REVIEW: Deterioration of Lyophilized Pharmaceutical Proteins
H. R. Costantino1,2, S. P. Schwendeman1,3, R. Langer1, and A. M. Klibanov4*
1 Department of Chemical Engineering and 4 Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; fax: 617-252-1609; E-mail: klibanov@mit.edu2 Current address: Pharmaceutical Research and Development, Genentech, Inc., 460 Pt. San Bruno Blvd., South San Francisco, CA 94080, USA
3 Current address: Division of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA
* To whom correspondence should be addressed.

|
|||
| H. R. Costantino | S. P. Schwendeman | R. Langer | A. M. Klibanov |
Received July 7, 1997
The successful use of proteins in pharmaceutical and other commercial applications requires close examination of their relative fragility. Because of the resultant enhanced stability, proteins are often formulated in the solid state, even though dehydration tends to alter their structure. Even in the solid form, however, proteins may become inactivated due to various deleterious processes, e.g., aggregation. This review focuses on such mechanisms, with an emphasis on case studies conducted in our laboratory. Proteins which have both disulfide bonds and free thiols may aggregate via thiol-disulfide exchange, and this process may be facilitated by lyophilization-induced structural perturbations. For proteins possessing disulfides but not free thiols, aggregation also may occur when native disulfides are beta-eliminated, thus giving rise to thiol species which can catalyze disulfide scrambling. Other deleterious processes have also been uncovered, including a formaldehyde-mediated aggregation of formalinized vaccines. It is illustrated how knowledge of such deterioration pathways makes possible the rational development of stable solid protein formulations.
KEY WORDS: lyophilized pharmaceutical proteins, stability
Proteins are finding ever-increasing use in therapeutic and other commercial applications. And yet, proteins are relatively fragile (compared to low-molecular-weight compounds) and prone to various pathways of deterioration, as reviewed elsewhere [1-4]. This fragility adversely impacts and potentially limits their development.
Therapeutic proteins are often formulated in the solid state, e.g., lyophilized, to yield a pharmaceutically acceptable shelf-life [5]. Furthermore, some novel controlled-delivery systems for pharmaceutical proteins, such as sustained-release from polymeric vehicles [6], also incorporate lyophilized proteins. In addition, solid enzymes suspended in organic solvents are valuable synthetic catalysts [7]. In order to successfully employ solid proteins for all these purposes, it is necessary to investigate and understand their deterioration in the lyophilized state. This topic, despite its practical significance, is foreign to classical biochemists who are used to dealing with proteins dissolved in water.
Protein deterioration in the solid state may take the form of both chemical and conformational. Griebenow and Klibanov [8] have quantified the (reversible) secondary structural alteration of numerous proteins upon lyophilization. This phenomenon may be relevant to chemical instability since in aqueous solution a change in protein structure often leads to increased susceptibility towards further deleterious processes, e.g., aggregation [1-3]. On the other hand, there have been reports that the alteration of protein structure upon dehydration may actually improve stability upon lyophilization [9] or during storage in the dried state [10]. However, in some instances there is no correlation between structural conservation and solid-state stability [11, 12].
This review presents several instructive case studies using model lyophilized proteins. We have focused on solid-state deterioration, in particular protein aggregation. Our approach has been to elucidate the mechanisms responsible for the deterioration of the protein in order to facilitate the subsequent development of rational strategies for their stabilization.
CASE STUDY OF ALBUMIN. AGGREGATION VIA THIOL-DISULFIDE
INTERCHANGE
In an early mechanistic study, Liu et al. [13] investigated the solid-state aggregation of bovine serum albumin (BSA) lyophilized from pH 7.3. Albumin is a pharmaceutically relevant molecule [14, 15] often chosen as a model for evaluation of sustained-release vehicles [16]. Following storage at elevated temperature (37°C) and water content (~40 g/100 g dry), a loss of BSA’s solubility was observed upon reconstitution. This solubility loss was attributed to protein aggregation occurring in the wetted solid.
