REVIEW: The Role of Active Site Flexibility in Enzyme Catalysis
Chen-Lu Tsou
National Laboratory of Biomacromolecules, Institute of Biophysics, Academia Sinica, Beijing 100101, China; fax: +86-10-6202-2026; E-mail: cltsou@sun5.ibp.ac.cn
 Chen-Lu Tsou |
Received July 24, 1997
It has been shown in this and other laboratories that during the unfolding of a number of enzymes inactivation generally precedes global unfolding of the enzyme molecule, leading to the suggestion that enzyme active sites are usually more "fragile" and more easily "perturbed" than the molecule as a whole and are therefore conformationally more flexible than the rest of the molecule. However, the role of active site flexibility in enzyme catalysis still remains to be explored. In the induced fit hypothesis originally proposed by Koshland, the presence of the substrate induces a conformational change at the active site so as to fit with the structure of the substrate. By X-ray crystallographic structural analysis of E. coli dihydrofolate reductase liganded with cofactors and substrates, Sawaya and Kraut showed the enzyme in different conformational states indeed while complexed with different ligands, suggesting that the enzyme molecule passes through different conformational states through the whole process of catalysis. Muscle lactate dehydrogenase can be stabilized either in concentrated ammonium sulfate or by cross-linking with glutaraldehyde together with a decrease in enzyme activity which can be restored to the original level in dilute guanidine hydrochloride possibly by increased flexibility at the active site. It is known that a number of enzymes can be activated by chaotropic agents such as urea or guanidine hydrochloride. The activation of dihydrofolate reductase by either urea or guanidine hydrochloride is accompanied by an increase in susceptibility to proteolysis. Isolation of the tryptic peptides of the activated enzyme and sequence analysis allowed identification of the sites of proteolysis to be at or near the active site of the enzyme, indicating an opening up of the active site conformation in the activated state. All the above indicate that active site flexibility plays an important role in enzyme catalysis. It is possible that during the catalytic cycle, the enzyme molecule passes through different stages and each stage requires the molecule to be in a different conformation, especially at the active site. Rapid transition between the different conformational states, and hence the flexibility of the active site, is therefore mandatory for the maximal expression of enzyme activity. KEY WORDS: enzyme catalysis, flexibility of active site, enzyme inactivation, dihydrofolate reductase, lactate dehydrogenase |
Abbreviations: DHFR) dihydrofolate reductase; GuHCl) guanidine hydrochloride; RNase) ribonuclease A.
In the lock and key theory proposed by Fischer in the 19th century [1], an enzyme active site was envisaged to have a rigid structure complementary exactly the structure of the substrate so as to facilitate substrate binding and the enzyme catalyzed reactions. It was not until the middle of the present century that Koshland advanced the induced fit theory to propose that the original structure of enzyme active sites does not fit the substrate exactly but the presence of the substrate induces changes in the active site structure to fit for substrate binding [2]. At the same time the structure of the substrate is also changed so as to fit the changed active site. The changed structure of the substrate is analogous to that of the transition state favorable to undergo the enzyme catalyzed reaction. The above theory has received considerable experimental support and is now widely accepted [3, 4]. Nevertheless, it is still implied that the changed active site is sufficiently structured so as to perform the catalytic function of the enzyme and is probably more stable than other parts of the enzyme molecule of less functional importance.
