REVIEW: Leukotrienes: Lipid Bioeffectors of Inflammatory Reactions
A. Sala*, S. Zarini, and M. Bolla
Center for Cardiopulmonary Pharmacology, University of Milan, Via Balzaretti 9, 20133 Milan, Italy; fax: (39) 2-29404961; E-mail: angelo.sala@unimi.it* To whom correspondence should be addressed.
Received July 22, 1997
The leukotrienes arise from oxidative metabolism of arachidonic acid through the action of the 5-lipoxygenase enzyme, leading to the unstable allylic epoxide leukotriene A4. This intermediate represents the substrate for two different specific enzymes, namely leukotriene A4-hydrolase and leukotriene C4-synthase, generating LTB4 and cysteinyl leukotrienes, respectively. The name "leukotriene" is referring to the cellular source (leukocytes are one of the major sources) as well as the conjugated triene that characterizes their structure. LTC4 and LTD4 are potent contracting agents of smooth muscle in airways and blood vessels; in addition, they induce mucus secretion and promote plasmatic exudation with direct action on endothelial cells. On the other side, LTB4 is known as a potent chemokinetic and chemotactic agent. A number of evidences reported in the literature underline the potential role of leukotrienes in the inflammatory responses that characterizes asthma and other pathological conditions. These potent lipid bioeffectors are synthesized during the course of inflammatory reactions and their pharmacological modulation is able to significantly attenuate the clinical manifestations associated with different inflammatory pathologies.
KEY WORDS: 5-lipoxygenase, leukotrienes, chemotaxis, bronchoconstriction, leukocytes, LTC4-synthase
Abbreviations: AA) arachidonic acid; LTA4, B4, C4, D4, E4) leukotriene A4, B4, C4, D4, E4; 5-LO, 15-LO) 5- and 15-lipoxygenase, respectively; 5-HPETE) 5-hydroperoxyeicosatetraenoic acid; PLA2) phospholipase A2; FLAP) five-lipoxygenase-activating protein; Cys-LTs) cysteinyl leukotrienes.
The leukotrienes can be divided into two different classes, based upon
their chemical structures and biological activity: 1) the cysteinyl
leukotrienes (Cys-LTs), namely leukotriene C4
(LTC4), leukotriene D4 (LTD4), and
leukotriene E4 (LTE4), containing different amino
acid residues, and 2) the dihydroxy-derivative leukotriene
B4 (LTB4). Both classes arise from the oxidative
metabolism of arachidonic acid (AA) through the action of the
5-lipoxygenase enzyme (5-LO) (EC 1.13.11.34), leading to the unstable
allylic epoxide leukotriene A4 (LTA4). This
intermediate represents the substrate for two different specific
enzymes, namely the leukotriene A4-hydrolase and the
leukotriene C4-synthase, generating LTB4 and
LTC4, respectively. Unlike cyclooxygenase, another important
enzyme catalyzing oxygenation of AA that is present in constitutive or
inducible form in most cell types, 5-LO presents a more limited
distribution. Leukocytes are one of the major sources of leukotrienes,
and the name "leukotriene" is indeed referring to the
cellular source as well as the conjugated triene that characterizes
their structure [1].
Cys-LTs were originally described as the slow reacting substance of anaphylaxis (SRS-A) [2, 3] and their biological activity has been known for over 50 years, well before their structural elucidation late in the seventies [4]. LTB4 was characterized as a major AA metabolite in rabbit polymorphonuclear leukocytes [5] and its biological properties described thereafter [6]. This review will consider the novel aspects of the biosynthesis and the biological activities of these important lipid bioeffectors, with special emphasis to their potential role in inflammatory reactions.
BIOSYNTHESIS
Free AA concentration is tightly controlled in inflammatory cells. AA is present mainly in ester form within endogenous stores located in membrane phospholipids, and the first step toward leukotriene generation is represented by the activation of specific phospholipases (for review see [7, 8]). In particular, both low-molecular-weight phospholipase A2 (PLA2) (14 kD, group I, II and III) and relatively high-molecular-weight PLA2 (110 kD) may participate in AA mobilization from phospholipid stores [9-11]. Together with AA mobilization, 5-LO activity requires cell activation and influx of extracellular calcium levels to produce leukotrienes [12]. Stimulation of neutrophils [13], rat basophilic leukemia cells as well as alveolar macrophages [14] results in translocation of 5-LO, a cytosolic or nuclear soluble enzyme [15], to the nuclear envelope where it colocalizes with five-lipoxygenase-activating protein (FLAP) an integral membrane protein of 18 kD necessary for leukotriene synthesis and for 5-LO translocation [16, 17]. The role of FLAP appears to be more complex than that of a docking protein for 5-LO and evidences have been reported that FLAP may lead to a very efficient conversion of the AA to leukotrienes by "handing" the substrate to the 5-LO [18, 19]. Recent data indicate that phosphorylation of 5-LO is associated with activation and translocation to the nuclear envelope [20], and that mitogen-activated protein kinase kinase I (MAPK kinase I) participates in the molecular processes governing activation and translocation of 5-LO from the cytosol to the nuclear membrane [21]. Concerning translocation of the 5-LO to the nuclear envelope, it is important to note that cytosolic PLA2 has also been shown to redistribute to the nuclear fraction upon activation in macrophages [22].
5-LO has been cloned from different cellular sources (reviewed in [23]) and its catalytic activity removes the pro-S hydrogen at C-7 by a redox mechanism involving non-heme iron, then inserting molecular oxygen at C-5, to yield 5(S)-hydroperoxy-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-HPETE); this unstable lipid hydroperoxide may be either reduced by peroxidases to the hydroxy analog or can be stereospecifically dehydrated to 5(S),6(S)-oxido-7E,9E,11Z,15Z-eicosatetraenoic acid or LTA4 by a second 5-LO-catalyzed step [24] (Fig. 1). Recently, conversion of 5-HPETE to LTA4 has been reported as the action of eosinophilic 15-lipoxygenase (15-LO) [25].