The mechanism responsible for moisture-induced deterioration of BSA was elucidated by dissolving the aggregates in specially designed aqueous solutions. Since strong denaturants such as 6 M urea or guanidine hydrochloride were incapable of solubilizing the aggregates, it was concluded that the aggregates were covalently bonded. Furthermore, it was shown that the covalent linkages were disulfides, since thiol agents such as dithioerythritol (DTE) were effective in solubilizing the aggregates. Aggregation via thiol-disulfide interchange is one of several typical mechanisms of deterioration uncovered for lyophilized proteins (see the table).
Some examples of deterioration of pharmaceutical proteins in the
lyophilized state
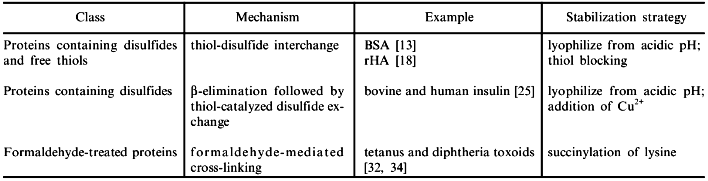
Subsequently, we examined the solid-state stability of recombinant human albumin (rHA), which has a high sequence homology to BSA [17]. It was found [18] that rHA, like its bovine counterpart, was susceptible to aggregation as a result of storage at 37°C and high moisture (96% relative humidity, where the water content was approximately 50 g/100 g dry protein). The time course of aggregation of rHA under these conditions is depicted in Fig. 1a.
The rHA aggregates were completely solubilized by DTE, indicating that they were disulfide-linked. The albumin molecule (both BSA and rHA) contains 17 cystine residues and one free thiol group of Cys-34 [17]. It was proposed and verified experimentally that the solid-state aggregation of albumin was due to a thiol-disulfide interchange [13, 18]. This process is triggered by a nucleophilic attack by a thiol (in the form of the thiolate ion) of one protein molecule on a sulfur atom of the cystine disulfide of another. The result is a new, intermolecular disulfide with conservation of the free thiolate ion. Since the process is catalytic with respect to the latter species, subsequent additional exchanges continue to take place, eventually yielding high-molecular-weight insoluble aggregates.Fig. 1. Deterioration of some lyophilized pharmaceutical proteins. Depicted are the time courses of the loss of solubility upon reconstitution (as compared to an unincubated control) for rHA stored at 37°C and 96% relative humidity (a), human insulin stored at 50°C and 96% relative humidity (b), and tetanus toxoid stored at 37°C and 84% relative humidity (c). All proteins were excipient-free and lyophilized from pH 7.3.
It was also found that the solid-state aggregation of albumin was dependent on the level of relative humidity, or more specifically, on the water content of the wetted protein. For example, for rHA the dependence of the extent of aggregation was bell-shaped with respect to the water content, with maximal aggregation occurring at about 50 g/100 g dry protein [18]. A similar bell-shaped dependency was also observed for the solid-state aggregation of BSA which also undergoes the thiol-disulfide exchange [13].
To understand the effect of water on albumin aggregation, one has to consider the fundamental role of water in the properties of the solid protein. An increase in the water content leads to increased reactivity for proteins due to several factors including [19]: 1) the ability of water to act as a "molecular lubricant" (increasing the conformational flexibility of proteins); 2) direct participation of water in deleterious processes; and 3) the ability of water to facilitate the diffusion of reactants. In the case of solid-state aggregation of albumin, the ability of water to increase the conformation mobility of the protein molecules should promote the intermolecular thiol-disulfide reaction, thus explaining the ascending part of the aforementioned bell-shaped curve.
