REVIEW: The Roles of Ceramide in the Regulation of Neuronal Growth and Development
A. H. Futerman
Department of Membrane Research and Biophysics, Weizmann Institute of Science, Rehovot, 76100, Israel; fax: 972-8-9344112; E-mail: bmfuter@weizmann.weizmann.ac.il
Received August 15, 1997
Ceramide can be formed by the activity of two general metabolic pathways, the anabolic pathway, in which ceramide is formed by acylation of a sphingoid long chain base, and the catabolic pathway, in which ceramide is formed by the degradation of either glycosphingolipids or of sphingomyelin (SM). The anabolic reactions take place in the early compartments of the secretory pathway (the endoplasmic reticulum and the Golgi apparatus) and the catabolic reactions take place either in lysosomes or at the plasma membrane. Work from our and other laboratories has shown that neuronal growth and development can be regulated by manipulating ceramide metabolism. Thus, synthesis of glucosylceramide from ceramide is required for axonal growth in cultured hippocampal neurons, but the formation of ceramide from SM, by a sphingomyelinase activity, stimulates the earliest stages of development in these cells, namely the formation of minor neuronal processes and the initial formation of the axon. Thus, ceramide and its metabolites play distinct roles in the same neuron, depending on the intracellular site of generation of ceramide and on the stage of neuronal development.
KEY WORDS: ceramide, glucosylceramide, inhibition, stereoisomers, neurons, neurotrophins, secretory pathway
Abbreviations: BDNF) brain-derived neurotrophic factor; ER) endoplasmic reticulum; FB1) fumonisin B1; bFGF) basic fibroblast growth factor; GSL) glycosphingolipids; SL) sphingolipids; SM) sphingomyelin; SMase) sphingomyelinase; PM) plasma membrane; HFA) 2-hydroxy fatty acid; NFA) normal fatty acid; NGF) nerve growth factor; C6-NBD-Cer) fluorescence labeled ceramide containing C6-acid.
Until the late 1980s--early 1990s, ceramide was considered to be little
more than an intermediate in the metabolic pathways of sphingolipid
(SL) metabolism, albeit an important intermediate since it is a
precursor for the synthesis of both sphingomyelin (SM) and the
glycosphingolipids (GSLs). A dramatic and unexpected shift of interest
in ceramide took place when it was observed that upon treatment of
cells with a variety of agents, ceramide was generated from SM,
presumably due to activation of a membrane-bound sphingomyelinase
(SMase). Interest in the role of ceramide as a lipid effector has
exploded during the past 6-7 years, with a large number of research
groups actively studying the roles of ceramide in various signaling
systems. It is now believed that ceramide plays important roles in
apoptosis, differentiation, cell cycle arrest, and senescence [1].
Despite the proliferation of studies examining ceramide as an intracellular signaling mediator in various cells, little work has been performed to analyze the role of ceramide as a modulator of cell development and growth in the nervous system. Some studies have analyzed the generation of ceramide in neuronal or glial cell lines upon stimulation by trophic factors, but less work has been done in either primary cultures of neurons, or in nervous tissue in situ. In the current manuscript, I will summarize work from both our laboratory and other laboratories that demonstrates that ceramide plays an important role in regulating neuronal growth and development, but that the exact role played by ceramide appears to depend in part on its intracellular site of generation and subsequent metabolism. As a prerequisite for understanding the roles of ceramide, I will discuss the intracellular sites of ceramide metabolism in the first part of this manuscript, in both the anabolic and catabolic pathways, and in the second part will proceed to discuss the possible roles of ceramide in regulating defined stages of neuronal and glial development.
THE INTRACELLULAR SITES OF CERAMIDE METABOLISM
Ceramide is a key intermediate in both the anabolic and catabolic pathways of SL metabolism. In general, most of the anabolic reactions take place in the early compartments of the secretory pathway, namely the endoplasmic reticulum (ER) and the Golgi apparatus (reviewed in [2]), whereas the catabolic reactions take place either in lysosomes (reviewed in [3]) or at the plasma membrane (PM). Most of the enzymes associated with ceramide catabolism have been purified, cloned, and sequenced, whereas only two of the enzymes involved in ceramide anabolism have been isolated. Much of the interest in ceramide catabolism is due to the lysosomal storage diseases, in which the immediate precursors of ceramide are not degraded, and thus ceramide is not generated, or ceramide itself is not degraded and thus accumulates. Interestingly, no human metabolic defects in SL synthesis have been reported, perhaps since such defects would be lethal.
The subcellular localization of metabolic reactions can be determined by a variety of techniques. Preeminent among these is immunohistochemical localization of a purified enzyme. However, this requires antibodies against the enzyme in question, which itself requires the isolation and purification of the enzyme. Due to inherent difficulties in isolating membrane-bound enzymes, little progress has been made towards purifying the enzymes of ceramide and SL synthesis. Thus, the intracellular sites of SL synthesis have for the most part been determined in highly purified subcellular fractions. This approach has been successfully employed in non-neuronal tissues (i.e., liver), but has been far less successful in neuronal tissues. Although there is no direct evidence that the intracellular sites of ceramide metabolism differ between neurons and non-neuronal tissues, nor any obvious rationale as to why they should differ, this issue has not been directly tested, so some caution is warranted in extrapolating data obtained from non-neuronal tissues to those of neuronal origin.
SL synthesis begins with the condensation of palmitoyl CoA and serine by serine palmitoyl transferase, which has been localized to the cytosolic surface of the ER in mouse liver subcellular fractions [4]. This condensation reaction yields 3-ketosphinganine, which is subsequently reduced to form sphinganine. It is now known that sphinganine (rather than its unsaturated counterpart, sphingosine) is the primary substrate for acylation by an N-acyl transferase, yielding dihydroceramide. The localization of dihydroceramide synthase to the ER has been unambiguously demonstrated using subcellular fractions from either mouse [4] or rat liver [5]. This reaction, and subsequent anabolic reactions in which ceramide is involved, are summarized in Table 1, which also documents progress in the isolation of some of the enzymes of the synthetic pathway.