FLEXIBILITY OF ENZYME ACTIVE SITES
Among the vast amount of literature on the unfolding of proteins, only a limited number of authors have paid some attention to the comparison of the conformation and activity changes during the course of enzyme denaturation. In the earlier years, Miller and Bolen [5] showed that guanidine hydrochloride (GuHCl) at 1.25 M inactivated pancreatic ribonuclease A (RNase A) without gross unfolding of the molecule, for which a much higher concentration of the denaturant was required. Similar results were reported by Yano and Irie for ATPase [6]. However, the majority of authors in that period reported concurrent processes for the inactivation and unfolding of enzyme molecules during denaturation [7-10] as would be predicted by the "all or none" model [11] for protein unfolding which was then the dominant model for protein unfolding and accepted by most workers in this field [12]. However, a detailed comparison of the conformation and activity changes of creatine kinase during denaturation by GuHCl or urea in this laboratory [13, 14] showed that inactivation occurs at much lower denaturant concentrations than required to bring about detectable global conformational changes of the molecule. Similar results have been since obtained for a large number of other enzymes in this and other laboratories [15, 16]. Inactivation before global molecular conformation change has also been found for proteins other than enzymes, for example, the inactivation of the choline transporter in erythrocytes by n-alkanols [17] and the effect of GuHCl on the binding of antibodies with antigens [18].
Kinetically, the inactivation rates of creatine kinase during GuHCl or urea denaturation are over three orders of magnitude faster than the rates of global molecular conformational changes under the same conditions [15]. The rates of inactivation of several enzymes by GuHCl so far examined vary over a wide range, with apparent first order rate constants ranging from 0.00054 sec-1 in 4 M GuHCl for papain to over 50 sec-1 for pancreatic RNase A in 2 M GuHCl [15], the latter is too fast for the rate constants to be accurately determined even by following the substrate reaction [19] in a stopped-flow apparatus because the viscosity of concentrated GuHCl solution makes the dead time of mixing upon dilution of the denatured enzyme in the denaturant considerably more than 10 msec. Three possibilities were initially considered [13, 14] for the observation that inactivation preceded molecular unfolding during the denaturation of creatine kinase, viz., 1) an inhibition of the enzyme by the denaturant; 2) dissociation of the active dimeric enzyme into inactive monomers; and 3) conformation perturbation of a relatively flexible enzyme active site. For this possibility, it is to be noted that during their observation on the effects of urea or GuHCl on the activity and dynamic structure of equine liver alcohol dehydrogenase, Strambini and Gonnelli [20] have suggested that the loss of enzyme activity is associated with a loosening of intramolecular interactions resulting in a greater fluidity of the interior region of the molecule.
The very fast rate of inactivation of pancreatic RNase in dilute GuHCl and the report in the literature that GuHCl inhibits RNase A [21] raises the question whether the decrease in activity in dilute GuHCl solutions for RNase A could be actually a reversible inhibition by GuHCl binding at the active site, as indeed suggested by Creighton [22], instead of a slight perturbation of the active site conformation produced by dilute GuHCl. However, results obtained for thermal denaturation studies show that thermal inactivation precedes thermally induced conformational change in a number of enzymes [23]. As no inhibitor is present during thermal denaturation, the inhibition by an inhibitor being responsible for the loss of enzyme activity can be excluded. The second possibility considered [14] for the observation that inactivation precedes general molecular unfolding of creatine kinase was that the enzyme has to be in its dimeric state to be active, and dissociation into the monomers, even without noticeable unfolding, could have led to inactivation. However, direct examination has shown that the dissociation of dimeric creatine kinase occurs at a much higher denaturant concentration than required to inactivate the enzyme [24]. Recent studies on the denaturation of a number of multimeric enzymes also show [25, 26] inactivation at lower GuHCl concentrations than required for dissociation of the multimeric enzyme molecules. From the above, it seems clear that the decrease in enzyme activity under mild denaturing conditions is not due either to inhibition by the denaturant or to dissociation for multimeric enzymes, but to conformation "perturbation" at the active site.
DIRECT DEMONSTRATION OF CONFORMATION CHANGES AT THE ACTIVE SITE
UNDER MILD DENATURING CONDITIONS
To show that the active site conformation is indeed "perturbed" under mild denaturation conditions leading thus to enzyme inactivation, conformational changes have been examined directly by fluorescent [27] or ESR [28] probes introduced to enzyme active sites. These probes show that under mild denaturation conditions, the active site conformation is indeed "perturbed" without detectable global changes of the enzyme molecules.