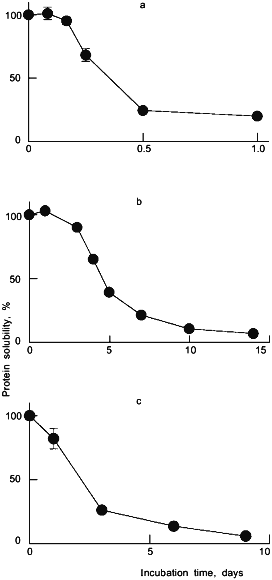
The descending portion of the albumin aggregation versus water content bell-shaped curve is most likely the result of a "dilution" effect [13] and rearrangement of the protein structure upon rehydration [18]. The latter scenario was proposed for rHA, which undergoes a significant reversible alteration of the secondary structure, as revealed by Fourier-transform infrared (FTIR) spectroscopy [8, 20]. FTIR spectroscopy has proven to be a useful technique for investigating the secondary structure of proteins in both the aqueous and solid states [8, 9]. Figure 2 depicts FTIR spectra for aqueous and lyophilized rHA in the amide III region. The spectra show significant differences between the aqueous and dehydrated samples, reflecting a difference in the protein secondary structure. Gaussian curve-fitting analyses of the data indicate that upon lyophilization the alpha-helix content of rHA dropped from 58% in aqueous solution to 25-35% with a concomitant rise in both beta-sheet and unordered structures. Thus, the rHA molecule is less ordered in the dried state. Such a disordering is likely to increase the accessibility of disulfides (which are otherwise buried in the native albumin structure [21]). This scenario is consistent with the observation that albumin aggregation via thiol-disulfide exchange in aqueous milieu is greatly accelerated under conditions which perturb the structure [22].
Fig. 2. Fourier-transform infrared (FTIR) spectra of rHA in the amide III region for aqueous solution, pH 7.3 (a), and powder lyophilized from pH 7.3 (b). The solid curves represent the superimposed original spectra and the Gaussian fit, while the dashed curves are individual Gaussian bands.
CASE STUDY OF INSULIN. beta-ELIMINATION FOLLOWED BY AGGREGATION VIA THIOL-CATALYZED DISULFIDE EXCHANGE
The foregoing example of albumin illustrates that an exchange between thiols and disulfides is a prominent deterioration pathway for proteins which contain these two moieties. Alternatively, a protein may contain an even number of cysteines which are all disulfide-bonded to form cystines (no free thiols). Such an example is provided by the classical therapeutic protein insulin, a therapeutically important hormone which is a strong candidate for use in a sustained-release device [23]. The native insulin molecule consists of two polypeptide chains, with one intrachain and two interchain disulfide bonds [24].
We have examined the stability of the insulin molecule (both bovine and human forms) for the lyophilized powder stored at elevated temperatures and humidities [25]. Figure 1b depicts the time course of aggregation for lyophilized human insulin stored at 50°C and 96% relative humidity. Closer examination of the moisture-induced insulin aggregates indicated that both covalent and non-covalent pathways could be responsible, depending on the conditions of preparation and storage of the hormone [25].
Noncovalent aggregation of insulin can occur in, and has been studied for, the aqueous solution where the deterioration is facilitated by unfolding of the protein molecule at a hydrophobic (e.g., air--liquid) interface [26]. The covalent aggregates were the result of intermolecular disulfide bonding, as revealed by dissolution in the presence of DTE [25]. The latter pathway dominates for protein lyophilized from neutral to alkaline pH. It has been shown independently that the insulin molecule is prone to lyophilization-induced structural alteration, namely a 20% increase in beta-sheet structure at the expense of alpha-helices and unordered motifs [8]. It is unknown whether alteration of insulin plays a role in its solid-state deterioration.
The finding that lyophilized insulin forms intermolecular disulfides was puzzling considering that their formation requires the existence of free thiols. The hypothesis was put forth [25] that beta-elimination of the disulfides in native had occurred, resulting in the formation of free thiols which could subsequently catalyze disulfide exchange. In the beta-elimination step, a hydroxyl ion facilitates the asymmetric cleavage of an intact S--S bond, yielding dehydroalanine and thiocysteine residues. Both of these are relatively unstable; the former can react with lysine to form a lysin-alanine cross-link, and the latter decomposes to various thiol products (e.g., HS-). The presence of such free thiols as a result of the high temperature and/or high humidity incubation of lyophilized insulin was directly confirmed experimentally [25].