Table 1. The metabolism of ceramide (see
text and references [2, 62]
for further details; key references are given in the table; for
abbreviations, see text)
| Enzyme | Substrate | Subcellular localization* | Metabolic disorder | Specific inhibitor** | Purified enzyme |
|---|---|---|---|---|---|
Synthetic (anabolic) pathway |
|||||
| Sphinganine N-acyl-transferase (dihydroceramide synthase) | sphinganine + fatty acyl CoA | cytosolic surface of ER [4, 5] | - | FB 1 and fumonisin B2, Alternaria toxin [63, 64] | - |
| Dihydroceramide dehydrogenase | dihydroceramide [65] | - | - | - | - |
| SM synthase | ceramide and phosphatidylcholine | lumenal surface [7] of cis and medial Golgi apparatus [7, 8, 66] | - | - | - |
| GlcCer synthase | ceramide and UDP-glucose | cytosolic surface of pre- or early Golgi apparatus [11-13, 67-69] | - | D-PDMP and PPMP [37, 70], N-butyldeoxynojirimycin [71] | [72, 73] |
| Ceramide kinase | ceramide and ATP | synaptic vesicles [16] and the PM [15] | - | - | - |
| UDP-galactose:ceramide galactosyltransferase (GalCer synthase) | ceramide and UDP-Gal | lumenal leaflet of ER (HFA) or cytosolic leaflet of Golgi (NFA) [13, 14] | - | - | [74, 75] |
Degradative (catabolic) pathway |
|||||
| Galactosyl-cerebrosidase | galactosylceramide | lysosomes | Krabbe disease | - | [76-78] |
| Glucocerebrosidase | glucosylceramide | lysosomes | Gaucher disease | conduritol B epoxide [79]; bromoconduritol B epoxide [80] | [81, 82] |
| Ceramide-1-phosphate phosphatase | ceramide-1-phosphate | PM [23] and synaptic vesicles [24] | - | - | - |
| Neutral and alkaline ceramidase | ceramide | - | - | (1S,2R)-D-erythro- 2-(N-myristoylamino)- 1-phenyl-1-propanol (D-erythro-MAPP) [83] | [25] |
| Acid ceramidase | ceramide | lysosomes | Farber disease | N-oleoylethanolamine [84, 85] | [86, 87] |
| Acid SMase | SM | lysosomes [88] | Niemann--Pick disease types A and B | - | [26, 89-92] |
| Neutral SMase | SM | PM [20, 21] | - | - | - |
| Neutral SMase | SM | cytosolic | - | - | [31]*** |
*In most cases, subcellular localization has been determined in non-neuronal cells.
**Excluded from this list are inhibitors that effect enzyme activity by non-specific mechanisms, such as detergents which disrupt membrane integrity.
***Partial purification.
Ceramide acts as substrate for a number of anabolic reactions. SM, the major phosphosphingolipid in animal cells, is produced by the transfer of phosphocholine from phosphatidylcholine to ceramide [6]. SM is synthesized at the lumenal surface of the cis and medial cisternae of the Golgi apparatus [7, 8], although there has been some discussion that SM may be made in endosomes or at the PM [9]; the hypothesis that endosomes can synthesis SM has recently been disproved [10], although small amounts of SM may indeed be synthesized at the PM [7]. Glucosylceramide (GlcCer) is formed by the glycosylation of ceramide, and is synthesized at the cytosolic surface of either an early Golgi apparatus compartment, or in a compartment that is intermediate in density between the ER and the Golgi apparatus [11-13]. It should be noted that a consistent but subtle difference has always been seen between the sites of synthesis of SM and GlcCer (see, for instance, reference [11]). Galactosylceramide (GalCer), which is abundant in the myelinating tissue of the nervous system, is formed by the galactosylation of ceramide, either at the lumenal leaflet of the ER (in the case of GalCer containing 2-hydroxy fatty acid (HFA)) or at the cytosolic leaflet of the Golgi apparatus (in the case of GalCer containing normal fatty acid (NFA)) [13, 14]. Finally, in addition to being metabolized to SM, GlcCer, and GalCer, two studies have suggested that ceramide can be phosphorylated at the plasma membrane [15], and in synaptic vesicles [16], by a calcium-dependent ceramide kinase, to yield ceramide-1-phosphate, which might be involved in terminating the modulatory effects of ceramide (see below). This suggestion has not been critically evaluated.
As mentioned previously, there is no reason to suspect that the site of synthesis of these lipids will be different in neurons compared to non-neuronal tissues. However, neurons possess unique morphologies, which results in some variation in the distributions of subcellular organelles. For instance, the smooth endoplasmic reticulum (sER), the major organelle involved in glycerolipid and sterol synthesis, is found not only in the cell body and dendrites, but also throughout the axon. Conceivably, this axonal sER could be a site of SL synthesis, at least of the synthesis of the sphingoid long chain bases, of ceramide, and of HFA-GalCer. If this is the case, then these precursors must be transported from the axon in a retrograde direction to the Golgi apparatus, which is found exclusively in the somatodendritic region of neurons. These issues have not been directly examined, although surprisingly, some SM synthesis has been detected in axons [17]. It appears unlikely that major amounts of SM, or of GSLs, are made in axons, since there is no evidence for Golgi apparatus elements in axons.
With the exception of ceramide kinase, all of the anabolic reactions discussed above are localized to the ER and the Golgi apparatus. After their synthesis in these locations, SM, GlcCer, and GalCer are either directly transported to the PM via vesicular transport mechanisms (via anterograde axonal transport in neurons), or in the case of GlcCer and GalCer, can be further glycosylated in the Golgi apparatus before being transported to the PM, also via vesicular transport. Since these lipids are either synthesized in the lumen of the Golgi apparatus, or translocated to the lumen after their synthesis, and since they do not undergo spontaneous transbilayer movement, they are restricted to the outer membrane leaflet of the PM. However, it should be emphasized that there are no reliable measurements accurately quantifying the ratio of endogenous SM, GlcCer, and GalCer between the outer and inner leaflets of the PM in neurons or in other tissues.
Another as yet unresolved questions concerns the half-life of these molecules prior to their endocytosis and degradation. That they can be rapidly internalized by endocytosis [18, 19] and hydrolyzed is in no doubt, but with one or two exceptions (see, for instance, references [20-22]), no data is available to permit quantitative analysis of the half lives of these lipids. Of more interest to this manuscript concerns their intracellular site(s) of degradation (Table 1). The major site of degradation of SM, GlcCer, GalCer, and ceramide is lysosomes, where they are degraded by acid hydrolases, but there is much debate about whether these lipids can also be degraded by hydrolases with a neutral or alkaline pH optimum at the PM, and about the topology of the enzymatic reactions at the PM. This is of crucial importance for understanding, for instance, the generation of ceramide by SMase in response to extracellular signals (see below).