Partial proteolysis, which has been shown to be a sensitive method to detect "subtle" conformational changes of protein molecules [29], has provided additional evidence for active site conformational changes under mild denaturation conditions. Inactivation of pancreatic RNase A occurs in GuHCl at low concentrations before the unfolding of the molecule as a whole can be detected [30]. It has been shown that concurrent to inactivation of RNase in dilute GuHCl, conformational change at the active site region of the RNase molecule does occur as indicated by increased susceptibility to digestion with either trypsin or proteinase K, and from the sequences of the peptide fragments liberated, the loci of proteolysis can be identified to be at or near the active site region. Binding of GuHCl as an inhibitor would have resulted, if anything, in protection against proteolysis, particularly at the active site region.
X-Ray diffraction studies have shown that enzyme active sites are usually situated at the hinge regions connecting two structural domains. For example, this has been shown by Artymiuk et al. for lysozyme as early as 1979 [31]. Similar results have been reported by comparative X-ray diffraction studies for the structure of thermolysin and other neutral proteases [32]. Studies of some other enzymes have likewise revealed structural flexibility of regions that are significant for activity, for instance, a comparison of the structure of alpha-lactalbumin from three difference mammalian species showed that these molecules have common regions of high thermal parameters (B-factors) and, hence, are likely to be flexible which is implicated in their lactose synthase activity [33]. The same conclusion has been reached by comparison of the structures of three mutants of the E. coli tryptophan synthase [34]. Porcine pepsin [35] and a mutant of RNase T1 [36] have also been reported to have flexible active site regions. It would seem likely, therefore, that the active sites of most enzymes are situated in such a region which is relatively more flexible or mobile than the molecule as a whole and consequently more sensitive to be perturbed by denaturants and physical factors.
FLEXIBILITY AT ACTIVE SITES REQUIRED FOR THE FULL EXPRESSION OF
ENZYME CATALYTIC POWER
For the consideration of the role of active site flexibility in enzyme catalysis, the well known "induced fit" hypothesis by Koshland [2] is to be recalled; it depicts a change in the active site conformation during substrate binding. It is possible that each intermediate step of the whole cycle of enzyme catalysis requires the enzyme molecule, especially the active site region, to be in a specific conformational states different from one another. Although direct experimental demonstration for the passage of the enzyme molecule through different conformational states at the active site during the catalytic cycle is still lacking, a recent X-ray crystallographic study of different forms of E. coli dihydrofolate reductase (DHFR), including the holo-enzyme, the Michaelis complex, the ternary product complex, the tetrahydrofolate binary complex as well as the tetrahydrofolate--NADPH complex yield structures which can be used to reconstruct a 2.1 Å resolution movie depicting the sequence of events during the catalytic cycle [37].
There are indications in the literature that the flexibility of enzyme active sites is required for maximal catalytic activity. It has long been known that some enzymes are activated by chaotropic agents or limited proteolysis and the activation has been suggested to be the result of a perfection of the active site structure as required for maximal catalytic efficiency. Dihydrofolate reductase can be markedly activated by salts, chaotropes including urea, GuHCl, thiourea, and formamide [38]. For fructose-1,6-bisphosphatase, alkaline phosphatase, and catalase, activation by denaturants or by partial proteolysis [39-41] have been observed. The activation of DHFR is accompanied by an increase in sensitivity of the enzyme molecule to heat and proteolysis and this is inconsistent with the perfection of the active site structure hypothesis but suggests instead a transition from a compact and stable to a relatively open and more extended structure [41]. Similar suggestions have been made for the activation of other enzymes. In this connection, it is to be noted that there is only relatively little difference between the structures at the active site region of chymotrypsin and chymotrypsinogen [42]. It is possible that the activation of the zymogen by partial proteolysis is not due to the perfection of a well structured active site with only subtle differences from that in the zymogen and required for catalysis, but to an increase in the flexibility at the active site region and it is the flexibility rather than a well formed structure of the active site which is responsible for the catalysis.
