REVIEW: GroE Chaperonin-Assisted Folding and Assembly of Dodecameric Glutamine Synthetase
M. T. Fisher
Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, Kansas 66160-7421, USA; fax: 913-588-7440; E-mail: mfisher1@kumc.edu
 M. T. Fisher |
Received October 23, 1997
The folding and assembly of Escherichia coli dodecameric glutamine synthetase is facilitated by the E. coli GroE chaperonins, GroEL and GroES. Since endogenous glutamine synthetase monomers are bound to GroEL immediately after cell lysis and are assembly competent, this strongly suggests that glutamine synthetase is an authentic substrate of the GroE chaperonins. At physiological temperatures, the in vitro reactivation of glutamine synthetase increases from 10 to 70-80% of the original activity when the chaperonin GroEL is included. Although nucleotide binding is sufficient to dissociate assembly competent glutamine synthetase monomers from GroEL, the addition of GroES substantially accelerates the dissociation, assembly, and reactivation. The interactions of glutamine synthetase monomers with the activated chaperonin are transient (t1/2 = 10 sec) and these monomers can be released from GroEL at high concentrations without misfolding or inappropriate aggregation. It has been found that the nucleotide-induced conformational change of GroEL is critical for folding success of glutamine synthetase because the simple displacement of glutamine synthetase monomers from the GroEL chaperonin with another protein substrate inhibits reactivation. During glutamine synthetase refolding, the "high affinity" nucleotide-free GroEL is most efficient in preventing initial folding intermediates from partitioning to off-pathway folding routes. Interestingly, the more physiologically relevant "low affinity" nucleotide-bound ((ATP/ADP) GroEL--GroES) complex is not as efficient at capturing the initial folding intermediates of glutamine synthetase. In contrast to glutamine synthetase, non-authentic "model" substrates such as mammalian mitochondrial rhodanese and mitochondrial malate dehydrogenase show no differences in folding efficiencies with either the "low affinity" or "high affinity" complexes. Besides the nature of the chaperonin complex itself, the mechanism of GroE-assisted folding is determined by the folding environment and, most importantly, by initial interactions of chaperonins with folding intermediates. Glutamine synthetase interacts only transiently with chaperonin complexes, while most of the "model" proteins exhibit relatively long interactions times. It may be indicative of a specific evolutionary selected mechanism of chaperonin-assisted folding (optimizing the folding kinetics), different from that observed with non-authentic chaperonin substrates. Since the kinetics of protein folding depends heavily on the solution environment, studies involving in vivo chaperonin substrates under conditions that closely mimic those found in the cell will be required to define and solve the physiologically relevant kinetic mechanism of chaperonin-assisted folding. KEY WORDS: glutamine synthetase from Escherichia coli, chaperonin-assisted folding, assembly |
Abbreviation: DHFR) dihydrofolate reductase.
Within the past decade, it has been discovered that cells contain
essential proteins called "molecular chaperones" that control
misfolding and inappropriate aggregation reactions in the cellular
milieu. Of these molecular chaperones, a special class called the
chaperonins have been more specifically implicated in controlling
assembly reactions (reviewed in [1]). The first
hint that chaperonins may aid in oligomer assembly came during
experiments characterizing bacteriophage lambda and T4 assembly
mutants [2, 3]. In these
particular studies, it was discovered that E. coli contains a
number of protein host factors that aid in the assembly of a coat
protein involved in the bacteriophage lambda life cycle but were
not a part of the assembled virus. In these first examples, it was
noted that lower levels of or subtle defects in these host factors
resulted in the misfolding of a bacteriophage lambda dodecameric
head coat protein, protein E. These host factors, now identified as
chaperonins, consist of a large (802 kD) protein binding ATPase, GroEL,
and a smaller (70 kD) accessory protein, GroES [4-6]. Although the chaperonins are
heat shock inducible, they are absolutely essential for growth at all
temperatures [7, 8].