GalCer is hydrolyzed by acid galactocerebrosidase in lysosomes (Table 1), but there is no evidence for a galactocerebrosidase activity at the PM with a neutral pH optimum. Defects in acid galactocerebrosidase result in the lysosomal storage disease, globoid-cell leukodystrophy (Krabbe disease). Since GalCer is a major lipid of myelin, there are severe neuronal dysfunction in Krabbe disease, particularly in the white matter. Lysosomal acid glucocerebrosidase hydrolyzes GlcCer, and defects in this enzyme activity result in Gaucher disease. Similarly, there is no evidence for a membrane-bound glucocerebrosidase that specifically hydrolyzes GlcCer at the PM with a neutral pH optimum. Gaucher disease, at least in its severe form, also displays significant neurological abnormalities, although nervous tissue is not the primary site of manifestation of the disease.
In contrast to galactocerebrosidase and glucocerebrosidase, hydrolases with a neutral pH optimum can degrade ceramide-1-phosphate, ceramide, and SM. In the case of ceramide-1-phosphate, rapid degradation to ceramide is catalyzed by ceramide-1-phosphate phosphatase, located at the PM in rat liver [23], and in synaptic vesicles from rat brain [24]. Whether this enzyme is a unique enzyme responsible for ceramide-1-phosphate hydrolysis, or whether phosphatidic acid phosphohydrolase also hydrolyzes ceramide-1-phosphate in addition to its preferred substrate, phosphatidic acid [16], has not yet been unambiguously established. In the case of ceramide, two hydrolases have been reported. Acid ceramidase is defective in Farber disease (Farber lipogranulomatosis), which also displays nervous system dysfunction. Ceramidases with alkaline and neutral pH optima have also been detected. Presumably the membrane-bound alkaline ceramidase (recently purified from guinea pig epidermis [25]) is localized at the PM, although this has not been definitely proven.
Despite the fact that the hydrolases discussed above can all directly regulate cellular levels of ceramide, none of them have been implicated in the signaling functions of ceramide. In contrast, many studies over the past few years have implied signaling functions for ceramide generated by the activity of SMase. SMases can be broadly divided into two classes, those with an acid pH optima (acid SMase, A-SMase) and those with a neutral pH optima (neutral SMase, N-SMase). A-SMases are localized at lysosomes, and defects in their activity result in Niemann--Pick disease types A and B, which shows some central nervous system neuropathy. This enzyme has been purified, cloned and sequenced from a variety of tissues (see, for instance, [26]), and recently, knock-out mice have been generated [27, 28]. A Zn2+-stimulated SMase is secreted by many cell types and appears to be a product of the A-SMase gene [29]. At least two N-SMases are known. The first, and presumably the one that is most relevant for the production of ceramide from SM at the cell surface, is activated by Mg2+ and is membrane-bound [30]; unfortunately, this enzyme has not yet been purified. A cytosolic, magnesium-independent, neutral sphingomyelinase has also been reported [31], which may also be activated during cell differentiation.
THE ROLE OF GlcCer IN REGULATING NEURONAL GROWTH AND DEVELOPMENT
Having defined the intracellular sites of ceramide metabolism, I will now proceed to discuss evidence that ceramide plays an important role in regulating cell development, and in particular, will focus on neuronal development. Studies in our laboratory on this issue began 5 or 6 years ago when we observed that inhibition of ceramide synthesis by the specific inhibitor, fumonisin B1 (FB1) (Table 1), completely blocked axon growth in hippocampal neurons between days 2 and 3 in culture. Thus, inhibiting ceramide synthesis in the ER (Table 1) had a pronounced effect on axon growth. These studies have advanced since this initial observation, and our data now indicate that ceramide must be metabolized to GlcCer in order to sustain both normal and accelerated axon growth (see below). Understanding the relationship between axon growth and GlcCer synthesis, which occurs in either a pre-Golgi apparatus compartment or in early cisternae of the Golgi apparatus (Table 1), is a major goal of our laboratory at present.
In all of these studies, we have used a unique neuronal culture system in which axons and dendrites develop by a stereotypic sequence of events giving rise to fully differentiated neurons, and in which axons and dendrites can be distinguished both biochemically and morphologically [32]. Neurons are obtained from the hippocampus of embryonic day 18 rats, and plated at low or high densities on glass cover slips that had been precoated with poly-L-lysine. Cover slips are subsequently transferred into culture dishes that contain a monolayer of astroglia, but cover slips are physically separated from the glial monolayer [33]. Importantly, cultures are maintained in serum-free medium.
This system of cultured hippocampal neurons has been extensively characterized over the past 20 years by the laboratory of Gary Banker and colleagues [33]. Growth has been classified into five distinct developmental stages [32]. In the initial stage of growth (stage 1), hippocampal neurons are characterized by the presence of many lamellipodia around the cell body. The second stage of development is marked by loss of lamellipodia and extension of a number of short processes, designated "minor processes" (stage 2). After some hours, one of the minor processes starts to grow rapidly (10-15 µm/h) and develops axonal characteristics (stage 3). Axons form branches as collaterals, and as each new branch emerges, the growth cone of the original axon loses its lamellipodial appearance and elongation stops. Dendrites develop from minor processes (stage 4), and the final stage of development (stage 5) is characterized by the formation of synaptic contacts between axons and dendrites.
In our initial study, cultured hippocampal neurons were incubated with FB1 on day 2 in culture, and axon lengths were quantified on day 3; no significant increase in axon length was observed upon incubation with FB1 between days 2 and 3 in culture whereas the axons of control cells grew by about 70 µm [34]. This was the first demonstration that axonal growth could be regulated by SL synthesis in primary neuronal cell cultures, although an earlier study had demonstrated an inhibitory effect of D-PDMP, an inhibitor of GlcCer synthesis (Table 1), on neurite growth in a murine neuroblastoma cell line [35], and a later study demonstrated that dendrite growth in Purkinje cells could also be inhibited by FB1 [36]. Subsequently, we demonstrated that D-PDMP also blocked axonal growth in hippocampal neurons, and that the effects of incubation with D-PDMP were indistinguishable from those of FB1 [37]. Statistical analyses of various parameters of neuronal growth suggested that the formation or stabilization of new collateral axonal branches was particularly sensitive to manipulating levels of SL synthesis, and that it was this specific facet of neuronal growth that required ongoing SL synthesis. In contrast to the effects of D-PDMP and FB1, incubation with conduritol B epoxide (CBE, an inhibitor of GlcCer degradation, Table 1) stimulated axonal growth by enhancing either the rate of formation or stabilization of axonal branches. The stimulatory effect of CBE could be completely abolished by co-incubation with FB1 [37], or by co-incubation with N-butyldeoxynojirimycin (Table 1) (Schwarz and Futerman, unpublished observations) implying that the ability of CBE to enhance axonal growth was a result of accumulation of a newly-synthesized GSL or of GlcCer.