Pretreatment with several different growth factors has been shown to affect leukotriene biosynthesis in polymorphonuclear leukocytes. Recombinant granulocyte-macrophage colony-stimulating factor increased the production of leukotrienes in eosinophiles and neutrophils challenged with A23187, the chemotactic peptides, and platelet-activating factor (PAF) [26-28]. Similar effects were observed using tumor necrosis factor and wholeblood[29], interleukin-3 and human basophils [30], and C-kit ligand and human lung mast cells [31]. The mechanism of this "priming" effect of cytokines toward 5-LO activity has not been elucidated, even if an increased AA bioavailability has been hypothesized [32].Fig. 1. Metabolism of the arachidonic acid by the 5-lipoxygenase enzyme and biosynthesis of leukotrienes. 5-HETE, 5-hydroxyeicosatetraenoic acid; 5-HPETE, 5-hydroperoxyeicosatetraenoic acid.
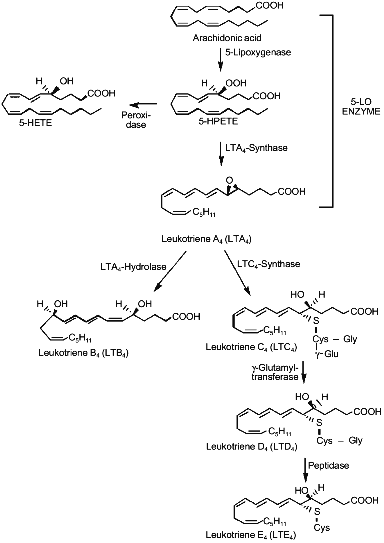
The activity of 5-LO declines during catalysis via a "suicide" inactivation process, involving irreversible binding of the highly reactive product LTA4 [33]. Enzymatic conversion of the relatively unstable allylic epoxide LTA4 has a half-life in physiological buffers below 1 sec [34] and represents the branching of the pathway leading from AA to either LTB4 or LTC4 and the other Cys-LTs.
Leukotriene C4-synthase has been cloned by two different research groups [35, 36] and, unlike cytosolic glutathione S-transferases, is an 18 kD microsomal protein. The protein sequence does not have similarities to other known glutathione-S-transferases, but it shows significant amino acid identities with FLAP, another integral membrane protein important for leukotriene biosynthesis. Leukotriene C4-synthase catalyzes the conjugation of LTA4 with glutathione leading to the formation of 5(S)-hydroxy-6(R)-S-glutathionyl-7E,9E,11Z,14Z-eicosatetraenoic acid (LTC4). LTC4 can be metabolized by a gamma-glutamyltransferase to the cysteinyl--glycyl conjugate LTD4, that in turn can be further metabolized by ubiquitous peptidases to the cysteinyl containing LTE4 (Fig. 1).
Leukotriene A4-hydrolase, the cytosolic enzyme catalyzing the stereospecific hydration of LTA4 to 5(S),12(R)-dihydroxy-6Z,8E,10E,14Z-eicosatetraenoic acid (LTB4) is a 68 kD protein containing zinc, and it is clearly distinct from epoxide hydrolase previously described in the liver [37, 38]. It is substrate-inactivated by covalent binding of LTA4 to a specific 21-residue peptide [39] and its inactivation represents the rate-limiting step in the synthesis of LTB4 in human neutrophils [40, 41].
TRANSCELLULAR BIOSYNTHESIS
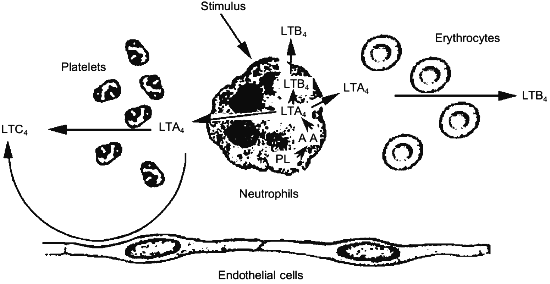
Since the structural elucidation of leukotrienes, a great deal of effort has been paid to the identification of the biosynthetic capacity of given cell types. This effort resulted in the characterization of different profiles of leukotriene biosynthesis depending upon the cell type considered. Neutrophils have been shown to synthesize large amounts of LTB4, possessing 5-LO and LTA4-hydrolase [42], while eosinophiles and mast cells preferentially synthesize LTC4, according to the presence of 5-LO and LTC4-synthase within these cells [43, 44]. Nevertheless, the co-incubation of different cell types was shown to greatly influence both the quantitative and the qualitative profile of the leukotrienes produced [45, 46]. These changes were demonstrated to depend upon the ability of neutrophils to export the unstable intermediate LTA4 to neighboring cells, that, although do not have the primary oxidative enzyme, namely the 5-LO, possess either LTA4-hydrolase (erythrocytes) [47] or LTC4-synthase (platelets, endothelial cells and vascular smooth muscle cells) [48-50] (Fig. 2). These findings point out to the important role in leukotriene biosynthesis of cells that are devoid of 5-LO activity.
This potentially relevant issue has been recently studied in detail in more complex systems, such as polymorphonuclear leukocyte-perfused isolated rabbit lung [51] and heart [52]. Interaction of neutrophils with the intact endothelium of these functioning organ systems resulted in a substantial diversion from the synthesis of LTB4, to the synthesis of relevant amounts of Cys-LTs. It is important to note that intact LTA4 has been described as the main metabolite released upon Ca2+-ionophore A23187 challenge of both bovine [53] and human [41] polymorphonuclear leukocytes and this observation provide further support to the potential relevance of the transcellular synthetic pathway of Cys-LTs.Fig. 2. Transcellular synthesis of leukotrienes by cellular cooperation between neutrophils and platelets, erythrocytes or endothelial cells.