Aggregation of lyophilized insulin was found to depend on the moisture level. Incubation of bovine insulin at 50°C and various humidities demonstrated that the extent of aggregation was greatly accelerated above 65% relative humidity [25]. Concomitantly, the water sorption by the lyophilized protein powder also began to rise at this point in the adsorption isotherm. These data illustrate the destabilizing effect of water on insulin, as was the case for albumin (discussed above). In the case of insulin, water not only increases the flexibility of the protein, thereby facilitating its aggregation, but it is also a direct participant, in the form of hydroxyl ion, involved in the beta-elimination step.
CASE STUDY OF TETANUS TOXOID. AGGREGATION VIA
FORMALDEHYDE-MEDIATED PATHWAY
A special class of proteins are those created when a naturally occurring macromolecule is covalently modified with formaldehyde to yield the formalinized species. Such is the case of a deadly neurotoxin, tetanus toxin, when it is detoxified by formaldehyde (diluted formalin) to create the vaccine for tetanus, tetanus toxoid (TT) [27]. The reactivity of proteins with formaldehyde is well-documented involving both stable and labile covalent formaldehyde linkages [28-30], which occur primarily with lysine, tyrosine, histidine, and cysteine residues [31]. Current efforts on developing a single-shot vaccine for tetanus have renewed the interest in the stability of this protein class [32].
As with insulin and albumin, we have observed that TT becomes insoluble when the lyophilized protein is exposed to moisture [12, 32-34]. In Fig. 1c, the rapid time course of aggregation of TT is seen under conditions of 86% relative humidity and 37°C. Unlike previous studies with unmodified proteins in our laboratory, insoluble aggregates formed under these conditions were held together by covalent non-disulfide bonds, as indicated by their insolubility in solvents containing 6 M urea and 10 mM DTE [32]. Acid hydrolysis of these aggregates revealed changes in the content of strongly formaldehyde-binding amino acid residues--Lys, Tyr, and His--implicating formaldehyde as the causal agent of the aggregation [32].
By considering the known chemistry of formaldehyde reactions with proteins, we devised a general mechanism to explain TT's aggregation behavior. The proposed mechanism is based on the premise that formaldehyde molecules are stored in the protein molecule as unstable linkages, such as hydroxymethylamine. Once water is removed, reactive Schiff base intermediates are formed that can combine with nucleophiles from a second protein molecule to form either stable or unstable covalent cross-links [32]. This mechanism was supported by inhibiting aggregation by either succinylating TT or reducing labile formaldehyde linkages with cyanoborohydride before lyophilization [32]. Furthermore, aggregation kinetics of another, unrelated, formalinized protein, diphtheria toxoid (DT), was found to be superimposable with those of TT when lyophilized from the same buffer solution [34]. Inhibition of the aggregation of both DT and TT by succinylation supports the view that the formaldehyde-mediated pathway is involved in both instances.
An additional intriguing finding was the nearly complete inhibition of aggregation of TT when co-lyophilized with small amounts of sorbitol [12, 32]. The stabilization effect could not be explained by structural or water content arguments because sorbitol does not change appreciably the water content in the protein powder, nor does it prevent the lyophilization-induced structural alteration appearing in the solid state as determined by FTIR [12]. However, when considering the formaldehyde-mediated pathway, the possibility of the plentiful hydroxyl groups of sorbitol out-competing the water molecules and neighboring protein molecules for the reactive Schiff base appears to be a plausible explanation for the stabilization effect [12].
OTHER PATHWAYS FOR DETERIORATION OF LYOPHILIZED PROTEINS
In addition to those described above in our case studies, other pathways for protein deterioration may take place [1-4, 6]. Some of them are intermolecular, e.g., aggregation, and others are intramolecular. It has been hypothesized that intermolecular pathways are dominant due to the close mutual proximity of protein molecules in the solid state [35].