At this point we did not know whether ongoing synthesis of higher-order GSLs (i.e., gangliosides) was required for axonal growth, or whether synthesis of one particular GSL, i.e., GlcCer, was sufficient. However, it was clear that the reduction in the rate of axonal growth upon inhibition of GSL synthesis was not due to a reduction in the total mass of GSLs, or of GlcCer, since short incubations with inhibitors (3-6 h) were able to block growth. We suggested that the reduction in axon growth was directly related to the synthesis and delivery of newly synthesized GSLs from the ER and Golgi apparatus to the axonal membrane (see [38]).
Studies within the past year or two have unambiguously shown that ongoing synthesis of GlcCer is required for axon growth during stage 3 [39], and for accelerated axon growth upon incubation with growth factors [40]. This is demonstrated most elegantly in studies using either laminin or basic fibroblast growth factor (bFGF) [40]. Both bFGF and laminin stimulate axonal growth by ~4-fold. Remarkably, the stimulatory effects of both factors could be completely abolished by either FB1 or D-PDMP. However, addition of a short-acyl chain derivative of ceramide, C6-NBD-D-erythro-ceramide (Table 2), together with FB1 antagonized the inhibitory effects of FB1 on bFGF-stimulated growth. In contrast, a short-acyl chain analog of another stereoisomer of ceramide, C6-NBD-L-threo-ceramide, was totally ineffective in antagonizing the effects of FB1. Analysis of the metabolism of the ceramide stereoisomers by TLC demonstrated that ~10-15% of C6-NBD-D-erythro-ceramide was converted to C6-NBD-D-erythro-GlcCer during a 3-h incubation, but C6-NBD-L-threo-ceramide was not metabolized at all to C6-NBD-L-threo-GlcCer [40]. Together with the observation that neither C6-NBD-D-erythro-ceramide or C6-NBD-L-threo-ceramide were able to antagonize the inhibitory effects of D-PDMP on bFGF-stimulated growth, these data demonstrated that the ability of bFGF and laminin to stimulate axonal growth requires the ongoing synthesis of GlcCer from ceramide. These data also demonstrate that the ability of exogenously-added ceramide to antagonize the inhibitory effects of FB1 are not due to activation of a ceramide-mediated signaling pathway (see below), since both D-erythro-ceramide and L-threo-ceramide are equally effective in ceramide-mediated signaling pathways [41, 42], but only C6-NBD-D-erythro-ceramide is able to antagonize the effects of FB1 (see Table 2 for the structures of the stereoisomers of ceramide).
Table 2. Stereoisomers of
C6-ceramide, their metabolism, and their biological activity
(the structures of the four stereoisomers of C6-ceramide and
the structure of C6-D-erythro-dihydroceramide are
shown, along with the R/S nomenclature for clarification; the
sphingoid long chain base is indicated by R; the extent of metabolism
of the stereoisomers to C6-GlcCer and their activities in
ceramide-mediated signaling pathways are taken from published data;
reproduced with permission from the Journal of Neuroscience [39])
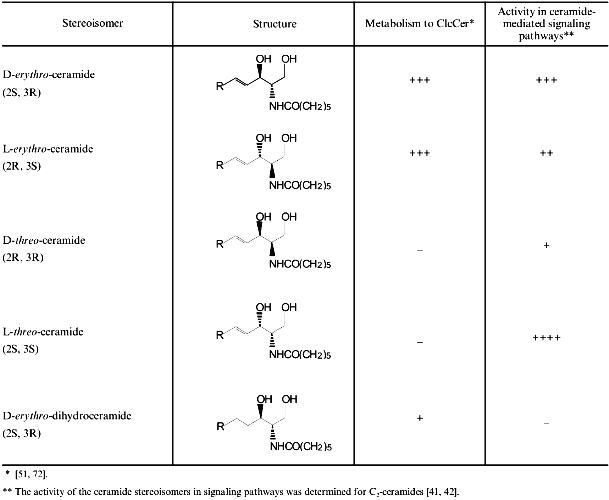
C6-NBD-D-erythro-ceramide is only metabolized to C6-NBD-D-erythro-GlcCer, and not to higher order complex GSLs in cultured hippocampal neurons [34, 40]. This suggests that ongoing synthesis of GlcCer, but not of higher-order glycosphingolipids, is required for bFGF to stimulate axon growth. Two general possibilities could explain the need for GlcCer synthesis in axonal growth. First, GlcCer might be required for bFGF and laminin to transduce signals after they bind to cell surface receptors; however, this possibility is unlikely since levels of cell surface GlcCer will not change significantly during a 3-h incubation with FB1 and D-PDMP (see [37]). The second possibility is that the requirement for ongoing GlcCer synthesis is related to the need to continually supply newly-synthesized GlcCer from its site of intracellular synthesis (Table 1) to the growing axonal membrane. In this scenario, the requirement for GlcCer synthesis could either be related to an event occurring at its sites of synthesis, or alternatively, could be related to an event occurring at the axonal PM after GlcCer is inserted into the PM. Inhibition of GlcCer synthesis might directly affect the formation of Golgi apparatus-derived vesicles, if GlcCer is a rate-limiting component in vesicle formation. However, recent studies have shown that D-PDMP does not effect the incorporation of C6-NBD-D-erythro-ceramide into Golgi apparatus-derived vesicles in hippocampal neurons, and that C6-NBD-L-threo-ceramide is also incorporated into Golgi apparatus-derived vesicles (Burack, Banker, and Futerman, unpublished observations), even though only C6-NBD-D-erythro-Cer is metabolized to GlcCer and able to reverse the inhibitory effects of FB1 on axonal growth. Alternatively, GlcCer synthesis might be required for the transport, sorting or insertion into Golgi-derived vesicles of a key protein that is involved in regulating axonal growth. In Chinese hamster ovary cells, inhibition of GlcCer synthesis blocks the delivery of a viral protein to the cell surface [43], although these effects were subsequently attributed to the accumulation of ceramide [44, 45] and not the depletion of GlcCer. This cannot be the reason for the disruption of axonal growth observed in hippocampal neurons, since exogenously-added ceramide does not disrupt growth at this stage of development, and ceramide must be metabolized to GlcCer to support growth. We are currently examining the transport and delivery of two viral proteins through the secretory pathway to the cell surface with and without inhibitors of SL synthesis at different stages of axonal growth.