Interestingly, a majority of the E. coli substrates whose
folding may require the aid of chaperonins in vivo tend to be
multisubunit proteins [9]. At the onset, the need
to understand how chaperonins aid in associative processes adds yet
another "layer of complexity" to the assembly of quaternary
structures. However, what is learned from studies into
chaperonin-assisted assembly reactions promises to aid in our
understanding of assembly reactions in ways that have not been realized
before.
This review specifically examines how the E. coli GroE chaperonins aid in the assembly of dodecameric E. coli glutamine synthetase. Far from being just an in vitro curiosity, there are numerous experimental observations supporting the contention that an interaction between GroEL and glutamine synthetase monomers actually occurs in vivo. Ironically, most current studies of GroEL--protein interactions in vitro use substrates that are not physiological substrates of the GroE chaperonins. Since GroEL is highly homologous to mitochondrial Hsp60, a majority of the researchers investigating the mechanism of chaperonin action have used mitochondrial proteins as model substrates. Although the in vitro folding of numerous mitochondrial proteins are most certainly facilitated by the bacterial GroE chaperonins, at a kinetic level, these substrates may not be entirely representative of those that normally interact with the chaperonins. In fact, as will be discussed later in this review, lifetimes of transient complexes between E. coli folding substrates and the E. coli GroE chaperonins may be much shorter than those lifetimes that are typically observed between “model” mitochondrial proteins and the E. coli GroE chaperonins. Thus, the study of interactions between E. coli glutamine synthetase and the E. coli GroE chaperonins may be relevant to the understanding of the mechanism of assisted folding because these proteins have co-evolved to fold and interact with each other. At the structural level, the assembly of glutamine synthetase is complicated because Mn2+ or Mg2+ ions are required for assembly and the individual monomers share extended regions of secondary elements with each other in the final quaternary structure [10]. Given these strict requirements for assembly, the formation of dodecameric glutamine synthetase is certainly not a simple associative process and may explain, in part, why chaperonins can aid the folding and assembly. Even though the assembly appears complex, the successful renaturation of glutamine synthetase can be accomplished by either varying the refolding solution conditions or by interacting with chaperonin proteins. Rather than directly aiding in the assembly process, chaperonins prevent misfolding reactions from dominating the folding process and control off-pathway kinetic partitioning reactions. At elevated protein concentrations and physiological temperatures, off-pathway kinetic partitioning reactions become more prevalent and the formation of active glutamine synthetase dodecamers is substantially diminished. Under these solution conditions, the GroE chaperonins exert their largest influence on the assembly of glutamine synthetase.
STRUCTURE AND ASSEMBLY OF DODECAMERIC GLUTAMINE SYNTHETASE
Glutamine synthetase is an essential enzyme involved in nitrogen metabolism. In the presence of ATP, this enzyme supplies nitrogen to the cell by catalyzing the condensation of ammonia and glutamate to produce glutamine. Glutamine serves as the intracellular nitrogen transporter for a wide array of biosynthetic reactions. Glutamine synthetase is a highly regulated system wherein numerous compounds can serve as competitive feedback inhibitors. In addition to this complex regulation process, glutamine synthetase shows enhanced inhibition through a very specific adenylation of tyrosine 397 [11, 12].
At the gross structural level, early electron micrographs of E. coli glutamine synthetase indicated that the 12 subunits were arranged in a double stack of hexamers with 622 point group symmetry [13, 14]. X-Ray crystallographic analysis of the glutamine synthetase dodecamers revealed not only the location and structures of the active sites but also uncovered some novel assembly interactions between subunits [15] (Figs. 1 and 2). These structures were determined for the Salmonella typhimurium enzyme, which is essentially the same as the E. coli enzyme, only 10 differences in the amino acid sequence being noted between the two enzymes [16].
Fig. 1. An isolated hexamer of glutamine synthetase is shown to point out the location of the active sites and the location of the metal ion-binding sites. These metal ions are required to facilitate proper assembly of glutamine synthetase (GS, glutamine synthetase).

