPC on the platelet response depends on the structure of the lysophospholipid. We discovered that lysophosphatidylethanolamine did not modulate PAF influence and did not prevent the inhibitory effect of LPC. The metabolic precursor of LPC, phosphatidylcholine, also had no effect on the PAF-stimulated increase in the concentration of intracellular Ca2+ in platelets and did not affect the inhibition by LPC. Thus, it is likely that the absence of fatty acid residue in position 2 and the presence of a choline residue in the hydrophilic part of the phospholipid molecule are essential for manifestation of the inhibitory effect of LPC [11].
It was shown that 10 µM LPC completely inhibited ADP- and thrombin-induced increase of the Ca2+ concentration in platelets. It is known that PAF, ADP, and thrombin realize their aggregation effect on platelets via activation of phosphoinositide exchange. This results in an increase in intracellular concentration of Ca2+ due to its influx into the cell from the external medium and release from intracellular stores [17]. In the platelets LPC evidently affects both the hormone-dependent Ca2+ influx to cells and hormone-dependent Ca2+ release from the intracellular stores. The effect of LPC on Ca2+ release from endoplasmic reticulum is probably associated with its influence of the signal transduction across the plasma membrane rather than with permeability of platelet intracellular membranes. This assumption is confirmed by experiments with thapsigargin, a specific inhibitor of endoplasmic reticulum Ca2+-ATPase, which induces a passive Ca2+ efflux from intracellular stores to the cytoplasm. We show that LPC only slightly decreased the effect of thapsigargin in Ca2+-containing medium and did not affect the thapsigargin-induced increase of intracellular Ca2+ concentration in platelets in Ca2+-free medium. It is well known that thapsigargin first induces Ca2+ release from intracellular stores and then, as the latter becomes exhausted, activates Ca2+ influx from the extracellular medium [20]. Thus, LPC suppresses exactly Ca2+ influx into the thapsigargin-treated platelets [11].
The study of aggregation of platelets has shown that LPC at the concentration of 10-30 µM completely removes aggregation induced by PAF, ADP, and thrombin. However, LPC itself did not induce aggregation of platelets. LPC also did not affect aggregation of platelets induced by phorbol myristate acetate and thapsigargin, whose mechanism of action involves neither membrane receptors nor G-protein-activated ionic channels. Therefore, it is most likely that the inhibitory effect of LPC on the hormone-stimulated increase in intracellular Ca2+ concentration in platelets is realized via its effect on the Ca-channel itself (inhibitory effect of LPC on thapsigargin-induced Ca2+ influx) or due to uncoupling the receptors and G-proteins [11].
REGULATION OF TRANSMEMBRANE SIGNALING CAUSED BY PKC
ACTIVATION
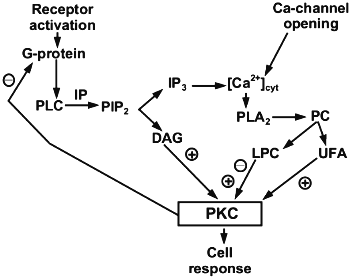
It is known that PKC participates in the cell response to different agonists including hormones, neurotransmitters, and growth factors. PKC is an enzyme family that comprises at least 12 isoforms in mammals. Activity of classic PKC isoform (cytosolic PKC alpha. beta. and gamma. depends on Ca2+ and phosphatidylserine. This enzyme is activated by an increase of DAG content in the membrane as a result of agonist-induced hydrolysis of phosphatidylinositol-4,5-bisphosphate. However, hydrolysis of other phospholipids, phosphatidylcholine in particular, may also affect PKC activity [18]. The effect of LPC on PKC activation and the mechanism underlying this process were studied on the enzyme isolated from rat brain. Its activity was assessed by the extent of phosphorylation of histone H1 (the major myelin protein) and two other myelin proteins with molecular masses of 35 and 47 kD, which were isolated from rat brain (Fig. 3) [19]. In these experiments LPC at low (less than 20 µM) and high (more than 30 µM) concentrations activated and inhibited PKC, respectively.