One prediction of our studies is that stimulation of axonal outgrowth by either bFGF or laminin may stimulate GlcCer synthesis, assuming that GlcCer synthesis is a rate-limiting step in axonal growth. Studies are presently underway to test this prediction, and preliminary data indicate that the rate of GlcCer synthesis is increased by ~2-fold in vivo upon incubation of hippocampal neurons with either bFGF or laminin (Boldin and Futerman, unpublished observations). If these experiments are validated, they will imply that GlcCer synthase is a regulated enzyme, and moreover, since regulation of enzyme activity occurs within short time periods, regulation must occur post-translationally, rather than by up-regulation of protein synthesis, as has been observed for GlcCer synthase under certain conditions [46, 47]. Interestingly, incubation with either cycloheximide (which inhibits protein synthesis) or brefeldin A (which disrupts the Golgi apparatus and consequently blocks Golgi-derived vesicle traffic) also blocks the ability of bFGF or laminin to stimulate axonal growth (Boldin and Futerman, unpublished observations), although the cause of this block is presumably due to a reduction in bulk membrane components rather than in a specific lipid, as is the case upon incubation with either FB1 or D-PDMP.
In summary, our studies demonstrate that ongoing synthesis of GlcCer in a pre- or early Golgi apparatus compartment is required for normal and accelerated axonal growth. This effect is quite distinct from those reported below, in which ceramide produced by degradation of SM is involved in regulating an earlier stage of neuronal development. Thus, at a different stages of neuronal development, newly-synthesized ceramide plays a different role to ceramide produced by the degradation of SM.
THE ROLE OF CERAMIDE GENERATED FROM THE DEGRADATION OF SM IN
NEURONAL DEVELOPMENT
The first demonstration indicating a relationship between the development and growth of cells in the nervous system and the SM cycle was not obtained in neurons, but rather in glia. Dobrowsky and colleagues [48] showed that the SM cycle could be activated in T9 glioma cells upon binding of nerve growth factor (NGF) to the low affinity neurotrophin receptor (p75NTR). NGF is known to control the survival, development and differentiation of neurons in the central and peripheral nervous system, and interacts with two classes of binding sites. The high affinity binding site requires expression of the product of the trk protooncogene, p140trk which is a receptor tyrosine kinase, whereas the low affinity receptor is a highly glycosylated 75-kD transmembrane protein that lacks kinase activity. Until the study by Dobrowsky et al., there was no evidence for the coupling of p75NTR to any known signaling pathways. Addition of NGF resulted in a 2-fold increase in the production of endogenous ceramide (from SM) and short-acyl chain, cell permeable ceramide analogs mimicked the effects of NGF on cell growth inhibition and process formation [48].
In a follow-up study, the same group reported that the death of oligodendrocytes could also be mediated by the interaction of NGF with the p75 receptor [49]. In these experiments, application of NGF to mature oligodendrocytes cultured from postnatal rat cerebral cortex resulted in their apoptotic cell death, but other neurotrophins such as brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) had no effect; exogenously added ceramide was also able to mimic the effect of NGF. In a final study, it was shown that central glial and neuronal populations displayed differential sensitivity to ceramide-dependent cell death [50]. Thus, whereas primary oligodendrocytes and precursor cells were affected by exogenously-added short-acyl chain ceramide, primary neuronal or astrocytic cultures were resistant to ceramide. Based on these studies, it was suggested that primary oligodendrocytes are much more susceptible to ceramide treatment than astrocytes and neuronal cells [50].
We have also analyzed the effect of ceramide generated from SM on the development of hippocampal neurons cultured according to the protocols described above [33]. The advantage of this culture system is that defined stages of growth can be examined, as can the contribution of glia. In our studies, we used both inhibitors of SL synthesis (Table 1), and also short-acyl chain derivatives of stereoisomers of ceramide (Table 2) to manipulate SL metabolism. Surprisingly, incubation of neurons with either FB1 or D-PDMP during the first 24 to 48 h in culture had no discernible effect on neuronal development [34, 37, 39], suggesting that SL synthesis is not required for either minor process formation (stage 2) or for the initial stages in axon growth (early stage 3), in contrast to the requirement during the later period of axon growth (see above). Presumably neurons contain a sufficiently large intracellular pool of SLs at this stage to sustain growth and development and do not therefore require ongoing synthesis. In contrast, C6-NBD-ceramide or C6-ceramide (Table 2) stimulated the transition of neurons from stage 1 to 2. Whereas the average length of the axon plexus of control neurons was 126 ± 3 µm after 24 h in culture, the average length of the axon plexus of neurons incubated with C6-NBD-ceramide was 183 ± 5 µm [39]. Analysis of the effects of ceramide stereoisomers on axon growth during the first 24 h in culture were consistent with the notion that ceramide is mediating its effects at this stage of development via a signaling pathway; naturally occurring ceramide occurs in the D-erythro-configuration, but three other stereoisomers exist, all of which are active to some extent in ceramide-mediated signaling pathways (see Table 2 and reference [39]). Both C6-NBD-D-erythro-Cer and C6-NBD-L-threo-Cer stimulated growth between days 0 and 1, but C6-D-erythro-dihydroCer, which is totally inactive in signaling pathways [41], had no effect [39]. Addition of D-PDMP together with C6-NBD-ceramide did not block the stimulatory effect of C6-NBD-ceramide [39], and even though C6-NBD-L-threo-Cer is not metabolized to C6-NBD-L-threo-GlcCer in hippocampal neurons [40] and in other cells [51], C6-NBD-L-threo-Cer stimulated growth as efficiently as C6-NBD-D-erythro-Cer [39], demonstrating that ceramide itself, and not an anabolic product, i.e., GlcCer, is responsible for mediating these effects on neuronal development.