The active sites of dodecameric glutamine synthetase are located between the intra-ring subunit interfaces. The substrates glutamate, ATP and ammonia can enter this active site through a channel in between the dimer interface. This active site channel is 10 Å in diameter and is parallel to the sixfold axis (Fig. 1). Within the active site, the two Mn2+ metal ions bind to specific amino acid residues to provide structural integrity as well as insure proper binding orientations for ATP and glutamate. Although Mn2+ ions are commonly used to store and stabilize glutamine synthetase and are also present in the X-ray crystal structures, the Mg2+ ion is the physiological metal in the biosynthetically active enzyme. When the transition state analog, methionine sulfoximine phosphate, binds at the active site, this subunit interface becomes resistant to dissociation by denaturant. As the active sites of glutamine synthetase are successively titrated with methionine sulfoximine phosphate, oligomeric intermediates such as tetramers, hexamers, octamers, decamers and the full dodecamer are generated. The minimal enzymatically active oligomeric unit of glutamine synthetase is a tetramer [17].Fig. 2. Up--down inter-ring dimer contacts of glutamine synthetase. Van der Waals representative structure of the inter-ring dimer contacts shows that there is sharing of secondary structural elements between the dimers. The locations of the shared alpha-helical C-terminal "thongs" from each monomer and the strand-turn-stand beta-loop structures are pointed out for an isolated inter-ring glutamine synthetase dimer.
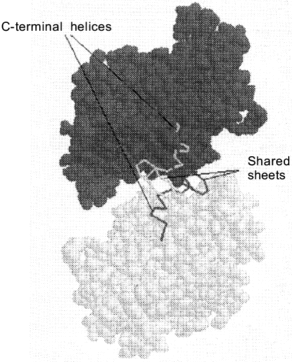
In the assembled dodecamer, the interactions between glutamine synthetase subunits involve up--down (inter-ring) and side--side (intra-ring) contacts. Intra-ring contacts are strengthened by metal ion binding. The innermost metal ion-binding site, called the n1 site, has the tightest binding affinity for the two metal ions and the binding free energy provided by this interaction has a large influence on overall structural stability of the dodecamer. Since the glutamine synthetase active site is located at heterologous subunit interfaces composed of side--side contacts, these contacts are strengthened when methionine sulfoximine phosphate binds to the active site and inactivates the enzyme. In the absence of this transition state analog, the up--down contacts appear to be stronger than the side--side interactions. Under solution conditions where mild dissociation of the dodecamer is observed, dimers dissociate from the oligomer yet they maintain their inter-ring up--down contacts [17, 18]. The X-ray crystal structure data show that the inter-ring contacts involve extensive sharing of secondary elements (domain swapping) between the inter-ring dimers. For example, the C-terminal of each glutamine synthetase subunit consists of a hydrophobic helix that is inserted into a hydrophobic cavity formed by the subunit below (Fig. 2). In addition, two anti-parallel beta-hairpins provided by each up--down subunit interact to form a small four-stranded beta-sheet structure.
It is clear from the structural data that the assembly of glutamine synthetase depends on specific hydrophobic interactions. Because of the extensive domain swapping in the assembled dodecamer, the final folded structure of the monomer in the final quaternary structure is almost certainly attained during or after oligomer formation. In addition to the domain swapping interactions, experiments outlined in the next section show that the Mn2+ or Mg2+ ions also play an important role in promoting assembly. As with most oligomeric proteins, the folding reactions that occur prior to glutamine synthetase assembly are critical for proper oligomer formation and final product yields are highly dependent on these initial folding reactions. Curiously, the most favorable in vitro folding conditions for glutamine synthetase are those that destabilize folding intermediates and slow the initial assembly reaction. Some aspects of these optimal in vitro conditions may mimic the mechanisms of the chaperonin-assisted folding and assembly reactions.
SPONTANEOUS REACTIVATION OF GLUTAMINE SYNTHETASE--FOLDING AND
ASSEMBLY WITHOUT CHAPERONINS
To obtain a reasonable mechanism that describes the chaperonin-assisted folding of glutamine synthetase, it is instructive to assess the spontaneous folding and assembly reactions of glutamine synthetase. With these folding and assembly steps defined, it is easier to determine where chaperonins contribute their largest influences in the assembly reaction. To achieve maximal refolding yields during oligomeric protein folding and assembly, many different solution conditions are commonly used in vitro [19, 20].