Such a two-phase mode of PKC regulation differs in LPC from the classic second messengers (i.e., DAG or UFA), which activate the enzyme over a wide range of concentrations and have no inhibitory effect even at very high concentrations. Other peculiarities of the LPC effect on PKC activity also are worth mention. First, the stimulatory effect of LPC can be registered only in the presence of phosphatidylserine and is associated with an increase in the affinity of the enzyme for this phospholipid. Second, similar to activation of the enzyme with DAG or phorbol myristate acetate, the LPC effect occurs in the presence of Ca2+. However, unlike DAG and phorbol esters, LPC decreased the affinity of the enzyme for Ca2+. Obviously, the mechanisms of action of the three listed classes of lipid second messengers (i.e., DAG, UFA, and LPC) are different. This assumption is confirmed by the fact that in vitro the effects of LPC and DAG on PKC activation were synergistic, while those of LPC and UFA were additive [19].Fig. 3. A possible role of LPC in PKC-dependent cell regulation (according to [21]). PIP2, IP3, and [Ca2+]cyt designate phosphatidylinositol-4,5-bisphosphate, inositol trisphosphate, and intracellular Ca2+, respectively; PC, phosphatidylcholine.
The mechanism of PKC activation by LPC is not completely understood. However, Oishi et al. reported on the high specificity of LPC towards PKC activation; they observed no activating effect of LPC on other protein kinases (i.e., myosin-light-chain kinase or cAMP-dependent protein kinase) [19]. Additionally, LPC displayed the ability to stimulate rat brain PKC in the case of phosphorylation of only the aforementioned myelin proteins, but not other proteins. This selectivity is possibly associated with the unique mode of binding of LPC to H1 or 35-kD and 47-kD proteins, as a result of which the affinity of LPC-protein complex for the enzyme increases.
The authors of [19] pointed out that DAG, UFA, and LPC the most efficiently activated PKC at concentrations of 5, 50, and 20 µM, respectively. Assuming that PKC activation is due to activation of only cytosolic PLA2, we find that LPC would be a more potent effector than UFA. At a moderate level of stimulation of cells according to this mechanism, LPC would be virtually the only activator, while at high levels of stimulation of cellular PLA2 the activating effect of UFA would be manifested along with the inhibitory effect of LPC. The latter at such a high concentration would play the role of negative effector of PKC regulation.
Nishizuka et al. obtained data that are different from those published previously [5, 7]. They reported that DAG pre-activation was necessary to reveal the LPC effect on PKC-dependent expression of interleukin-2 receptor and thymidine accumulation in T-lymphocytes. A similar result was observed when DAG was substituted with phorbol ester [7]. LPC stimulated PKC-dependent differentiation of HL-60 cells to macrophages, which was evaluated by the expression of a specific cell marker, CD11b. In either case the LPC effect was manifested only in the presence of DAG. This indicates its participation in the transmembrane signaling by a PKC-dependent mechanism. These results could not be adequately explained by the hypothesis of Oishi et al. [19]. For this reason, the authors of [20] also studied the effect of LPC on rat brain PKC activity in vitro. Unlike Oishi et al. [19], they studied regulation of PKC subclasses. Their results showed that 10 µM LPC markedly increased DAG-induced activation of alpha-. beta-. and gamma-isoforms of rat brain PKC. In the absence of DAG no effect of LPC on PKC activity was observed. It appeared that LPC either had no effect on other PKC isoforms or suppressed their activity. Similar to Oishi et al. [19], the authors of [20] also could not propose the exact mechanism of PKC activation. The results of this study imply that LPC may induce translocation of the regulatory part of the PKC molecule from the cytosol to the membrane, i.e., it performs the first necessary stage of signal transduction. When reviewing his studies on the effect of LPC on signal transduction via PKC-mediated pathway as well as the studies of other researchers of this phenomenon, Nishizuka concluded that LPC and UFA play an auxiliary role in the process of PKC activation. He suggests that these compounds prolong the effects of direct PKC activators (DAG or phorbol esters) [21].
The authors of [22] studied the effect of LPC on the activation of PKC from rabbit aorta. The effect of LPC was evaluated by the increase in the content of phosphorylated endogenous proteins in aorta and in the production of superoxide anion O2-, which result from PKC activation in artery smooth muscle. A specific inhibitor of PKC, calphostin C, abolished this effect of LPC. Additional evidence in favor of LPC-induced PKC activation is the similarity of the effects of LPC and the classic PKC activator, phorbol ester.