METABOLISM
Cysteinyl leukotrienes. Bioconversion of LTC4 into LTD4 and LTE4 does not appear as a catabolic inactivation because LTD4 is at least as potent as LTC4 with respect to most biological activities, and LTE4 appears to be only slightly less potent. Infusion of radiolabeled LTC4 and LTE4 in normal subjects results in rapid disappearance from the bloodstream, associated with the detection of fractional amounts of LTE4 in urine [54] during the first 2 h. Substantial amounts of omega- and beta-oxidized metabolites of LTE4 are detected in urine at later times [55]. Several-fold increase in urinary LTE4 excretion observed in patients with important liver dysfunction suggests that the liver may represent the site of catabolism of Cys-LTs [56-58].
Leukotriene B4. On the contrary to what is observed for Cys-LTs, no urinary metabolites of LTB4 have been identified so far. LTB4 undergoes rapid metabolism in purified polymorphonuclear leukocytes preparations to give 20-hydroxy-LTB4 and 20-carboxy-LTB41 [59]. This conversion is catalyzed by a specific cytochrome P-450 enzyme [60], but occurs mainly after release of intact LTB4 and re-uptake by neighboring cells [61, 41]. In light of the lower biological activity of the 20-carboxy metabolite, omega-oxidation may represent a mechanism to locally inactivate the LTB4. On the other end, this metabolism is not observed in monocytes, eosinophiles, and macrophages, and there are limited evidences that it may occur in vivo.
1 Although the generally applied 20-carboxy-derivative should be termed 19-carboxy-one from the chemical point of view.
BIOLOGICAL ACTIVITY
Cysteinyl leukotrienes. LTC4 and LTD4 are among the most potent bronchoconstricting agents, being over 10,000-fold more potent, on molar basis, than histamine. They are active on isolated human airways [62] as well as in vivo (reviewed in [63]). Correct stereochemistry at C-5 and C-6 is essential for spasmogenic activity, while either changes in the chirality of cysteine or substitution of glycine for alanine are relatively inconsequential in effect [64, 65]. LTE4 is relatively less potent than LTC4 and LTD4 in this respect, but substantial hyperreactivity toward LTE4 has been observed in asthmatic patients [66]. In addition Cys-LTs are able to induce mucus secretion from human bronchial mucosa [67, 68], that may contribute to the obstruction of airway lumen in asthma.
LTC4 and LTD4 cause contraction of both venous and arterial vascular smooth muscle preparation [69-72] and Cys-LTs synthesis through neutrophil--endothelial cell cooperation results in marked coronary vasoconstriction, impaired functions and morphological modifications of isolated rabbit heart preparations [73]. In isolated human pulmonary veins and arteries, LTD4 induces contraction above basal tone but also nitric oxide-dependent relaxation of vessels previously contracted with noradrenaline [74].
In the microcirculation, Cys-LTs promote plasmatic exudation with a direct action on the endothelial cells [75, 76]. Intradermal injection of LTC4 and LTD4 induces vasodilation in man but may cause vasoconstriction in the guinea-pig [77]; this latter effect may relate to a secondary production of thromboxane, as observed for the bronchoconstriction both in vivo and in vitro in this specie [78].
Biological activities of Cys-LTs are mediated through specific membrane receptors. At least two different receptors have been pharmacologically characterized (Cys-LT1 and Cys-LT2) (for review see [79]), but a Cys-LT3 receptor has been hypothesized as responsible for the effect observed in guinea pig lung [80]. Receptor antagonists developed from the structure of LTD4 mainly antagonize the effects mediated by the Cys-LT1 receptor, that appears to represent the receptor responsible for the contraction of isolated human bronchi. Activation of Cys-LT1 receptors seems coupled to at least two different G-proteins, both pertussis toxin-sensitive and insensitive, and causes intracellular calcium mobilization through different mechanisms [81, 82].
Leukotriene B4. LTB4 stimulates human and rabbit neutrophil locomotion at subnanomolar concentrations [83, 84]. Chemokinetic and chemotactic activity of LTB4 [85] are dependent on the stereochemistry of the hydroxyl groups at C-5 and C-12 [86-88]. In vivo biological activity of LTB4 has been shown upon injection in the rat peritoneal cavity, resulting in substantial accumulation of macrophages and polymorphonuclear leukocytes [89]. Accumulation of neutrophils at the site of injection has been shown by direct application of LTB4 either intradermally or into the interior chamber of rabbit eye [90, 91]. Similar effects of LTB4 were shown using the skin chamber technique on the rabbit back and the human forearm [92].
Intravital microvascular microscopy showed rapid adhesion of leukocytes upon superfusion with LTB4, followed by progressive diapedesis into extravascular tissues [93, 94]. Adhesion and migration of leukocytes was accompanied by increased microvascular permeability that was totally leukocyte-dependent.
LTB4 has been shown to increase, in an autocrine fashion, the 5-LO activation observed upon challenge of polymorphonuclear leukocytes [95]. Taken together, the effect of LTB4 on 5-LO and the increased microvascular permeability observed upon LTB4-induced leukocyte adhesion, may be seen as a typical example of transcellular synthesis of Cys-LTs, where leukocyte activation causes adhesion to endothelial cells and transfer of LTA4 resulting in the biosynthesis of the LTC4, possessing a direct effect on vascular permeability. LTB4 is able to enhance the adhesion of neutrophils to nylon wool, Sephadex G-25 [96] and endothelial cells [97]; enhancement of endothelial cell--neutrophil adhesion is nevertheless the result of effects on endothelial cells and not on the neutrophils [98].
LTB4, and to a lesser extent other lipoxygenase products, is able to induce neutrophil aggregation and degranulation [99, 100], and it has been demonstrated that the secretion of azurophilic granules in human polymorphonuclear leukocytes is largely mediated through an autocrine effect of LTB4 [101].