Optimal Conditions for Unfolding of Glutamine Synthetase
As with most proteins, the highest refolding yields for in vitro glutamine synthetase assembly depends on the specific denaturation solution conditions used to initially unfold the protein. The proper denaturation of glutamine synthetase is a critical procedure for insuring proper reassembly of oligomers because the nature of the unfolded conformer has a dramatic influence on the properties, lifetimes, and structure of the initial folding intermediates prior to assembly. The rapid removal of the denaturation conditions generates initial monomeric folding intermediates and these intermediates initially interact with the chaperonin proteins. For in vitro folding of oligomeric proteins, the assembly reaction is secondary.
The denaturation solution conditions that result in the highest yields of reactivated glutamine synthetase were largely defined by Stadtman and coworkers [21]. Complete dissociation and unfolding of dodecameric glutamine synthetase commonly requires a high concentration of denaturant (6 M guanidine-HCl or 7 M urea). As with most proteins containing reduced cysteines, the oxidation of these residues during the unfolding of glutamine synthetase is avoided. The dissociation process and the unfolding reaction are accelerated if the structural Mg2+ (or Mn2+) ions are removed by EDTA chelation. If refolding is attempted from glutamine synthetase samples that have been incubated at intermediate denaturant concentrations (3 M guanidine-HCl or 4 M urea), the overall yields decline to undetectable levels. Presumably, residual structures of unfolded proteins in this denaturation range dramatically influence the degree of aggregation and misfolding [22]. When refolding is attempted from these denaturing conditions, the resulting intermediate(s) form large stable inactive aggregates after the denaturant is removed [23]. At higher denaturant concentrations, the aggregates formed at intermediate denaturant concentrations dissociate and complete unfolding is observed. The highest refolding and reactivation yields are achieved when renaturation is initiated from this completely unfolded state. The short-lived glutamine synthetase folding intermediates that are formed after unfolded subunits are rapidly diluted into refolding conditions are the optimal substrates for in vitro chaperonin-assisted folding.
Folding of Glutamine Synthetase in vitro
In vitro folding yields depend on initial concentration and temperature. Since glutamine synthetase subunits form inappropriate aggregates within a particular denaturant concentration range (1-3 M guanidine-HCl or 3-5 M urea), the subunits are more successfully refolded using large scale "concentration jumps" where the denaturant concentration is rapidly decreased from denaturing to renaturing conditions. This simple procedure avoids long term exposure of the refolding subunits to adverse denaturant concentrations. During refolding, the protein can potentially equilibrate with many different intermediate populations. In general, the highest yields are obtained when “on-pathway” intermediate populations that are kinetically committed to fold correctly are preferentially formed. If, however, the folding protein shows favorable kinetic partitioning with misfolded intermediates, the yields of correctly folded protein decrease and in some instances, approach zero. As with most oligomeric proteins, the refolding yields of glutamine synthetase show a pronounced decline as the initial concentration of the refolding subunits increases [24, 25]. It has been repeatedly demonstrated that an increase in the concentration of folding protein will increase the non-productive aggregation reactions because bimolecular collisions between misfolded intermediates become more frequent [26, 27]. This “off-pathway” partitioning diminishes the amount of “committed” native intermediates leading to the observed decrease in the refolding yields. To minimize misfolding, the most successful strategy is to decrease the concentration of the folding protein thus avoiding or limiting inappropriate collisional reactions [27]. For optimal in vitro refolding of glutamine synthetase, the concentration range of 0.45-0.55 µM subunits repeatedly leads to the highest yields of active enzyme. Below these optimal concentrations, the recoverable activities once again decline presumably because of diminished interactions between refolded subunits.