The results obtained in this laboratory on muscle lactate dehydrogenase H4 are highly suggestive [43]. Muscle lactate dehydrogenase can be considerably stabilized against unfolding during denaturation in GuHCl by either cross-linking with glutaraldehyde or in concentrated ammonium sulfate solutions as shown by the shift of the GuHCl concentrations required for both inactivation and unfolding of the enzyme molecule (Fig. 1). In both cases, the enzyme loses about 1/3 its original activity in the absence of the denaturant. However, dilute GuHCl can restore the activity back to its original level. Further increase in GuHCl concentration results in unfolding of the molecule and loss of activity. It appears reasonable to suggest that the stabilization with either glutaraldehyde cross-linking or in concentrated ammonium sulfate is accompanied by decreased flexibility at the active site which in turn leads to a decrease in enzyme activity and the reactivation by dilute GuHCl is due to the restoration of the original flexibility at the active site which is required for the full expression of enzyme activity. The inability of crystalline RNase A for substrate or inhibitor binding below the dynamic transition temperature at 220 K is also highly suggestive that flexibility of the enzyme is essential for its activity [44].
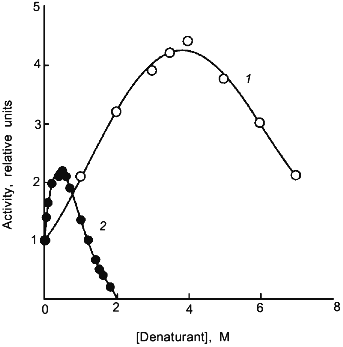
The loosening of the active site structure during enzyme activation by denaturants has recently been demonstrated for DHFR [45]. Figure 2 shows the activation of this enzyme by urea and GuHCl. The activation can be as high as up to 4- and 2-fold in 4 M urea and 1 M GuHCl, respectively, with greatly increased susceptibility to proteolysis by trypsin; the tryptic peptides have been separated and sequenced. By comparison of the N-terminal sequences of the peptide fragments thus obtained with the known sequence of the enzyme allows identification of the loci of tryptic proteolysis which, as can be seen in Fig. 3, are all situated at or near the active site region. It is therefore clear that the activation of this enzyme by the denaturants is accompanied by a loosening of the active site structure. The recent X-ray crystallographic structure study of different forms of E. coli DHFR mentioned above [37] depicts loop and subdomain movements during the catalytic cycle suggesting the requirement of flexibility in substrate binding and in proton transfer.Fig. 1. Activity and conformational changes of lactate dehydrogenase H4 cross-linked with glutaraldehyde during denaturation in GuHCl. Curves 1, 2, 3, and 4 represent, respectively, relative changes in ellipticity at 220 nm, fluorescence emission intensity at 340 nm, enzyme activity (left ordinate), and emission maximum shift (right ordinate).
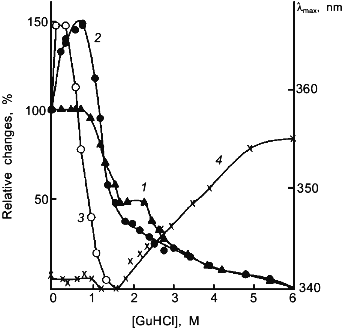
Fig. 2. Activation of dihydrofolate reductase by urea (1) and GuHCl (2). The reaction mixture contained the indicated concentrations of urea or GuHCl.