Reactive oxygen species production upon LTB4 challenge has been reported in some, but not all, studies, suggesting that lipoxygenase metabolites are poor activators of the respiratory burst. An enhancing effect of LTB4 toward chemotactic peptide-stimulated oxidative metabolism was observed in human neutrophils [102]. Recently LTB4 has been reported to be a potent activator of the peroxisome proliferator-activated receptor alpha [103], a transcription factor that plays a key role in lipid homeostasis by regulating the oxidative degradation of fatty acids and their derivatives, such as LTB4 itself. This observation generated the fascinating hypothesis that activation of its own metabolism through omega-oxidation may act as a negative feedback, contributing to limit the pro-inflammatory effect of LTB4 [104]; this hypothesis is also providing new grounds for the interpretation of the beneficial effects of lipid dietary manipulation in pathologies associated with the synthesis of lipid inflammatory mediators (for review see [105]).
The biological activities of LTB4 are mediated by the B-LT receptor characterized initially as specific high-affinity binding sites on human neutrophils [106]. Partial purification of B-LT receptor has been performed by different groups [107, 108] and cloning has been recently reported [109]. The B-LT receptor belongs to the family of seven transmembrane-spanning proteins and shows the highest overall homologies to somatostatin receptor type 3, human interleukin-8 receptor and human formyl-peptide related receptor II. It possibly couples to different G-proteins, as suggested by different sensitivities to pertussis toxin of inhibition of adenylate cyclase activity and increase of intracellular calcium concentrations observed in CHO cells stably transfected with the B-LT receptor [109].
LEUKOTRIENES IN INFLAMMATION
Leukotrienes are potent lipid bioeffectors and their synthesis is tightly controlled in 5-LO bearing cells. Since their structural characterization, their biological activities suggested a potential involvement of both Cys-LTs and LTB4 in inflammatory responses [110]. Cys-LTs are found in bronchial lavages [111], as well as in nasal secretions [112] obtained after local specific challenge in atopic subjects. Urinary excretion of LTE4 has been widely used as an index of systemic production of Cys-LTs and increased urinary LTE4 has been reported after antigen challenge of atopic asthmatics [113] and either oral or inhaled aspirin challenge of aspirin-sensitive subjects [114, 115].
The clinical evaluation of both Cys-LTs receptor antagonists and 5-LO inhibitors shed some more light on the potential pathogenetic role of Cys-LTs in bronchoconstriction (for review see [116]). The inhalation of potent, second generation Cys-LT1 receptor antagonists resulted both in the improvement of basal respiratory functions in asthmatics [117] and the prevention of exercise-induced bronchoconstriction [118]. Zileuton, the first selective 5-LO inhibitor approved by the U.S. Federal Drug Administration for use in asthma, has shown to improve symptoms and use of beta-agonist in mild to moderate asthmatics [119], and to prevent the development of bronchial, nasal, gastrointestinal and dermal symptoms following aspirin ingestion in aspirin-sensitive subjects [120].
Recently, increasing attention is being paid to the possible genetic modifications underlying the pathophysiology of asthma; increased expression of LTC4-synthase was observed in lung tissues from aspirin-sensitive asthmatics [121]. DNA obtained from normal and asthmatic subjects showed a family of three mutations of the transcription factor binding regions of the 5-LO gene; this correlated with the responsiveness to the treatment with the 5-LO inhibitor zileuton [122].
Asthma is not the only inflammatory pathological condition that appears to involve Cys-LTs production. LTC4 and LTD4 have been detected in pulmonary lavages from children showing essential pulmonary hypertension [123]. Enhanced urinary excretion of LTE4 was observed in acute respiratory distress syndrome patients [124] and in subjects with systemic lupus erythematosus [125]. In these latter subjects, the treatment with a 5-LO inhibitor also resulted in a significant improvement of the overall SLAM (Systemic Lupus Activity Measure) when compared with placebo [126]. Increased level of Cys-LTs both in sputum, that correlated with the severity of the pulmonary pathology, and in urine were found in cystic fibrotic patients [127, 128].
Percutaneous transluminal coronary angioplasty (PTCA), a rapidly diffusing method for treatment of symptomatic coronary artery disease, may result in subacute restenosis as well as in abrupt closure. Plaque rupture by PTCA has been reported to trigger intracoronary biosynthesis of Cys-LTs [129] that may contribute to coronary vasospasm routinely occurring after PTCA [130] as well as other PTCA-related complications. Interestingly, increased excretion of urinary LTE4 have been reported in cardiac ischemic patients [131].
Much less information is available concerning the involvement of LTB4 in inflammatory reactions. LTB4 is generated and released in vitro from colonic mucosa obtained from patients with ulcerative colitis or Crohn's disease [132] and in vivo [133]. Although several pilot studies have been conducted on the effect of 5-LO inhibition in ulcerative colitis, no data are available to date to support a causative role of LTB4 in the clinical manifestations of inflammatory bowel disease. LTB4 has also been found to be present in high concentrations in psoriatic scales [134] and a clinical controlled multicenter study involving patients affected by psoriasis vulgaris showed significant improvement of scaling and erythema in patients treated with a topical 5-LO inhibitor versus a control group [135]. Furthermore attenuation of dermal inflammation has been observed with a specific LTB4-antagonist [136].
CONCLUSIONS
Taken together, the evidences reported above underline the potential role of leukotrienes in the inflammatory responses that characterizes asthma and other pathological conditions. These potent lipid bioeffectors are synthesized during the course of inflammatory reactions and their pharmacological modulation is able to significantly attenuate the clinical manifestations associated with different inflammatory pathologies.
The development of new leukotriene receptor antagonists or synthesis inhibitors possessing higher potencies and good safety profiles, as well as novel therapeutic approaches to different targets, such as the leukotriene C4-synthase or nuclear transcription factors of corresponding enzymes, represent an important task that might help to provide a better understanding of the role of these lipids in physiology and pathology.
REFERENCES
1.Samuelsson, B., Borgeat, P., Hammaström, S.,
and Murphy, R. C. (1979) Prostaglandins, 17,
785-787.
2.Kellaway, C. H., and Trethewie, W. R. (1940) Q.
J. Exp. Physiol. Cogn. Med. Sci., 30, 121-145.
3.Brocklehurst, W. E. (1953) J. Physiol.