Another potential pathway for protein cross-linking in the solid state is the reaction between asparagine or glutamine and lysine, as hypothesized for the moisture-induced aggregation of lyophilized ribonuclease A [36] and recombinant bovine somatotropin (rbSt) [35]. The latter protein also undergoes noncovalent aggregation upon heating of its lyophilized powder [37]. Increasing the moisture level exacerbates the aggregation of both ribonuclease A [38] and rbSt [35, 37]. Enzymes suspended in organic solvents upon heating also undergo a similar type of deterioration [39]. Furthermore, a mechanistic study [40] demonstrated the dimerization of solid insulin (lyophilized from pH 4) involving the C-terminal asparagine of the A chain and the N-terminal amino group of the A or B chain of another molecule via a cyclic anhydride intermediate.
In addition to intermolecular pathways, intramolecular pathways may also be responsible for protein deterioration in the solid state. These mechanisms include: 1) deamidation of asparagine and glutamine residues, particularly the former adjacent to a glycine residue; 2) hydrolysis of peptide bonds, e.g., cleavage at the C- or N-terminal of Asp residues; and 3) oxidation, especially of methionine residues. For a more complete description of these and other mechanisms for protein deterioration, including examples for lyophilized peptides and proteins refer to [6].
RATIONAL STRATEGIES FOR STABILIZATION OF LYOPHILIZED
PROTEINS
Understanding the mechanisms responsible for deterioration of proteins in the lyophilized state is more than merely a challenging intellectual exercise, for obtaining such information is essential in rational formulation development. The table lists examples of deterioration pathways and of mechanism-based, rational approaches to stabilization.
For proteins which undergo thiol-disulfide interchange, several prevention strategies can be used. One simple approach is to lower the pH of the aqueous solution prior to lyophilization to ensure the protonated state of the thiol group of cysteine [13, 18]. The rationale for this approach is that the ionogenic groups in proteins tend to retain their ionization state upon lyophilization, as reported in a recent study of model compounds [41]. Indeed, lyophilization of BSA [13] or rHA [20] from acidic solutions completely stabilizes the proteins against solid-state aggregation during the high temperate and high humidity storage.
Another rational way to stabilize proteins against thiol-disulfide exchange is to chemically block the thiol group(s) involved in the process. For example, S-alkylating the Cys-34 of albumin stabilizes the protein not only during high temperature and high humidity storage [13, 42], but also when loaded within a polymeric matrix such as poly(fatty acid dimer:sebacic acid) [42] and poly(lactide-co-glycolide) [43].
In the case of insulin, which is liable to beta-elimination followed by thiol-catalyzed disulfide interchange, several stabilization strategies were proposed and verified [25]. These include lowering the pH prior to lyophilization (to both reduce beta-elimination and protonated free thiols) and addition of Cu2+ to the formulation to catalyze the oxidation of free thiols.
For formalinized proteins, whose moisture-induced aggregation involves lysine residues, a useful approach is succinylation of the latter. The generality of this approach was verified with both tetanus and diphtheria toxoids [32, 34].
For any deleterious process involving intermolecular protein--protein interactions, dilution of the solid protein should be effective. This can be achieved by lyophilizing the protein from an aqueous solution containing an inert polymer [13], e.g., poly(ethylene glycol). Also, since increased conformational mobility is usually conducive to faster deleterious reactions in solid proteins, and this mobility is enhanced by hydration, controlling the latter’s extent by optimizing the humidity and moisture content in the system is yet another powerful tool for stabilizing solid proteins.
CONCLUDING REMARKS
In summary, studies on lyophilized pharmaceutical proteins demonstrate their tendency to deteriorate, particularly via aggregation in the presence of moisture. Thus, while it is true that lyophilized proteins are usually more stable than those dissolved in water, this extra stability is not unlimited.
Numerous deleterious mechanisms occurring in proteins have been elucidated by us and others. The examples above illustrate how a mechanistic approach is useful in solid protein formulation development. This approach is fundamentally distinct from a simple screening for a combination of operational and formulation conditions which provide suitable stability. Some rules-of-thumb have been proposed, as considered elsewhere [3-5, 44]. It is unlikely that a universal strategy will be developed to formulate solid proteins, but it is possible to develop some general strategies that are likely to be effective against a variety of protein deterioration pathways. These include the use of cryoprotectors (to protect against freezing) and lyoprotectors (to protect against drying) which provide an amorphous, glassy matrix for the protein where chemical reactivity can be kept to a minimum. As additional, specific pathways for solid-state protein deterioration are uncovered, it will become possible to develop further, rational approaches to ensure high stability.