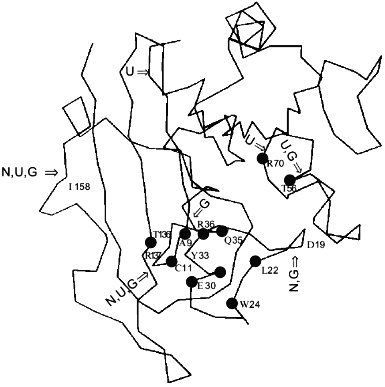
Similar findings by different methods for a number of enzymes have likewise led to the suggestion of active site flexibility being involved in enzyme activity. For example, Yang and Miles [46], reported that by mutation studies, the flexible loop residues in the tryptophan synthase alpha-subunit have critical roles in modulating its enzymatic activities of the beta-subunit in the alpha2beta2 complex. From a study of ligand induced conformational changes of E. coli ornithine transcarbamylase and its specific mutants, Goldsmith and Kuo have suggested the utilization of flexibility in conformational isomerization of the enzyme molecule as required for substrate binding, and catalysis [47].Fig. 3. Cleavage positions in the DHFR molecule by proteolysis with trypsin in various conditions. "N" indicates the peptide bonds in native DHFR hydrolysed by trypsin, "U" the hydrolysed bonds in 2 M urea activated form, and "G" the hydrolysed bonds in 1 M GuHCl. The active site residues Ala-9, Cys-11, Leu-22, Trp-24, Glu-30, Tyr-31, Glu-34, Gln-35, Thr-56, Arg-70, and Thr-136 are marked with black circles.
DISCUSSION
It must be emphasized that the flexibility believed to be essential for enzyme activity can only be relative and very likely limited to specified molecular regions. It is well known that some surface regions of protein molecules are highly mobile so as to escape characterization by structural studies. There is no apparent evidence to suggest that such highly mobile surface regions are directly involved in enzyme catalysis. The dynamic nature of protein structure and the importance of flexibility for protein function are now generally recognized [48], and the importance of molecular flexibility for protein function in general has been recently emphasized by Dobson [49] and by Kurzynski [50]. For examples, flexibility has been proposed to be the key for the function of the p53 protein, a tumor suppressor that arrests the growth of cells containing damaged DNA [51]. Binding site flexibility appears to be essential for the binding of GroEL to both GroES [49] and to the target protein [52] and for the binding of actin with myosin [53].
Cross-linking to fix the C-terminal segment of the B-chain of insulin on the N-terminal of A-chain reduces the activity to about 5% of the native protein. Although the mobility of the C-terminal region of the B-chain is thereby considerably hindered, the structure of the molecule remains essentially the same. On the other hand, the highly active despentapeptide insulin, with the C-terminal 5 residues of the B-chain truncated, has essentially the same structure as native insulin except that the C-terminal segment of the B-chain becomes highly mobile so as to reveal a structural region which is originally hidden in the native structure, and this region is proposed to be responsible for receptor binding of the insulin molecule [54]. Consequently the flexibility of the C-terminal segment of the B-chain is essential for insulin binding to its receptor.
The immense increase in protein structure studies in recent years have been partly motivated by the possible use in the design of enzyme targeted drugs. Previous studies on drug design were mostly based on models of rigid enzyme structures. The proposal of flexible a active site being essential for enzyme activity would definitely have implications on the design of enzyme targeted drugs [55].
The initial inactivation of some proteins at low denaturant concentrations is usually easily reversible, involves limited conformational changes and can be compared to the molten globule state as initially proposed for the partially denatured state of alpha-lactalbumin and now found to be a common feature for the initial denatured state of many proteins. The consensus on the concept of the molten globule state has been defined as high contents of secondary structure, considerable molecular compactness, non-specific tertiary structure, and significant flexibility [56]. The molecule of enzymes inactivated in dilute GuHCl is partly loosened especially in the active site region and thus open to attack by proteolysis without noticeable changes in its secondary structure. For the inactivation of a multimeric enzyme, creatine kinase, in dilute GuHCl, increased flexibility has been revealed by a fluorescent probe introduced at the active site and general compactness of the molecule as a whole by viscosity measurements [24]. All the above are consistent with the concept of the molten globule. However, the initial state of inactivation involves conformation changes mostly at the active site region only, and for the molten globule state global molecular changes are considered. Further unfolding is accompanied by changes of the enzyme molecules as a whole, which can then be detected by the commonly employed physical methods. It would be interesting to examine whether the above holds generally for other molten globule state enzymes and proteins which have been shown to lose their activities without noticeable conformational changes of the molecules as a whole during the initial stage of denaturation by chemical denaturants and physical factors. In recent years, a "pre-molten globule state" has been proposed to precede the molten globule state in the initial stage of unfolding of proteins [57]. Unfortunately, the initial stages of unfolding has only been but rarely correlated to the activity changes during protein denaturation. The relation of this "pre-molten globule state" to the initial functionally inactivated state is to be further investigated.