The data reported in [22] are inconsistent with pharmacological studies performed on pig aorta fragments [16]. Similar to the studies on platelets, in this study it was shown that the mechanism of PLC action is associated with uncoupling the signal transduction chain via G-proteins [11, 16]. However, these contradictions are probably due to the fact that in aorta tissue from different animal species and in human aorta the process of relaxation is mediated by different factors that are secreted by endothelial cells in response to hormonal effects. For instance, in the case of rabbit aorta, the endothelium-dependent relaxation is induced mainly by NO. This process in pig coronary artery and human aorta is induced not only by NO, but also by endothelium-derived hyperpolarizing factor [10]. In contrast to NO, which increases the level of intracellular Ca2+ in smooth muscle cells, hyperpolarizing factor opens K+-channels in these cells. Additionally, mechanism of LPC effects probably depends on the hormone or agonist, whose system of signal transduction is affected by LPC. Finally, the fact that PKC activation suppresses the pathway of signal transduction via G-proteins also should not be overlooked [17]. Activation of G-proteins results in an increase in Ca2+ level, which, in turn, would inevitably activate cytosolic PLA2 and produce additional amounts of LPC (Fig. 1). Therefore, it cannot be ruled out that LPC formed in the membrane may act as a metabolite which provides feedback in the hormonal response of the cell [21].
The above reasoning is corroborated in a study that revealed the inhibitory effect of LPC on the synthesis and secretion of endothelium-dependent relaxation factor by bradykinin-stimulated bovine aorta endothelial cells [23]. It was shown that LPC suppresses both bradykinin-induced production of inositol-1,4,5-trisphosphate and increase in the level of intracellular Ca2+ in aorta cells. The authors of [23] note that the mechanism of negative feedback, which blocks Ca2+-dependent signal transduction from agonist, is triggered via PKC activation. Therefore, any possible PKC activation in endothelial cells would suppress receptor-mediated processes associated with the synthesis of the relaxation factor. Since LPC suppresses both intermediate and final effects of bradykinin, the authors of [23] suggest that LPC as a well-known PKC activator is most likely to trigger the mechanism of negative feedback. Kugiyama et al. [24] in a study on human endothelial cell culture directly showed that 1 µM LPC in a dose-dependent manner inhibits bradykinin-, thrombin-, and histamine-induced increase in the concentration of inositol-1,4,5-trisphosphate and Ca2+ in these cells as well as moderately activates PKC. They suggested that it is precisely PKC activation which can be partially involved (on the principle of negative feedback) in the inhibition of early stage of transmembrane signal transduction which is observed during the effect of LPC on endothelial cells [24].
PHYSIOLOGICAL ROLE
Currently it is suggested that physiological role of the increase in LPC level in tissues is associated with its effect on contraction and relaxation abilities of smooth muscle (see the table). Several authors independently detected an increase in the level of this lipid in myocardium as a result of experimental ischemic lesion of the myocardium in animals. Quantitative data vary over a wide range (the average level of LPC in the case of ischemia increases 2-3 times) [25-29]. Study of the effect of exogenous LPC on isolated myocardium showed that it influences opening of Na-channels. As a result, the tonus of heart contractions sharply changes. There is a hypothesis on the relationship between disruption of heart rhythm during ischemia and accumulation of LPC in the myocardium. Because the rhythm of contractions depends on Ca2+ level in cardiomyocytes of myocardium, the effect of LPC on cardiomyocytes in tissue culture was studied. The data showed that exogenous LPC increases intracellular Ca2+ level due to its effect both on influx of extracellular Ca2+ and mobilization of Ca2+ from intracellular stores [30-33].
We revealed a decrease in the sensitivity of the myocardium of different animals to acetylcholine in the presence of LPC [1]. In further studies on rabbit atrial cell membranes we demonstrated that LPC affects the binding of acetylcholine antagonist to the muscarinic receptor. Analysis of the results showed that the mechanism of this effect is associated with the ability of muscarinic receptors to form oligomeric complexes in the presence of LPC, which manifest positive cooperativity in binding to ligand. These data suggest that LPC production the myocardium stimulated by hormone-sensitive PLA2 may play an important role in the regulation of cholinergic process [34].