Although a measure of success has been achieved towards understanding protein stability in the solid, e.g., lyophilized state, a number of important issues remain to be fully understood. For example, the question has not yet been definitively answered whether protein structure, or rather the perturbation of protein structure in the dried state, is necessarily related to the stability. Another significant area is that of sustained delivery from polymeric matrices, and assessing the utility of stabilization strategies (developed for unincorporated protein) for the case of the protein formulation in vivo. Such key areas are the focus of current and future research towards understanding the deterioration of lyophilized proteins. Moreover, with the advent of gene therapy, the problem of viral instability, particularly acute for retroviruses, emerges as one of the major bottlenecks [45]. For example, retroviruses lose their activity during freezing, lyophilization, storage, ultracentrifugation, and transfection. It remains to be seen to what extent the protein stability/stabilization issues and strategies are applicable to more complex, nucleic-acid-containing macromolecular assemblies such as viruses.
This project has been financially supported by the National Institutes of Health (grant GM26698) and the Biotechnology Process Engineering Center at M.I.T.
REFERENCES
1.Volkin, D. B., and Klibanov, A. M. (1989) in
Protein Function: A Practical Approach (Creighton, T. E., ed.)
Oxford University Press, Oxford, pp. 1-24.
2.Manning, M. C., Patel, K., and Borchardt, R. T.
(1989) Pharm. Res., 6, 903-918.
3.Cleland, J. L., Powell, M. F., and Shire, S. J.
(1993) Crit. Rev. Therapeutic Drug Carrier Systems, 10,
307-377.
4.Costantino, H. R., Langer, R., and Klibanov, A. M.
(1994) J. Pharm. Sci., 83, 1662-1669.
5.Pikal, M. J. (1997) in Peptide and Protein
Delivery (Lee, V. H. L., ed.) Marcel Dekker, New York.
6.Schwendeman, S. P., Costantino, H. R., Gupta, R.
K., Klibanov, A. M., and Langer, R. (1997) in Controlled Drug
Delivery. Challenges and Strategies (Park, K., ed.) ACS Books,
Washington, DC, pp. 229-267.
7.Klibanov, A. M. (1990) Acc. Chem. Res.,
23, 114-120.
8.Griebenow, K., and Klibanov, A. M. (1995) Proc.
Natl. Acad. Sci. USA, 92, 10969-10976.
9.Prestrelski, S. J., Tedeschi, N., Arakawa, T., and
Carpenter, J. F. (1993) Biophys. J., 65, 661-671.
10.Prestrelski, S. J., Pikal, K. A., and Arakawa, T.
(1995) Pharm. Res., 12, 1250-1259.
11.Costantino, H. R., Griebenow, K., Mishra, P.,
Langer, R., and Klibanov, A. M. (1995) Biochim. Biophys. Acta,
1253, 69-74.
12.Costantino, H. R., Schwendeman, S. P., Griebenow,
K., Klibanov, A. M., and Langer, R. (1996) J. Pharm. Sci.,
85, 1290-1293.
13.Liu, W. R., Langer, R., and Klibanov, A. M.
(1991) Biotech. Bioeng., 37, 177-184.
14.He, X. M., and Carter, D. C. (1992)
Nature, 358, 209-215.
15.Geisow, M. J. (1992) Trends Biotechnol.,
10, 335-337.
16.Langer, R., Rhine, W. D., Hsieh, D. S. T., and
Bawa, R. S. (1980) in Controlled Release of Bioactive Materials
(Baker, R., ed.) Academic Press, New York, pp. 83-98.
17.Peters, T. (1985) Adv. Protein Chem.,
37, 161-245.
18.Costantino, H. R., Langer, R., and Klibanov, A.
M. (1995) Bio/Technol., 13, 493-496.
19.Hageman, M. J. (1992) in Stability of Protein
Pharmaceuticals. Part A. Chemical and Physical Pathways of Protein
Degradation (Ahern, T. J., and Manning, M. C., eds.) Plenum Press,
New York, pp. 273-309.