(London), 129, 16P-17P.
4.Murphy, R. C., Hammarström, S., and
Samuelsson, B. (1979) Proc. Natl. Acad. Sci. USA, 76,
4275-4279.
5.Borgeat, P., and Samuelsson, B. (1979) J. Biol.
Chem., 254, 2643-2646.
6.Ford-Hutchinson, A. W., Bray, M. A., Doig, M. V.,
Shipley, M. E., and Smith, M. J. (1980) Nature, 286,
264-265.
7.Waite, M. (1990) Biochemistry, Molecular Biology
and Physiology of Phospholipase A2 and Its Regulating
Factors, Plenum Press, New York, pp. 1-22.
8.Dennis, E. A. (1994) J. Biol. Chem.,
269, 13057-13060.
9.Fonteh, A. N., Bass, D. A., Marshall, L. A., Seeds,
M., Somet, J. M., and Chilton, F. H. (1994) J. Immunol.,
152, 5438-5446.
10.Marshall, L. A., Winkler, J. D., Groswold, D. E.,
Bolognese, B., Roshak, A., Sung, C. M., Webb, E. F., and Jacobs, R.
(1994) J. Pharmacol. Exp. Ther., 268, 709-717.
11.Leslie, C. C. (1991) J. Biol. Chem.,
266, 11366-11371.
12.Wong, A., Cook, M. N., Foley, J. J., Saran, H.
M., Marshall, P., and Hwang, S. M. (1991) Biochemistry,
30, 9346-9351.
13.Rouzer, C. A., and Kargman, S. (1988) J. Biol.
Chem., 263, 10980-10988.
14.Brock, T. G., McNish, R. W., and Peters-Golden,
M. (1995) J. Biol. Chem., 270, 21652-21658.
15.Brock, T. G., Paine, R., III, and Peters-Golden,
M. (1994) J. Biol. Chem., 269, 22059-22066.
16.Miller, D. K., Gillard, J. W., Vickers, P. J.,
Sadowski, S., Leveille, C., Mancini, J. A., Charleson, P., Dixon, R.
A., Ford-Hutchinson, A. W., Fortin, R., et al. (1990) Nature,
343, 278-281.
17.Dixon, R. A. F., Diehl, R. E., Opas, E., Rands,
E., Vickers, P. J., Evans, J. F., Gillard, J. W., and Miller, D. K.
(1990) Nature, 343, 282-284.
18.Abramovitz, M., Wong, E., Cox, M. E., Richardson,
C. D., Li, C., and Vickers, P. J. (1993) Eur. J. Biochem.,
215, 105-111.
19.Mancini, J. A., Abramovitz, M., Cox, M. E., Wong,
E., Charleson, S., Perrier, H., Wang, Z., Prasi, T. P., and Vickers, P.
J. (1993) FEBS Lett., 318, 277-281.
20.Lepley, R. A., Muskardin, D. T., and Fitzpatrick,
F. A. (1996) J. Biol. Chem., 271, 6179-6184.
21.Lepley, R. A., and Fitzpatrick, F. A. (1996)
Arch. Biochem. Biophys., 331, 141-144.
22.Peters-Golden, M., and McNish, R. W. (1993)
Biochem. Biophys. Res. Commun., 196, 147-153.
23.Samuelsson, B., and Funk, C. D. (1989) J.
Biol. Chem., 264, 19469-19472.
24.Shimizu, T., Rådmark, O., and Samuelsson, B.
(1984) Proc. Natl. Acad. Sci. USA, 81, 689-693.
25.MacMillan, D. K., Hill, E., Sala, A., Sigal, E.,
Shuman, T., Henson, P. M., and Murphy, R. C. (1994) J. Biol.
Chem., 269, 26663-26668.
26.Silberstein, D. S., Owen, W. F., Gasson, J. C.,
DiPersio, J. F., Golde, D. W., Bina, J. C., Soberman, R., Austen, K.
F., and David, J. R. (1986) J. Immunol., 137,
3290-3294.
27.Dahinden, C. A., Zingg, J., Maly, F. E., and de
Weck, A. L. (1988) J. Exp. Med., 167, 1281-1295.
28.McColl, S. R., Krump, E., Naccache, P. H., and
Borgeat, P. (1989) Agents and Actions, 27, 465-468.
29.Palmantier, R., Surette, M. E., Sanchez, A.,
Braquet, P., and Borgeat, P. (1994) Lab. Invest., 70,
696-704.
30.Kurimoto, Y., De Weck, A. L., and Dahinden, C. A.
(1991) Eur. J. Immunol., 21, 361-368.
31.Bischoff, S. C., and Dahinden, C. A. (1994) J.
Exp. Med., 175, 237-244.
32.DiPersio, J. F, Billing, P., Williams, R., and
Gasson, J. C. (1986) J. Immunol., 140, 4315-4320.
33.Lepley, R. A., and Fitzpatrick, F. A. (1994)
J. Biol. Chem., 269, 2627-2631.
34.Fitzpatrick, F. A., Morton, D. R., and Wynalda,
M. A. (1982) J. Biol. Chem., 257, 4680-4683.
35.Lam, B. K., Penrose, J. F., Freeman, G. J., and
Austen, K. F. (1994) Proc. Natl. Acad. Sci. USA, 91,
7663-7667.
36.Welsch, D. J., Creely, D. P., Hauser, S. D.,
Mathis, K. J., Krivi, G. G., and Isakson, P. C. (1994) Proc. Natl.
Acad. Sci. USA, 91, 9745-9749.
37.Radmark, O., Shimizu, T., Jornvall, H., and
Samuelsson, B. (1984) J. Biol. Chem., 259,
12339-12345.
38.Evans, J. F., Dupuis, P., and Ford-Hutchinson, A.
W. (1985) Biochim. Biophys. Acta, 840, 43-50.
39.Mueller, M. J., Wetterholm, A., Blomster, M.,
Jörnvall, H., Samuelsson, B., and Haeggström, J. Z. (1995)
Proc. Natl. Acad. Sci. USA, 92, 8383-8387.