In this review we considered papers which describe effects of LPC on relaxation of arteries in experimental animals because the authors of these papers sought to reveal the mechanism of the physiological effect of LPC. This problem is of great interest because a significant increase in the level of LPC (bound to oxidized forms of lipoproteins, low-density lipoproteins in particular) in the blood serum in the case of atherosclerosis was unambiguously demonstrated [35]. Oxidized low-density lipoproteins can disrupt endothelium-dependent relaxation of arteries induced by such physiological effectors as thrombin, serotonin, or acetylcholine [36-38]. At present, all accumulated experimental data confirm the assumption that it is LPC that provides the inhibitory effect of oxidized low-density lipoproteins on endothelium-dependent relaxation. This is one of the main lesions in the case of atherosclerosis [39]. Data obtained by Men'shikovskii on the increase in PLA2 content in atherosclerotic lesions of arteries [40] suggest that LPC may be produced in the vessel wall due to PLA2 activation stimulated by the development of atherosclerotic lesions of the vessel. Comparison of all mentioned data shows that LPC participates in the regulation of arterial vascular tonus under normal conditions and especially in the case of atherosclerosis as a second messenger, which provides transduction of the signal across the plasma membrane.
In this review we focused on those LPC effects that affect transmembrane signal transduction. However, at concentrations higher than the critical concentration of micelle formation, LPC can cause rearrangements of the lipid bilayer, which result in the occurrence of nonspecific permeability of membranes. Therefore, in terms of physiological LPC effects, we should mention only its reversible effect on the mechanisms of receptor-mediated signal transduction, gating of ionic channels, and activation of membrane enzymes. We should also note that LPC concentrations in cells and tissues (such as blood plasma, myocardium in the case of ischemia, or platelets) are not always the operating physiologically active concentrations, because LPC, which is characterized by a high affinity for hydrophobic proteins (e.g., albumin) can form inactive pools.
The data considered unambiguously show that LPC, which is produced during activation of hormone-sensitive PLA2 and affects signal transduction via G-protein or PKC activation, may act as a second messenger in transmembrane signaling.
REFERENCES
1.Zvezdina, N. D., Prokazova, N. V., Vaver, V. A.,
Bergelson, L. D., and Turpaev, T. M. (1978) Biochem. Pharmacol.,
27, 2793-2801.
2.Nakano, T., Raines, E. W., Abraham, J. A.,
Klagsburn, M., and Ross, R. (1994) Proc. Natl. Acad. Sci. USA,
91, 1069-1073.
3.Kume, N., Cybulsky, M. I., and Gimborne, M. A.
(1992) J. Clin. Invest., 90, 1138-1144.
4.Yokote, K., Morisaki, N., Zenibayashi, M., Ueda,
S., Kanzaki, T., Saito, Y., and Yoshida, S. (1993) Eur. J.
Biochem., 217, 723-729.
5.Asaoka, Y., Yoshida, K., Sasaki, Y., and Nishizuka,
Y. (1993) Proc. Natl. Acad. Sci. USA, 90, 4917-4921.
6.Sakai, M., Miyazaki, A., Hakamata, H., Sasaki, T.,
Yui, S., Yamazaki, M., Shichiri, M., and Horiuchi, S. (1994) J.
Biol. Chem., 269, 31430-31435.
7.Asaoka, Y., Oka, M., Yoshida, K., Sasaki, Y., and
Nishizuka, Y. (1992) Proc. Natl. Acad. Sci. USA, 89,
6447-6451.
8.Quinn, M. T., Kondratenko, N., and Parthasarathy,
S. (1991) Biochim. Biophys. Acta, 1082, 293-302.
9.Murohara, T., Kugiyama, K., Ohgushi, M., Sugiyama,
S., Ohta, Y., and Yasue, H. (1994) Am. J. Physiol., 267,
H2441-H2449.
10.Eizawa, H., Yui, Y., Inoue, R., Kosuga, K.,
Hattori, R., Aoyama, T., and Sasayama, S. (1995) Circulation,
92, 3520-3526.
11.Korotaeva, A. A., Cheglakov, I. V., and
Prokazova, N. V. (1997) Platelets, 8, 43-51.
12.Axelrod, J., Burch, R. M., and Jelsema, C. L.
(1988) Trends Neurosci., 3, 117-123.
13.McKean, M., Smith, J. B., and Silver, M. Y.
(1981) J. Biol. Chem., 256, 1522-1524.
14.Parthasarathy, S., and Barnett, J. (1990)
Proc. Natl. Acad. Sci. USA, 87, 9741-9745.
15.Besterman, J. M., and Dmanico, P. L. (1992)
Biochemistry, 31, 2046-2056.
16.Flavahan, N. A. (1993) Am. J. Physiol.,
264, H722-H727.
17.Avdonin, P. V., and Tkachuk, V. A. (1994)
Receptors and Intracellular Calcium [in Russian], Nauka, Moscow,
pp. 29-42.