As the temperature of the refolding mixture is increased to the physiological levels (37°C), spontaneous glutamine synthetase reactivation yields drop to 10% of the native activity. Under these restrictive temperature conditions, the inclusion of the GroE chaperonins in the refolding solutions dramatically improves glutamine synthetase folding yields. In addition, the spontaneous renaturation yield can be slightly improved from 10 to 30% when bovine serum albumin is added to the refolding mixture [24]. Compared with other 'inert” proteins, bovine serum albumin has also been observed to improve folding of mitochondrial citrate synthase [28]. Although the molecular origins for these slight improvements in folding efficiency are not known, bovine serum albumin may have some intrinsic chaperoning activity of its own (i.e., binding to hydrophobic intermediates) because it can bind to exposed hydrophobic surfaces [28]. Interestingly, if one replaces bovine serum albumin with an equivalent (w/w) concentration of ovalbumin as the 'inert” control protein, the spontaneous folding falls to its original low level (10-15%) (P. A. Voziyan and M. T. Fisher, unpublished data).
The metal ligands, Mn2+ or Mg2+, are required for glutamine synthetase assembly and reactivation. Earlier work by Stadtman and coworkers had established that glutamine synthetase reactivation depends on the presence of Mn2+ or Mg2+ ions [21, 29]. No reactivation was observed if these metals ions were absent. Early experimental results could not determine whether assembly preceded or was induced by metal ion binding. When the assembly of glutamine synthetase was followed by glutaraldehyde cross-linking and right angle light scattering measurements, these data clearly indicated that the addition of Mn2+ or Mg2+ ions induce the assembly of glutamine synthetase [23]. With respect to metal specificity, it has been shown that lower concentrations of Mn2+ ion can result in substantial reactivation of glutamine synthetase compared to similar concentrations of the more physiological Mg2+ ion ligand. For example, under optimal in vitro conditions for spontaneous folding, the presence 0.5 mM Mn2+ was sufficient to refold glutamine synthetase to high yields (~70% of original activity) whereas the addition of 3 mM Mg2+ to the refolding buffer did not result in any observable reactivation (M. T. Fisher, unpublished data). The concentration requirements of Mn2+ ions appear to correlate with the higher binding affinity that this ion has for metal-free glutamine synthetase compared with the binding affinity of Mg2+ ion [30].
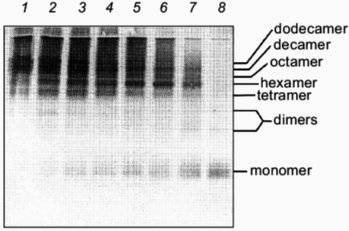
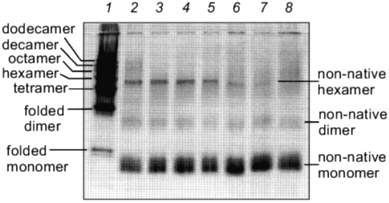
The kinetics of in vitro assembly reaction of glutamine synthetase can be followed by rapidly cross-linking assembling subunits with glutaraldehyde as a function of time. The results from these cross-linking experiments and in vitro reactivation reactions indicate that kinetics of glutamine synthetase assembly is directly dependent on the addition of Mn2+ or Mg2+ ions [24] (compare Figs. 3 and 4). The oligomeric intermediates that are generated from incomplete cross-linking of native glutamine synthetase with glutaraldehyde serve as excellent molecular mass markers for the intermediates that form during refolding and assembly. The initially cross-linked subunits found early in the assembly process indicate that monomers form dimers. The dimers associate to form tetramers which may contain at least two active sites and the formation of tetramers correlates directly with the emergence of glutamine synthetase activity. By this method of analysis, two dimer forms of glutamine synthetase can be resolved (Fig. 3). The higher order oligomeric intermediates (hexamers, octamers, decamers, and dodecamers) appear to form by successive dimer addition. Odd oligomeric intermediates (trimers, pentamers, heptamers, nanomers, and hendecamers) are not observed throughout the time course of assembly [31]. If either Mn2+ or Mg2+ are absent during the assembly, no active glutamine synthetase is recovered. Curiously, in the absence of these metal ions, inactive dimers and hexamers can still form but they do not migrate at the same positions on SDS-PAGE as their native counterparts (Fig. 4).