40.Nadeau, M., Fruteau de Laclos, B., Picard, S.,
Braquet, P., Corey, E. J., and Borgeat, P. (1984) Can. J. Biochem.
Cell Biol., 62, 1321-1326.
41.Sala, A., Bolla, M., Zarini, S.,
Müller-Peddinghaus, R., and Folco, G. C. (1996) J. Biol.
Chem., 271, 17944-17948.
42.Borgeat, P., and Samuelsson, B. (1979) Proc.
Natl. Acad. Sci. USA, 76, 2148-2152.
43.Weller, P. F., Lee, C. W., Foster, D. W., Corey,
E. J., Austen, K. F., and Lewis, R. A. (1983) Proc. Natl. Acad. Sci.
USA, 80, 7626-7630.
44.MacGlashan, D. W., Jr., Schleimer, R. P., Peters,
S. P., Schulman, E. S., Adams, G. K., III, Newball, H. H., and
Lichtenstein, L. M. (1982) J. Clin. Invest., 70,
747-751.
45.McGee, J. E., and Fitzpatrick, F. A. (1986)
Proc. Natl. Acad. Sci. USA, 83, 1349-1353.
46.Maclouf, J. A., and Murphy, R. C. (1988) J.
Biol. Chem., 263, 174-181.
47.McGee, J. E., and Fitzpatrick, F. A. (1985) J.
Biol. Chem., 260, 12832-12837.
48.Soderstrom, M., Mannervik, B., Garkov, V., and
Hammarstrom, S. (1992) Arch. Biochem. Biophys., 294,
70-74.
49.Maclouf, J., Murphy, R. C., and Henson, P. M.
(1989) Blood, 74, 703-707.
50.Feinmark, S. J., and Cannon, P. J. (1987)
Biochim. Biophys. Acta, 922, 125-135.
51.Grimminger, F., Kreusler, B., Schneider, U.,
Becker, G., and Seeger, W. (1990) J. Immunol.,
144,1866-1872.
52.Sala, A., Rossoni, G., Buccellati, C., Berti, F.,
Folco, G., and Maclouf, J. (1993) Br. J. Pharmacol.,
110, 1206-1212.
53.Sala, A., Testa, T., and Folco, G. (1996) FEBS
Lett., 388, 94-98.
54.Maclouf, J., Antoine, C., De Caterina, R.,
Sicari, R., Murphy, R. C., Patrignani, P., Loizzo, S., and Patrono, C.
(1992) Am. J. Physiol., 263, H244-H249.
55.Sala, A., Voelkel, N., Maclouf, J., and Murphy,
R. C. (1990) J. Biol. Chem., 265, 21771-21778.
56.Sala, A., Armetti, L., Piva, A., and Folco, G.
(1994) Prostaglandins, 47, 281-292.
57.Uemura, M., Buchholz, U., Kojima, H., Keppler,
A., Hafkemeyer, P., Fukui, H., Tsujii, T., and Keppler, D. (1994)
Hepatology, 20, 804-812.
58.Mayatepek, E., Lehmann, W. D., Fauler, J.,
Tsikas, D., Frolich, J. C., Schutgens, R. B., Wanders, R. J., and
Keppler, D. (1993) J. Clin. Invest., 91, 881-888.
59.Shak, S., and Goldstein, I. M. (1984) J. Biol.
Chem., 259, 10181-10187.
60.Soberman, R. J., Harper, T. W., Murphy, R. C.,
and Austen, K. F. (1985) Proc. Natl. Acad. Sci. USA, 82,
2292-2295.
61.Cluzel, M., Undem, B. J., and Chilton, F. H.
(1989) J. Immunol., 143, 3659-3665.
62.Dahlén, S.-E., Hedqvist, P.,
Hammarström, S., and Samuelsson, B. (1980) Nature,
288, 484-486.
63.Dahlen, S.-E. (1988) in Asthma: Basic
Mechanisms and Clinical Management (Barnes, P. J., Roger, I. W.,
and Thomson, N. C., eds.) Academic Press, London, pp. 213-230.
64.Drazen, J. M., Lewis, R. A., Austen, K. F., Toda,
M., Brion, F., Marfat, A., and Corey, E. J. (1981) Proc. Natl. Acad.
Sci. USA, 78, 3195-3198.
65.Lewis, R. A., Lee, C. W., Levine, L., Morgan, R.
A., Weiss, J. W., Drazen, J. M., Oh, H., Hoover, D., Corey, E. J., and
Austen, K. F. (1983) in Advances in Prostaglandin, Thromboxane and
Leukotriene Research (Samuelsson, B., Ramwell, P. W., and Paoletti,
R., eds.) Vol. 11, Raven Press, New York, pp. 15-26.
66.Arm, J. P., O'Hickey, S. P., Hawksworth, R. J.,
Fong, C. Y., Crea, A. E., Spur, B. W., and Lee, T. H. (1990) Am.
Rev. Resp. Dis., 142, 1112-1118.
67.Coles, S. J., Neill, K. H., Reid, L. M., Austen,
K. F., Nii, Y., Corey, E. J., and Lewis, R. A. (1983)
Prostaglandins, 25, 155-170.
68.Marom, Z., Shelhamer, J. H., Bach, M. K., Morton,
D. R., and Kaliner, M. (1982) Am. Rev. Resp. Dis., 126,
449-451.
69.Hanna, C. J., Bach, M. K., Pare, P. D., and
Schellenberg, R. R. (1981) Nature, 290, 343-344.
70.Schellenberg, R. R., and Foster, A. (1984)
Prostaglandins, 27, 475-482.
71.Michelassi, F., Landa, L., Hill, R. D.,
Lowenstein, E., Watkins, W. D., Petkau, A. J., and Zapol, W. M. (1982)
Science, 217, 841-843.
72.Roth, D. M., and Lefer, A. M. (1983)
Prostaglandins, 26,573-581.