18.Exton, J. H. (1990) J. Biol. Chem.,
265, 1-4.
19.Oishi, K., Raynor, R. L., Charp, P. A., and Kuo,
J. F. (1988) J. Biol. Chem., 263, 6865-6871.
20.Sasaki, Y., Asaoka, Y., and Nishizuka, Y. (1993)
FEBS Lett., 320, 47-51.
21.Nishizuka, T. (1992) Science, 258,
607-614.
22.Ohara, Y., Peterson, T. E., Zheng, B., Kuo, J.
F., and Harrson, D. G. (1994) Atheroscler. Thromb., 14,
1007-1013.
23.Inoue, N., Hirata, K., Tamada, M., Hamamori, Y.,
Matsuda, Y., Akita, H., and Yokoyama, M. (1992) Circ. Res.,
71, 1410-1421.
24.Kugiyama, K., Ohgusi, M., Sugiyama, S., Murohara,
T., Fukunaga, K., Miyamoto, E., and Yasue, H. (1992) Circ. Res.,
71, 1422-1428.
25.Shaikh, N. A., and Downar, E. (1981) Circ.
Res., 49, 316-325.
26.Pogowizd, S. M., Onufer, J. R., Kramer, J. B.,
Sobel, B. E., and Corr, P. B. (1986) Circ. Res., 59,
416-426.
27.Mock, T., and Man, R. Y. K. (1990) Lipids,
25, 357-362.
28.Tanaka, H., Ikeda, U., and Shigenobu, K. (1993)
Gen. Pharmacol., 1, 239-241.
29.Sato, T., Isheda, H., Nakazawa, H., and Arita, M.
(1996) J. Mol. Cell. Cardiol., 28, 2183-2194.
30.Chen, M., Hashizume, H., and Abiko, Y. (1996)
Br. J. Pharmacol., 118, 865-870.
31.Chen, M., Hashizume, H., Xiao, C. Y., Hara, A.,
and Abiko, Y. (1997) Life Sci., 60, 57-60.
32.Kang, J. X., and Leaf, A. (1996) Eur. J.
Pharmacol., 297, 97-100.
33.Undrovinas, A. I., Fleidervish, I. A., and
Makielski, J. C. (1992) Circ. Res., 71, 1231-1241.
34.Suslova, I. V., Korotaeva, A. A. and Prokazova,
N. V. (1995) Dokl. Ros. Akad. Nauk, 342, 273-276.
35.Steinberg, D., Parthasarathy, S., Carew, T. E.,
Khoo, J. C., and Witztum, J. M. (1989) New Engl. J. Med.,
320, 915-924.
36.Mangin, E. L., Kugiyama, K., Nguy, J. H., Kerns,
S. A., and Henry, P. D. (1993) Circ. Res., 72,
161-166.
37.Kugiyama, K., Kerns, S. A., Morrisett, J. D.,
Roberts, R., and Henry, P. D. (1990) Nature, 344,
160-162.
38.Fukao, M., Hattori, Y., Kanno, M., Sakuma, I.,
and Kitabatake, A. (1995) Br. J. Pharmacol., 116,
1541-1544.
39.Flavahan, N. A. (1992) Circulation,
85, 1927-1938.
40.Menschikowski, M., Kasper, M., Lattke, P.,
Schiering, A., Schiefer, S., Stockinger, H., and Jaross, W. (1995)
Atherosclerosis, 118, 173-181.
41.Quinn, M. T., Parthasarathy, S., and Steinberg,
D. (1988) Proc. Natl. Acad. Sci. USA, 85, 2805-2809.
42.Murohara, T., Scalia, R., and Lefer, A. M. (1996)
Circ. Res., 78, 780-789.
43.Ikeuchi, Y., Nishiizaki, T., and Matsuoka, T.
(1995) Biochem. Biophys. Res. Commun., 217, 811-816.
44.Sugiyama, S., Kugiyama, K., Ohgushi, M.,
Fujimoto, K., and Yasue, H. (1994) Circ. Res., 74,
565-575.
45.Hoque, A. N. E., Hoque, N., Hashizume, H., and
Abiko, Y. (1994) Jpn. J. Pharmacol., 67, 233-241.
46.Hara, S., Shike, T., Takasu, N., and Mizui, T.
(1997) Arterioscler. Thromb. Vasc. Biol., 17,
1258-1266.