20.Costantino, H. R., Griebenow, K., Mishra, P.,
Langer, R., and Klibanov, A. M. (1995) Biochim. Biophys. Acta,
1253, 69-74.
21.Habeeb, A. F. S. A. (1978) Adv. Exp. Med.
Biol., 98, 101-117.
22.Andersson, L.-O. (1969) Arch. Biochem.
Biophys., 133, 277-285.
23.Costantino, H. R., Liauw, S., Mitragotri, S.,
Langer, R., Klibanov, A. M., and Sluzky, V. (1997) ACS Symp.
Ser., in press.
24.Brange, J. (1987) Gaplenics of Insulin: The
Physicochemical and Pharmaceutical Aspects of Insulin and Insulin
Preparations, Springer-Verlag, Berlin.
25.Costantino, H. R., Langer, R., and Klibanov, A.
M. (1994) Pharm. Res., 11, 21-29.
26.Sluzky, V., Tamada, J. A., Klibanov, A. M., and
Langer, R. (1991) Proc. Natl. Acad. Sci. USA, 88,
9377-9381.
27.Bizzini, B. (1984) Bacterial Vaccines,
Academic Press, New York.
28.Fraenkel-Conrat, H., and Olcott, H. S. (1948)
J. Am. Chem. Soc., 70, 2673-2684.
29.Fraenkel-Conrat, H., and Olcott, H. S. (1948)
J. Biol. Chem., 174, 827-843.
30.Fraenkel-Conrat, H., and Mecham, D. K. (1949)
J. Biol. Chem., 177, 477-486.
31.Means, G. E., and Feeney, R. E. (1971)
Chemical Modification of Proteins, Holden-Day, San
Francisco.
32.Schwendeman, S. P., Costantino, H. R., Gupta, R.
K., Siber, G. R., Klibanov, A. M., and Langer, R. (1995) Proc. Natl.
Acad. Sci. USA, 92, 11234-11238.
33.Schwendeman, S. P., Tobio, M., Jaworowicz, M.,
Alonso, M. J., and Langer, R. (1997) J. Microencapsulation, in
press.
34.Schwendeman, S. P., Costantino, H. R., Gupta, R.
K., Tobio, M., Chang, A. C., Alonso, M. J., Siber, G. R., and Langer,
R. (1996) Dev. Biol. Stand., 87, 293-306.
35.Hageman, M. J., Bauer, J. M., Possert, P. L., and
Darrington, R. T. (1992) J. Agric. Food Chem., 40,
348-355.
36.Townsend, M. W., and DeLuca, P. P. (1991) J.
Pharm. Sci., 80, 63-66.
37.Bell, L. N., Hageman, M. J., and Bauer, J. M.
(1995) Biopolymers, 35, 201-209.
38.Townsend, M. W., and DeLuca, P. P. (1990) J.
Pharm. Sci., 79, 1083-1086.
39.Volkin, D. B., Staubli, A., Langer, R., and
Klibanov, A. M. (1991) Biotech. Bioeng., 37,
843-853.
40.Strickley, R. G., and Anderson, B. D. (1996)
Pharm. Res., 13, 1142-1153.
41.Costantino, H. R., Griebenow, K., Langer, R., and
Klibanov, A. M. (1997) Biotech. Bioeng., 53,
345-348.
42.Costantino, H. R., Shieh, L., Klibanov, A. M.,
and Langer, R. (1997) J. Control. Release, 44,
255-261.
43.Crotts, G., and Park, T. G. (1997) J. Control.
Release, 44, 123-134.
44.Nail, S. L. (1997) in Controlled Drug
Delivery. Challenges and Strategies (Park, K., ed.) ACS Books,
Washington, DC, pp. 185-203.
45.Kotani, H., Newton, P. B., Zhang, S., Chiang, Y.
L., Otto, E., Weaver, L., Blaese, R. M., Anderson, W. F., and
McGarrity, G. J. (1994) Human Gene Therapy, 5,
19-28.