Fig. 3. Glutaraldehyde cross-linking of glutamine synthetase assembly intermediates in the presence of Mg2+. After initiation of glutamine synthetase assembly, 0.5% glutaraldehyde was added to cross-link existing assembly intermediates at various times in the assembly reaction. After each addition, the reaction was stopped after 30 sec. The cross-linked intermediates were concentrated and separated by SDS-PAGE and visualized by silver staining. Glutaraldehyde was added at 20 sec, 5, 10, 15, 20, 30, and 60 min (lanes 8-2). Lane 1 represents the cross-linked product for native glutamine synthetase as a comparison. Note the two separate dimer species.
Small molecular additives such as 1 M urea and 400 mM KCl increase in vitro reactivation yields of glutamine synthetase. Not only can enhanced yields be observed when optimal protein concentrations, temperatures, and metal ion concentrations are used, it was found that the addition of 1 M urea and 400 mM KCl to the refolding solution results in a significant increase in spontaneous glutamine synthetase reactivation [24]. It is not known how these additives improve the renaturation yields of glutamine synthetase at the molecular level but their presence has some interesting effects on the renaturation kinetics of the dodecamer. The most noticeable change is the appearance of an extended lag in the reactivation kinetics [24]. Since the minimal active unit of dodecameric glutamine synthetase is a tetramer, this lag in reactivation may reflect a number of “activity silent” reactions such as monomer folding, dimer, and tetramer formation. It is clear that these additives initially influence the kinetics of the formation of active sites. Although the reactivation rates have declined under these conditions, the overall yields are consistently higher. Thus, it appears that in this case, controlling the kinetics of glutamine synthetase assembly can actually improve the final folding yield.Fig. 4. Glutaraldehyde cross-linking of glutamine synthetase assembly intermediates in the absence of Mg2+. The same procedure outlined in Fig. 3 was followed. Glutaraldehyde was added at 5, 10, 15, 20, 30, 60 min and 4 h (lanes 8-2). Lane 1 is a sample of incompletely cross-linked native glutamine synthetase where the various intermediates were used as molecular mass markers.
Such improvements in refolding yields in the presence of low molecular weight additives have been observed before. For example, a number of refolding reactions of other unrelated proteins show enhanced folding yields and rates in the presence of non-denaturing urea concentrations [28, 32, 33]. Camacho and Thirumalai have proposed that lower concentrations of denaturants such as urea or guanidine hydrochloride destabilize initial folding intermediates without altering the final native conformation [34]. The addition of non-denaturing urea concentrations (with respect to the native protein) is thought to lower the activation barriers between metastable states and native conformations by unfolding less stable misfolded or native-like intermediates. Curiously, molecular chaperones may aid protein folding in much the same way; i.e., by partially unfolding misfolded intermediates driven through binding interactions [35, 36]. It is reasoned that the energy to unfold misfolded intermediates is provided by the binding interactions between the intermediate and the chaperone protein [35].
For glutamine synthetase, the combination of lower protein concentrations, lower temperatures, low denaturant concentrations and high ionic strength all prevent misfolding and deleterious aggregation. At present, no direct evidence exists to suggest that the folding intermediates that commit to a native state following chaperonin interaction have the same conformation as the committed intermediates that may be produced without chaperonins. However, it is conceivable that the solution effects that promote favorable folding conditions without chaperone proteins (i.e., unfolding misfolded proteins) may mirror certain elements of the chaperone protein mechanism in vivo.