Another advantage of this culture system is that effects on both glia and neurons can be examined, as can the contribution of glia towards neuronal growth, since neurons are grown in culture dishes that contain a monolayer of glia although they are not in physical contact with the glia (see above, and reference [33]). We observed that C6-NBD-D-erythro-Cer also stimulated the transition of neurons from stage 1 to 2 even when cells were cultured in the absence of glia, but at much lower concentrations than in the presence of glia; thus, incubation with 5 µM C6-NBD-D-erythro-Cer resulted in a 50-60% increase in the number of stage 3 cells at 24 h in the presence of glia but in the absence of glia, a similar increase in the number of stage 3 cells was obtained using 0.05-0.1 µM C6-NBD-D-erythro-Cer [37]. The concentration differences required to see an effect were not due to the uptake of C6-NBD-D-erythro-Cer by glial when neurons were cultured with a glial monolayer, and thus a reduction in the effective concentration of C6-NBD-D-erythro-Cer in the medium, since similar amounts of NBD-fluorescence were recovered from the culture medium of neurons grown with or without glia. Endogenous ceramide (generated by incubation with N-SMase) could also stimulate neuronal development when neurons were cultured in the absence of glia. Incubation with 50-75 mU/ml N-SMase resulted in a significant increase in the percent of neurons in stage 3 after the first 24 h in culture.
These data demonstrate that neurons are indeed sensitive to the effects of either exogenously-added ceramide, or ceramide generated from SMase, and at the concentrations discussed above, neuronal development is enhanced. In contrast, at higher concentrations of either N-SMase (>250 mU/ml) or of C6-NBD-D-erythro-Cer, apoptotic cell death was induced at both early (days 0-1) and later stages of development (days 2-3). The lack of specificity of ceramide stereoisomers in inducing apoptosis, and in regulating the transition from stage 1 to 2, together with the lack of effect of dihydroceramide [41], is consistent with the possibility that ceramide acts via an intracellular signaling pathway in the regulation of these events. Our data have therefore shown that in hippocampal neurons, minor process formation and apoptosis can be regulated by ceramide-dependent signaling pathways, and that the decision whether to enter these diametrically opposed pathways depends on intracellular ceramide concentrations.
In proliferating cells, ceramide suppresses growth and stimulates differentiation [52]. For instance, ceramide inhibits proliferation of neuroblastoma Neuro2a cells and induces their differentiation [53], and stimulates T9 glioma differentiation and processes formation (see above, and reference [48]). However, ceramide cannot act via arrest of proliferation and stimulation of differentiation in hippocampal neurons since these neurons are post-mitotic at their time of removal from the hippocampus. Rather, ceramide acts by accelerating the transition from stage 1 to 2 at low concentrations, and induces apoptotic cell death at higher concentrations.
At present, we do not know the molecular mechanisms by which ceramide accelerates the transition from stage 1 to 2, nor do we know whether an endogenous N-SMase activity is involved in the physiological regulation of this early stage of development. However, using a short-acyl chain derivative of sphingomyelin (SM), C6-NBD-SM [7, 20, 21] to detect activity, we have recently demonstrated that hippocampal neurons contain significant levels of endogenous N-SMase activity (Brann and Futerman, unpublished observations). Moreover, neuronal development can be stimulated by various neurotrophins, including NGF and BDNF. As shown by others, binding of neurotrophins to the p75NTR receptor results in generation of ceramide [48], that may in turn (via an as yet unknown mechanism) induce gene expression, in some cases perhaps via the transcription factor NF-kB [54]. RNAse protection assays demonstrate that hippocampal neurons express the p75NTR receptor (Brann, Fainzilber, and Futerman, unpublished observations), and we are currently examining whether this pathway is involved in the acceleration of neuronal development induced by ceramide, and whether a down-stream effect results in alterations in cytoskeletal organization and consequently minor process formation.
Our studies, together with a number of others (reviewed in [55]) imply that ceramide generated from SM is involved in regulating neuronal development, either by stimulating growth (differentiation) or by inducing apoptosis, depending on the signal by which SMase is activated, and on the receptors (p75, trks, or others) present in the cells in question. However in all cases, ceramide levels are probably elevated by stimulation of SM hydrolysis by an SMase presumably located at the PM, and not by an increase in the synthesis of ceramide at the ER (compare with [56]). Two issues are raised by these observations: (1) What is the source and topology of SM that is degraded upon activation of SMase, and (2) which of the various SMases (Table 1) is involved in generating ceramide? Analyzing the source of SM has been problematic. In some studies, exogenously added N-SMase is able to induce apoptosis or differentiation [39], but not in others (reviewed in [57]) suggesting that the pool of SM involved in signaling is inaccessible or different from the bulk pool of SM. Since SM is synthesized in the lumen of the Golgi apparatus, it is found at the external leaflet of the PM (Table 1), yet it has been shown that there is a pool of SM that is resistant to degradation by exogenously added SMase [58]; whether this implies that the resistant SM is on the inner leaflet of the membrane bilayer is open to debate. However, a recent study has shown that there may indeed be a distinct pool of SM that is involved in apoptosis, at least in non-neuronal cells [57]. Likewise, although it is normally assumed that N-SMase is involved in ceramide generation in signaling, there is some evidence, from A-SMase deficient mice, that A-SMase is required for apoptosis [59], whereas other studies reach the opposite conclusion (i.e., see reference [60]). Clearly, these ambiguities will only be resolved by definitive analysis of the distribution of SM at the cell surface, and by the isolation and cloning of N-SMase. It is to be hoped that there will be considerable progress in these two areas in the near future.
Finally, one fascinating issue which has yet to be studied is ceramide signaling in cells from patients suffering from Farber lipogranulomatosis (Table 1). Unfortunately, no animal models of this disease exist, and ceramidase-deficient mice have yet to be generated. However, since the concentrations of ceramide that are required for signaling functions are similar to those that exist in skin fibroblasts from patients with Farber disease, it might be predicted that ceramide signaling is altered in cells with defective or reduced acid ceramidase. In particular, it will be interesting to analyze the storage of ceramide in neurons [61], and to examine whether there are changes in the rate of neuronal growth and development, and effects on apoptotic cell death.
Work from the author's laboratory was supported by the United States-Israel Binational Science Foundation (Jerusalem, Israel), the German-Israel Foundation for Scientific Research and Development, and the Mizutani Foundation for Glycosciences.
REFERENCES
1.Hannun, Y. A. (1996) Science, 274,
1855-1859.
2.Futerman, A. H. (1994) Curr. Topics
Membranes, 40, 93-110.
3.Sandhoff, K., and Kolter, T. (1996) Trends Cell
Biol., 6, 98-103.
4.Mandon, E. C., Ehses, I., Rother, J., Van Echten,
G., and Sandhoff, K. (1992) J. Biol. Chem., 267,
11144-11148.
5.Hirschberg, K., Rodger, J., and Futerman, A. H.
(1993) Biochem. J., 290, 751-757.
6.Voelker, D. R., and Kennedy, E. P. (1982)
Biochemistry, 21, 2753- 2759.