I wish to acknowledge the financial support by the Panden Projects of the Chinese Commission of Science and Technology for this series of studies and to Ms. J. H. Liu for her help in the preparation of the manuscript.
REFERENCES
1.Price, N. C., and Stevens, L. (1982)
Fundamentals of Enzymology, Oxford University Press, Oxford, UK,
p. 2.
2.Koshland, D. E., Jr. (1958) Proc. Natl. Acad.
Sci. USA, 44, 98-104.
3.Stryer, L. (1991) Biochemistry, Freeman and
Co., New York, pp. 119-110.
4.Horton, H. R., Moran, L. A., Ochs, R. S., Rawn, J.
D., and Sacrimgeour, K. G. (1992) Principles of Biochemistry,
Neil Patterson Publishers, Prentice Hall, NJ, pp. 124-125.
5.Miller, J. F., and Bolen, D. W. (1978) Biochem.
Biophys. Res. Commum., 81, 610-615.
6.Yano, Y., and Irie, M. (1975) J. Biochem.,
78, 1001-1011.
7.Nieto, M., and Ayala, J. A. (1977) Biochem.
J., 161, 321-331.
8.Engelhard, M., Rudolph, R., and Jaenicke, R. (1976)
Eur. J. Biochem., 67, 447-453.
9.Busby, T. F., Atha, D. H., and Ingham, K. C. (1981)
J. Biol. Chem., 256, 12140-12147.
10.Doster, W., and Hess, B. (1981)
Biochemistry, 20, 772-780.
11.Hartley, R. W. (1968) Biochemistry,
7, 2401-2408.
12.Schellman, J. A. (1987) Annu. Rev. Biophys.
Biophys. Chem., 16, 115-117.
13.Yao, Q. Z., Zhou, H. M., Hou, L. X., and Tsou, C.
L. (1982) Sci. Sin., 25B, 484-493.
14.Yao, Q. Z, Tian, M., and Tsou, C. L. (1984)
Biochemistry, 23, 2740-2744.
15.Tsou, C. L. (1993) Science, 282,
380-381.
16.Tsou, C. L. (1995) Biochim. Biophys. Acta,
1253, 141-162.
17.Deves, R., and Krupka, R. M. (1990) Biochim.
Biophys. Acta, 1032, 3-40.
18.Wang, S. D., Luo, I., Guo, A. Q., Zhou, J. M.,
and Tsou, C. L. (1997) Biochem. J., in press.
19.Tsou, C. L. (1988) Adv. Enzymol.,
61, 381-436.
20.Strambini, G. B., and Gonnelli, M. (1986)
Biochemistry, 25, 2471-2476.
21.Anfinsen, C. B., Harrington, W. F., Hvidt, A.,
Lindstrom-Lang, K., and Ottesen, M. (1955) Biochim. Biophys.
Acta, 17, 141-142.
22.Creighton, T. E. (1990) Biochem. J.,
270, 1-16.
23.Zhang, Y. L., Zhou, J. M., and Tsou, C. L. (1993)
Biochim. Biophys. Acta, 1164, 61-67.
24.Liu, G. H., Hou, L. X., and Tsou, C. L. (1989)
Chin. Sci. Bull., 34, 1036-1040.
25.Lin, Y. Z., Liang, S. J., Zhou, J. M., Tsou, C.
L., Wu, P. Q., and Zhou, Z. K., Biochim. Biophys. Acta,
1038, 247-252.
26.Jiang, R. F., and Tsou, C. L. (1994) Biochem.
J., 303, 241-245.
27.Zhou, H. M., Zhang, X. H., Yong, Y., and Tsou, C.