CHAPERONIN-ASSISTED ASSEMBLY OF GLUTAMINE SYNTHETASE
In vivo, the cell does not have the luxury of changing certain solution conditions (e.g., lowering temperature or protein concentrations) in order to achieve maximum folding yields. Instead, cells can both utilize and vary the amounts of numerous molecular chaperones to improve protein folding efficiencies, aid in protein transport, and clear the cell of misfolded proteins. As with most oligomeric proteins, in vitro refolding yields of glutamine synthetase drop dramatically as the temperature increases to 37°C [24]. At these higher temperatures, the in vitro loss in recoverable glutamine synthetase activity can be avoided by adding non-denaturing amounts of urea (~0.8-1.0 M) (M. T. Fisher, unpublished data). These low concentrations of urea prevent the development of very large amorphous aggregates. In vivo, instead of urea, cells use molecular chaperones to prevent the inappropriate aggregation of folding proteins. The smaller molecular chaperone Hsp70 (Heat Shock Protein 70) prevents aggregation by binding to extended hydrophobic regions that would normally be buried in the interior of the protein [37]. In the case of Hsp25, these chaperones bind to partially folded intermediates to control the size of the protein aggregate and maintain solubility [38]. The GroE chaperonins also prevent large-scale aggregation but appear to bind intermediates that have a substantial amount of native folded structure (reviewed in [39]).
There is still significant debate about the mechanism by which GroE chaperonins prevent inappropriate aggregation. While some investigators suggest that chaperonins prevent aggregation by directing progressive accumulation of the native fold through sequestered folding [40, 41], others argue that chaperonins function by reversing misfolding of highly structured off-pathway intermediates [35, 36, 42]. While the former mechanism focuses on folding reactions that occur after the protein dissociates from its bound position on the chaperonin surface, the latter mechanism emphasizes the importance of the initial binding interaction and the reversal of misfolded conformations. Of relevance to sequestered folding, it has been repeatedly shown that folding can occur in the encapsulated state created by the formation of a GroEL--GroES--nucleotide complex. Most of these observations involve altered chaperonins or chaperonin conditions where the lifetime of the GroEL--GroES complex is extended. This complicates the interpretations because it becomes hard to distinguish between what is observed in vitro and what actually occurs in vivo. In the case of quaternary protein assembly, it is generally agreed that the dissociation of the GroEL--substrate complex is crucial for folding and assembly [43]. It has been determined that a large amount of non-native protein dissociates from the chaperonin complex both in vitro and in vivo [44-46]. This observation indicates that substrates will dissociate from the chaperonin and will not rebind if they have attained structure which is no longer recognized by the chaperonin. These protein structures not only include native folded proteins, but non-native structures as well [47]. For both mechanisms, the important discrimination step that determines when a folding protein will enter a chaperonin-assisted pathway is ultimately determined by the initial binding (or rebinding) reaction.
A large number of genetic studies using GroE mutants indicate that chaperonins assist the assembly of oligomeric proteins [9]. It appears that chaperonins can increase folding yields by releasing assembly competent monomers. Using the dodecameric glutamine synthetase as the model oligomeric protein, our laboratory has begun to define the critical reactions where E. coli chaperonins influence folding and assembly of an E. coli oligomeric protein. The major goal is to characterize the components of chaperonin-assisted reactions at the moment when substrate--chaperonin interactions occur. We believe that defining how and where chaperonins shunt kinetic intermediates away from aggregation-prone conformers in the folding and assembly reactions is a key to understanding of the mechanism of chaperonin-assisted assembly.