7.Futerman, A. H., Stieger, B., Hubbard, A. L., and
Pagano, R. E. (1990) J. Biol. Chem., 265, 8650-8657.
8.Jeckel, D., Karrenbauer, A., Birk, R., Schmidt, R.
R., and Wieland, F. (1990) FEBS Lett., 261, 155-157.
9.Allan, D., and Kallen, K.-J. (1994) Trends Cell
Biol., 4, 350-353.
10.Van Helvoort, A., Stoorvogel, W., van Meer, G.,
and Burger, K. N. J. (1997) J. Cell Sci., 110,
781-788.
11.Futerman, A. H., and Pagano, R. E. (1991)
Biochem. J., 280, 295-302.
12.Jeckel, D., Karrenbauer, A., Burger, K. N. J.,
van Meer, G., and Wieland, F. (1992) J. Cell Biol., 117,
259-267.
13.Burger, K. N. J., Vanderbijl, P., and van Meer,
G. (1996) J. Cell Biol., 133, 15-28.
14.Vanderbijl, P., Strous, G. J., Lopes-Cardozo, M.,
Thomasoates, J., and van Meer, G. (1996) Biochem. J.,
317, 589-597.
15.Kolesnick, R. N., and Hemer, M. R. (1990) J.
Biol. Chem., 265, 18803-18808.
16.Bajjalieh, S. M., Martin, T. F. G., and Floor, E.
(1989) J. Biol. Chem., 264, 14354-14360.
17.Vance, J. E., Pan, D. B., Campenot, R. B.,
Bussiere, M., and Vance, D. E. (1994) J. Neurochem., 62,
329-337.
18.Sofer, A., Schwarzmann, G., and Futerman, A. H.
(1996) J. Cell Sci., 109, 2111-2119.
19.Sofer, A., and Futerman, A. H. (1996) J.
Neurochem., 67, 2134-2140.
20.Koval, M., and Pagano, R. E. (1989) J. Cell
Biol., 108, 2169-2181.
21.Koval, M., and Pagano, R. E. (1990) J. Cell
Biol., 111, 429-442.
22.Mayor, S., Presley, J. F., and Maxfield, F. R.
(1993) J. Cell Biol., 121, 1257-1269.
23.Boudker, O., and Futerman, A. H. (1993) J.
Biol. Chem., 268, 22150-22155.
24.Shinghal, R., Scheller, R. H., and Bajjalieh, S.
M. (1993) J. Neurochem., 61, 2279-2285.
25.Yada, Y., Higuchi, K., and Imokawa, G. (1995)
J. Biol. Chem., 270, 12677-12684.
26.Newrzella, D., and Stoffel, W. (1992) Biol.
Chem. Hoppe Seyler, 373, 1233-1238.
27.Horinouchi, K., Erlich, S., Perl, D. P., Ferlinz,
K., Bisgaier, C. L., Sandhoff, K., Desnick, R. J., Stewart, C. L., and
Schuchman, E. H. (1995) Nature Genetics, 10,
288-293.
28.Otterbach, B., and Stoffel, W. (1995)
Cell, 81, 1053-1061.
29.Schissel, S. S., Schuchman, E. H., Williams, K.
J., and Tabas, I. (1996) J. Biol. Chem., 271,
18431-18436.
30.Hostetler, K. Y., and Yazaki, P. J. (1979) J.
Lipid Res., 20, 456-463.
31.Okazaki, T., Bielawska, A., Domae, N., Bell, R.
M., and Hannun, Y. A. (1994) J. Biol. Chem., 269,
4070-4077.
32.Dotti, C. G., Sullivan, C. A., and Banker, G. A.
(1988) J. Neurosci., 8, 1454-1468.
33.Goslin, K., and Banker, G. (1991) in Culturing
Nerve Cells, MIT Press, Cambridge, pp. 251-281.
34.Harel, R., and Futerman, A. H. (1993) J. Biol.
Chem., 268, 14476-14481.
35.Uemura, K., Sugiyama, E., and Taketomi, T. (1991)
J. Biochem. (Tokyo), 110, 96-102.
36.Furuya, S., Ono, K., and Hirabayashi, Y. (1995)
J. Neurochem., 65, 1551-1561.
37.Schwarz, A., Rapaport, E., Hirschberg, K., and
Futerman, A. H. (1995) J. Biol. Chem., 270,
10990-10998.
38.Futerman, A. H., and Banker, G. A. (1996)
Trends Neurosci., 19, 144-149.
39.Schwarz, A., and Futerman, A. H. (1997) J.
Neurosci., 17, 2929-2938.
40.Boldin, S., and Futerman, A. H. (1997) J.
Neurochem., 68, 882-885.
41.Bielawska, A., Crane, H. M., Liotta, D., Obeid,
L. M., and Hannun, Y. A. (1993) J. Biol. Chem., 268,
26226-26232.
42.Fishbein, J. D., Dobrowsky, R. T., Bielawska, A.,
Garrett, S., and Hannun, Y. A. (1993) J. Biol. Chem.,
268, 9255-9261.
43.Rosenwald, A. G., Machamer, C. E., and Pagano, R.
E. (1992) Biochemistry, 31, 3581-3590.
44.Rosenwald, A. G., and Pagano, R. E. (1993) J.
Biol. Chem., 268, 4577-4579.
45.Chen, C. S., Rosenwald, A. G., and Pagano, R. E.
(1995) J. Biol. Chem., 270, 13291-13297.
46.Abe, A., Radin, N. S., and Shayman, J. A. (1996)
Biochim. Biophys. Acta, 1299, 333-341.
47.Sando, G., Howard, E. J., and Madison, K. C.
(1996) J. Biol. Chem., 271, 22044-22051.
48.Dobrowsky, R. T., Werner, M. H., Castellino, A.
M., Chao, M. V., and Hannun, Y. A. (1994) Science, 265,
1596-1599.
49.Casaccia-Bonnefil, P., Carter, B. D., Dobrowsky,
R. T., and Chao, M. V. (1996) Nature, 383, 716-719.
50.Casaccia-Bonnefil, P., Aibel, L., and Chao, M. V.
(1996) J. Neurosci. Res., 43, 382-389.
51.Pagano, R. E., and Martin, O. C. (1988)
Biochemistry, 27, 4439-4445.
52.Pushkareva, M., Obeid, L. M., and Hannun, Y. A.
(1995) Immunol. Today, 16, 294-297.