L. (1993) Biochem. J., 291, 103-107.
28.Liu, Z. J., and Zhou, J. M. (1995) Biochim.
Biophys. Acta, 1253, 63-68.
29.Wilson, J. E. (1991) Meth. Biochem. Anal.,
35, 207-250.
30.Yang, H. J., and Tsou, C. L. (1995) Biochem.
J., 305, 371-384.
31.Artymiuk, P. J., Blake, C. C. F., Grace, D. E.
P., Oatley, S. J., Phillips, D. C., and Sternberg, M. J. E. (1979)
Nature, 280, 563-568.
32.Holland, D. R., Tronrud, D. E., Pley, H. W.,
Flaherty, K. M., Stark, W., Jansonius, J. N., Mckay, D. B., and
Matthews, B. W. (1992) Biochemistry, 31,
11310-11316.
33.Pike, A. C., Brew, K., and Acharya, K. R. (1996)
Structure, 4, 691-703.
34.Yang, X. J., and Miles, E. W. (1992) J. Biol.
Chem., 267, 7520-7528.
35.Abad-Zapatero, C., Rydel, T. J., and Erickson, J.
(1990) Proteins, 8, 62-81.
36.Koellner, G., Choe, H. W., Heinemann, U.,
Grunert, H. P., Zouni, A., Hahn, U., and Saenger, W. (1992) J. Mol.
Biol., 224, 701-713.
37.Sawaya, M. R., and Kraut, J. (1997)
Biochemistry, 36, 586-603.
38.Blakley, R. L. (1984) in Folates and
Pterins (Blakley, R. L., and Benkovic, S. J., eds.) Vol. 1, Wiley,
New York, pp. 191-253.
39.Zhao, F. K., Shi, J. P., and Xu, G. J. (1986)
Sci. Sin., 28B, 599-607.
40.Rao, N. M., and Nagaraj, R. (1991) J. Biol.
Chem., 266, 5018-5024.
41.Duffy, T. H., Beckman, S. B., Peterson, S. M.,
Vitols, K. S., and Huennekens, M. (1987) J. Biol. Chem.,
262, 7028-7033.
42.Wang, D. C., Bode, W., and Huber, R. (1985) J.
Mol. Biol., 185, 595-624.
43.Ma, Y. Z., and Tsou, C. L. (1991) Biochem.
J., 277, 207-211.
44.Rasmussen, B. F., Stock, A. M., Ringe, D., and
Petsko, G. A. (1992) Nature, 357, 423-424.
45.Fan, Y. X., Ju, M., Zhou, J. M., and Tsou, C. L.
(1996) Biochem. J., 315, 97-102.
46.Yang, X. J., and Miles, E. W. (1992) J. Biol.
Chem., 267, 7520-7528.
47.Goldsmith, J. G., and Kuo, L. C. (1993) J.
Biol. Chem., 268, 18481-18484.
48.Jencks, W. P. (1975) Adv. Enzymol.,
43, 219-410.
49.Dobson, C. M. (1993) Curr. Biol.,
3, 530-532.
50.Kurzynski, M. (1997) Biophys. Chem.,
65, 1-28.
51.Milner, J. (1995) Trends Biochem. Sci.,
20, 49-51.
52.Fenton, W. A., and Horwich, A. L. (1997)
Protein Sci., 6, 743-760.
53.Hummel, Z. (1992) J. Theor. Biol.,
157, 331-334.
54.Liang, D. C., Chang, W. R., and Wan, Z. L. (1994)
Biophys. Chem., 50, 63-71.
55.Engh, R. A., Brandstetter, H., Sucher, G.,
Eichinger, A., Baumann, U., Bode, W., Huber, R., Poll, T., Rudolph, R.,
and von der Saal, W. (1996) Structure, 4, 1353-1362.
56.Ptitsyn, O. B. (1995) Adv. Protein Chem.,
47, 83-229.
57.Uversky, V. N., and Ptitsyn, O. B. (1996) J.
Mol. Biol., 255, 215-228.