73.Sala, A., Aliev, G. M., Rossoni, G., Berti, F.,
Buccellati, C., Burnstock, G., Folco, G. C., and Maclouf, J. (1996)
Blood, 87, 1824-1832.
74.Ortiz, J. L., Gorenne, I., Cortijo, J., Seller,
A., Labat, C., Sarria, B., Abram, T. S., Gardiner, P. J., Morcillo, E.,
and Brink, C. (1995) Br. J. Pharmacol., 115,
1382-1386.
75.Dahlen, S.-E., Bjork, J., Hedqvist, P., Arfors,
K. E., Hammarström, S., Lindgren, J. A., and Samuelsson, B. (1981)
Proc. Natl. Acad. Sci. USA, 78, 3887-3891.
76.Drazen, J. M., Austen, K. F., Lewis, R. A.,
Clark, D. A., Goto, G., Marfat, A., and Corey, E. J. (1980) Proc.
Natl. Acad. Sci. USA, 77, 4354-4358.
77.Williams, T. J., and Piper, P. J. (1980)
Prostaglandins, 19, 779-789.
78.Omini, C., Folco, G. C., Viganò, T.,
Rossoni, G., Brunelli, G., and Berti, F. (1981) Pharmacol. Res.
Comm., 13, 633-640.
79.Metters, K. M. (1995) J. Lipid Mediators Cell
Signalling, 12, 413-427.
80.Tudhope, S. R., Cuthbert, N. J., Abram, T. S.,
Jennings, M. A., Maxey, R. J., Thompson, A. M., Norman, P., and
Gardiner, P. J. (1994) Eur. J. Pharmacol., 264,
317-323.
81.Chan, C. C., Ecclestone, P., Nicholson, D. W.,
Metters, K. M., Pon, D. J., and Rodger, I. W. (1994) J. Pharmacol.
Exp. Therap., 269, 891-896.
82.Howard, S., Chan-Yeung, M., Martin, L., Phaneuf,
S., and Salari, H. (1992) Eur. J. Pharmacol., 227,
123-129.
83.Ford-Hutchinson, A. W., Bray, M. A., Doig, M. V.,
Shipley, M. E., and Smith, M. J. (1980) Nature, 286,
264-265.
84.Palmer, R. M., Stepney, R. J., Higgs, G. A., and
Eakins, K. E. (1980) Prostaglandins, 20, 411-418.
85.Goetzl, E. J., and Pickett, W. C. (1980) J.
Immunol., 125, 1789-1791.
86.Lewis, R. A., Goetzl, E. J., Drazen, J. M.,
Soter, N. A., Austen, K. F., and Corey, E. J. (1981) J. Exp.
Med., 154, 1243-1248.
87.Dahinden, C. A., Clancy, R. M., and Hugli, T. E.
(1984) J. Immunol., 133, 1477-1482.
88.Palmblad, J., Uden, A. M., Lindgren, J. Å.,
Rådmark, O., Hansson, G., and Malmsten, C. L. (1981) FEBS
Lett., 144, 81-84.
89.Smith, M. J., Ford-Hutchinson, A. W., and Bray,
M. A. (1980) J. Pharm. Pharmacol., 32, 517-518.
90.O'Flaherty, J. T., Hammett, M. J., Shewmake, T.
B., Wykle, R. L., Love, S. H., McCall, C. E., and Thomas, M. J. (1981)
Biochem. Biophys. Res. Commun., 103, 552-558.
91.Bhattacherjee, P., Hammond, B., Salmon, J. A.,
Stepney, R., and Eakins, K. E. (1981) Eur. J. Pharmacol.,
73, 21-28.
92.Bray, M. A., Ford-Hutchinson, A. W., and Smith,
M. J. (1981) Prostaglandins, 22, 213-222.
93.Bjork, J., and Arfors, K. E. (1982) Agents and
Actions, 11, 63-72.
94.Bjork, J., Hedqvist, P., and Arfors, K. E. (1982)
Inflammation, 6, 189-200.
95.McDonald, P. P., McColl, S. R., Braquet, P., and
Borgeat, P. (1994) Br. J. Pharmacol., 111, 852-860.
96.Goetzl, E. J., Brindley, L. L., and Goldman, D.
W. (1983) Immunology, 50, 35-41.
97.Gimbrone, M. A., Jr., Brock, A. F., and Schafer,
A. I. (1984) J. Clin. Invest., 74, 1552-1555.
98.Hoover, R. L., Karnovsky, M. J., Austen, K. F.,
Corey, E. J., and Lewis, R. A. (1984) Proc. Natl. Acad. Sci.
USA, 81, 2191-2193.
99.O'Flaherty, J. T., Wykle, R. L., Lees, C. J.,
Shewmake, T., McCall, C. E., and Thomas, M. J. (1981) Am. J.
Pathol., 105, 264-269.
100.Bokoch, G. M., and Reed, P. W. (1981) J.
Biol. Chem., 256, 5317-5320.
101.Hatzelmann, A., Fruchtmann, R., Mohrs, K. H.,
Raddatz, S., and Müller-Peddinghaus, R. (1994) Biochem.
Pharmacol., 48, 31-39.
102.Gay, J. C., Beckman, J. K., Brash, A. R.,
Oates, J. A., and Lukens, J. N. (1984) Blood, 64,
780-785.
103.Devchand, P. R., Keller, H., Peters, J. M.,
Vazquez, M., Gonzalez, F. J., and Wahli, W. (1996) Nature,
384, 39-43.
104.Serhan, C. N. (1996) Nature,
384, 24-25.
105.Weber, P. C., and Sellmayer, A. (1990) in
Advances in Prostaglandin, Thromboxane and Leukotriene Research
(Samuelsson, B., Ramwell, P. W., Paoletti, R., Folco, G., and
Granstrom, E., eds.) Vol. 21A, Raven Press, New York, pp. 217-224.
106.Lin, A. H., Ruppel, P. L., and Gorman, R. R.
(1984) Prostaglandins, 28, 837-849.