53.Riboni, L., Prinetti, A., Bassi, R., Caminiti,
A., and Tettamanti, G. (1995) J. Biol. Chem., 270,
26868-26875.
54.O'Neill, L. A. J., and Kaltschmidt, C. (1997)
Trends Neurosci., 20, 252-258.
55.Dechant, G., and Barde, Y.-A. (1997) Curr.
Opin. Neurobiol., 7,413-418.
56.Bose, R., Verheij, M., Haimovitz-Friedman, A.,
Scotto, K., Fuks, Z., and Kolesnick, R. (1995) Cell, 82,
405-414.
57.Zhang, P., Liu, B., Jenkins, G. M., Hannun, Y.
A., and Obeid, L. M. (1997) J. Biol. Chem., 272,
9609-9612.
58.Linardic, C. M., and Hannun, Y. A. (1994) J.
Biol. Chem., 269, 23530-23537.
59.Santana, P., Pena, L. A., Haimovitz Friedman, A.,
Martin, S., Green, D., McLoughlin, M., Cordon-Cardo, C., Schuchman, E.
H., Fuks, Z., and Kolesnick, R. (1996) Cell, 86,
189-199.
60.Zumbansen, M., and Stoffel, W. (1997) J. Biol.
Chem., 272, 10904-10909.
61.Levade, T., Moser, H. W., Fensom, A. H., Harzer,
K., Moser, A. B., and Salvayre, R. (1995) J. Neurol. Sci.,
134, 108-114.
62.Futerman, A. H. (1994) Trends Glycosci.
Glycotechnol., 6, 143-153.
63.Merrill, A. H., Wang, E., Gilchrist, D. G., and
Riley, R. T. (1993) Adv. Lipid Res., 26, 215-234.
64.Merrill, A. H., Liotta, D. C., and Riley, R.
(1996) Trends Cell. Biol., 6, 218-223.
65.Rother, Y., van Echten, G., Schwarzmann, G., and
Sandhoff, K. (1992) Biochem. Biophys. Res. Commun., 189,
14-20.
66.Van Helvoort, A., Stoorvogel, W., van Meer, G.,
and Burger, K. N. J. (1997) J. Cell Sci., 110,
781-788.
67.Coste, H., Martel, M.-B., Azzar, G., and Got, R.
(1985) Biochim. Biophys. Acta, 814, 1-7.
68.Coste, H., Martel, M.-B., and Got, R. (1986)
Biochim. Biophys. Acta, 858, 6-12.
69.Trinchera, M., Fabbri, M., and Ghidoni, R. (1991)
J. Biol. Chem., 266, 20907-20912.
70.Inokuchi, J., and Radin, N. S. (1987) J. Lipid
Res., 28, 565-571.
71.Platt, F. M., Neises, G. R., Reinkensmeier, G.,
Townsend, M. J., Perry, V. H., Proia, R. L., Winchester, B., Dwek, R.
A., and Butters, T. D. (1997) Science, 276, 428-431.
72.Paul, P., Kamisaka, Y., Marks, D. L., and Pagano,
R. E. (1996) J. Biol. Chem., 271, 2287-2293.
73.Ichikawa, S., Sakiyama, H., Suzuki, G., Hidari,
K., and Hirabayashi, Y. (1996) Proc. Natl. Acad. Sci. USA,
93, 4638-4643.
74.Schaerenwiemers, N., Vanderbijl, P., and Schwab,
M. E. (1995) J. Neurochem., 65, 2267-2278.
75.Schulte, S., and Stoffel, W. (1993) Proc.
Natl. Acad. Sci. USA, 90, 10265-10269.
76.Sakai, N., Inui, K., Fujii, N., Fukushima, H.,
Nishimoto, J., Yanagihara, I., Isegawa, Y., Iwamatsu, A., and Okada, S.
(1994) Biochem. Biophys. Res. Commun., 198, 485-491.
77.Sakai, N., Inui, K., Tatsumi, N., Fukushima, H.,
Nishigaki, T., Taniike, M., Nishimoto, J., Tsukamoto, H., Yanagihara,
I., Ozono, K., and Okada, S. (1996) J. Neurochem., 66,
1118-1124.
78.Chen, Y. Q., Rafi, M. A., de Gala, G., and
Wenger, D. A. (1993) Hum. Mol. Genet., 2, 1841-1845.
79.Legler, G. (1977) Meth. Enzymol.,
46, 368-381.
80.Legler, G., and Bieberich, E. (1988) Arch.
Biochem. Biophys., 260, 437-442.
81.Dale, G. L., and Beutler, E. (1976) Proc.
Natl. Acad. Sci. USA, 73, 4672-4674.
82.Pentchev, P. G., Brady, R. O., Blair, H. E.,
Britton, D. E., and Sorrell, S. H. (1978) Proc. Natl. Acad. Sci.
USA, 75, 3970-3973.
83.Bielawska, A., Greenberg, M. S., Perry, D.,
Jayadev, S., Shayman, J. A., McKay, C., and Hannun, Y. A. (1996) J.
Biol. Chem., 271, 12646-12654.
84.Coroneos, E., Martinez, M., McKenna, S., and
Kester, M. (1995) J. Biol. Chem., 270, 23305-23309.
85.Sugita, M., Dulaney, J. T., and Moser, H. W.
(1972) Science, 178, 1100-1102.
86.Bernardo, K., Hurwitz, R., Zenk, T., Desnick, R.
J., Ferlinz, K., Schuchman, E. H., and Sandhoff, K. (1995) J. Biol.
Chem., 270, 11098-11102.
87.Koch, J., Gartner, S., Li, C. M., Quintern, L.
E., Bernardo, K., Levran, O., Schnabel, D., Desnick, R. J., Schuchman,
E. H., and Sandhoff, K. (1996) J. Biol. Chem., 271,
33110-33115.
88.Furst, W., and Sandhoff, K. (1992) Biochim.
Biophys. Acta, 1126, 1-16.
89.Quintern, L. E., and Sandhoff, K. (1991) Meth.
Enzymol., 197, 536-540.
90.Schuchman, E. H., Levran, O., Pereira, L. V., and
Desnick, R. J. (1992) Genomics, 12, 197-205.
91.Lansmann, S., Ferlinz, K., Hurwitz, R.,
Bartelsen, O., Glombitza, G., and Sandhoff, K. (1996) FEBS
Lett., 399, 227-231.
92.Ferlinz, K., Hurwitz, R., Vielhaber, G., Suzuki,
K., and Sandhoff, K. (1994) Biochem. J., 301,
855-862.