107.Sherman, J. W., Mendelson, M. A., Boggs, J. M.,
Koo, H., and Goetzl, E. J. (1992) J. Cell Biochem., 48,
367-372.
108.Miki, I., Watanabe, T., Nakamura, M., Seyama,
Y., Ui, M., Sato, F., and Shimizu, T. (1990) Biochem. Biophys. Res.
Commun., 166, 342-348.
109.Yokomizo, T., Izumi, T., Chang, K., Takuwa, Y.,
and Shimizu, T. (1997) Nature, 387, 620-624.
110.Samuelsson, B. (1983) Science,
220, 568-575.
111.Miadonna, A., Tedeschi, A., Brasca, C., Folco,
G. C., Sala, A., and Murphy, R. C. (1990) J. Allergy Clin.
Immunol., 85, 906-913.
112.Miadonna, A., Tedeschi, A., Leggieri, E.,
Lorini, M., Folco, G. C., Sala, A., Qualizza, R., Froldi, M., and
Zanussi, C. (1987) Am. Rev. Resp. Dis., 136,
357-362.
113.Manning, P. J., Rokach, J., Malo, J. L.,
Ethier, D., Cartier, A., Girard, Y., Charleson, S., and O'Byrne, P. M.
(1990) J. Allergy Clin. Immunol., 86, 211-220.
114.Christie, P. E., Tagari, P., Ford-Hutchinson,
A. W., Charlesson, S., Chee, P., Arm, J. P., and Lee, T. H. (1991)
Am. Rev. Resp. Dis., 143, 1025-1029.
115.Sestini, P., Armetti, L., Gambaro, G., Pieroni,
M. G., Refini, R. M., Sala, A., Folco, G. C., Bianco, S., and Robuschi,
M. (1996) Am. J. Resp. Critical Care Med., 153,
572-575.
116.Folco, G. C., Buccellati, C., Testa, T., Bolla,
M., and Sala, A. (1996) in Eicosanoids: from Biotechnology to
Therapeutic Applications (Folco, G. C., Samuelsson, B., Maclouf,
J., and Velo, G. P., eds.) Plenum Publishing Corporation, New York, pp.
165-182.
117.Gaddy, J. N., Margolskee, D. J., Bush, R. K.,
Williams, V. C., and Busse, W. W. (1992) Am. Rev. Resp. Dis.,
146, 358-363.
118.Manning, P. J., Watson, R. M., Margolskee, D.
J., Williams, V. C., Schwartz, J. I., and O'Byrne, P. M. (1990) New
Engl. J. Med., 323, 1736-1739.
119.Israel, E., Rubin, P., Kemp, J. P., Grossman,
J., Pierson, W., Siegel, S. C., Tinkelman, D., Murray, J. J., Busse,
W., Segal, A. T., et al. (1993) Ann. Internal Med., 119,
1059-1066.
120.Israel, E., Fischer, A. R., Rosenberg, M. A.,
Lilly, C. M., Callery, J. C., Shapiro, J., Cohn, J., Rubin, P., and
Drazen, J. M. (1993) Am. Rev. Resp. Dis., 148,
1447-1451.
121.Sampson, A. P., Cowburn, A. S., Sladek, K.,
Adamek, L., Nizankowska, E., Szczeklik, A., Lam, B. K., Penrose, J. F.,
Austen, K. F., and Holgate, S. T. (1997) Int. Arch. Allergy
Immunol., in press.
122.Drazen, J. M., Yandava, C., Pillari, K. H.,
Swanson, L., Dydo, M., Szczerbak, N., Bube, L., and the Zileuton Study
Group (1997) Am. J. Resp. Crit. Care Med., 155,
A257.
123.Stenmark, K. R., James, S. L., Voelkel, N. F.,
Toews, W. H., Reeves, J. T., and Murphy, R. C. (1983) New Engl. J.
Med., 309, 77-80.
124.Westcott, J. Y., Thomas, R. B., and Voelkel, N.
F. (1991) Prostaglandins Leukotrienes Essent. Fatty Acids,
43, 151-158.
125.Hackshaw, K. V., Voelkel, N. F., Thomas, R. B.,
and Westcott, J. Y. (1992) J. Rheumatol., 19,
252-258.
126.Hackshaw, K. V., Shi, Y., Brandwein, S. R.,
Jones, K., and Westcott, J. Y. (1995) J. Rheumatol., 22,
462-468.
127.Sampson, A. P., Spencer, D. A., Green, C. P.,
Piper, P. J., and Price, J. F. (1990) Br. J. Clin. Pharmacol.,
30, 861-869.
128.Spencer, D. A., Sampson, A. P., Green, C. P.,
Costello, J. F., Piper, P. J., and Price, J. F. (1992) Pediatric
Pulmonol., 12, 90-94.
129.Brezinski, D. A., Nesto, R. W., and Serhan, C.
N. (1992) Circulation, 86, 56-63.
130.Fischell, T. A., Derby, G., Tse, T. M., and
Stadius, M. L. (1988) Circulation, 78, 1323-1334.
131.Carry, M., Korley, V., Willerson, J. T.,
Weigelt, L., Ford-Hutchinson, A. W., and Tagari, P. (1992)
Circulation, 85, 230-236.
132.Sharon, P., and Stenson, W. F. (1985)
Gastroenterology, 88, 55-63.
133.Lauritsen, K., Laursen, L. S., Bukhave, K., and
Rask-Madsen, J. (1986) Gastroenterology, 91,
837-844.
134.Ruzicka, T., Simmet, T., Peskar, B. A., and
Ring, J. (1986) J. Invest. Dermatol., 86, 105-108.
135.Degreef, H., Dockx, P., De Doncker, P., De
Beule, K., and Cauwenbergh, G. (1990) J. Am. Acad. Dermatol.,
22, 751-755.
136.Fretland, D. J., Gokhale, R., Mathur, L.,
Baron, D. A., Paulson, S. K., and Stolzenbach, J. (1995)
Inflammation, 19, 333-346.