Evidence for in vivo Relevance of Chaperonin-Assisted Glutamine Synthetase Assembly
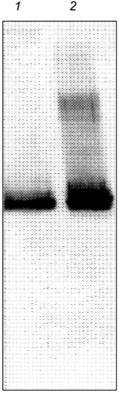
A number of observations suggest that glutamine synthetase is an authentic substrate for GroE-assisted protein folding in vivo. The first documented interactions between glutamine synthetase and chaperonins were found in plant chloroplasts. Glutamine synthetase subunits were transiently associated with resident chloroplast GroEL-like chaperonins after import into the chloroplasts [48]. The evidence that the E. coli GroEL chaperonin interacts with E. coli glutamine synthetase comes from analysis of proteins that are bound to GroEL after cell lysis. Following rapid lysis, incubation of the extract with affinity purified anti-GroEL antibodies immobilized on protein A-attached Sepharose beads [49] results in the co-immunoprecipitation of GroEL and glutamine synthetase subunits. When these complexes are specifically disrupted by GroES and ATP, the eluant was found to contain full length glutamine synthetase subunits (Fig. 5). More importantly, in the presence of Mg2+ ions, these dissociated subunits were assembly competent and slowly form active glutamine synthetase oligomers. In light of this observation, it is also important to note that native dodecameric glutamine synthetase does not readily dissociate under normal physiological conditions and is highly stable [50]. Therefore, it is highly unlikely that glutamine synthetase subunits actually dissociate from the glutamine synthetase dodecamer and bind to GroEL. As a test for such a possibility, unbound GroEL and native dodecameric glutamine synthetase were incubated together at 37°C with 1 mM Mg2+ or Mn2+. After long incubations (24 h), no glutamine synthetase subunits became bound to GroEL (M. T. Fisher, unpublished data). Based on these data, one can conclude that the active subunits that remain bound to GroEL after cell lysis do not originate from fully formed dodecamers. They are, most likely, products of GroE-assisted folding of nascent glutamine synthetase polypeptide chains.
It is interesting to note that chaperonins were first identified as mysterious proteins that copurified with ribulose bisphosphate decarboxylase (rubisco) subunits [51]. Ultimately, the folding and assembly of rubisco has since been shown to require chaperonins in vivo [52]. If chaperonin--substrate interactions observed in vitro after cell lysis have relevance to in vivo interactions, then it may be a general method for identifying potential substrates of the chaperonin system. By this criterion, the experimental results strongly suggest that glutamine synthetase is a substrate for chaperonins in vivo.Fig. 5. Western-blot analysis of eluant from freshly isolated GroEL shows that glutamine synthetase is one of the chaperonin substrates. After rapid lysis of E. coli and removal of ADP and ATP with apyrase, GroEL was immunoprecipitated with immobilized, affinity-purified anti-GroEL antibodies. After extensive washing with refolding buffer, the immobilized GroEL--substrate complexes were incubated with GroES and ATP for 5 min. The antibodies were removed and the proteins released from GroEL were separated on SDS-PAGE, transferred to nitrocellulose, and probed with substrate specific antibodies. In lane 1, antibodies to glutamine synthetase show the presence of glutamine synthetase compared to a purified glutamine synthetase preparation (lane 2).
Specific Roles of Chaperonins in Folding and Assembly of Glutamine Synthetase
Initial interactions between folding glutamine synthetase subunits and GroEL. The initial interactions between folding proteins and the chaperonins are dependent on the dynamic nature of the folding intermediate and the affinity of the chaperonin species. The affinities of both the protein substrate and the chaperonins change throughout the binding reaction. Because GroEL can bind ATP and GroES and hydrolyze ATP to ADP, there are many interconverting conformations of the GroEL chaperonin that exhibit a variety of binding affinities for folding proteins [53]. The affinities between the protein substrate and GroEL also vary because the folding substrate is not a static structure. For example, if a particular protein folds more rapidly than the time it would take for its folding intermediate to collide with the chaperonin, very little or no interaction will be observed. Because of the dynamic nature of the chaperonin--substrate interactions, a substantial population of the chaperonin complex must have the correct binding affinity(ies) and the transient population of the folding substrate must exist within the collision times to allow for adequate binding to occur. At present, experimental results suggest that numerous folding intermediates from the same protein can bind to chaperonin [45, 54-56]. Of these intermediates, the more unfolded the protein intermediate, the tighter its binding affinity for the chaperonin [53-55].
Many investigators (including the author) have used the nucleotide-free GroEL to generate GroEL--protein complexes because this form of the chaperonin is most efficient at capturing the transient folding intermediates [57-59]. Indeed, the nucleotide-free form of GroEL has been repeatedly shown to possess the highest affinity for folding substrates [24, 53, 54, 60]. When this GroEL species is used at concentrations in excess of the folding protein concentration, folding or assembly reactions are either completely halted or are significantly slowed due to direct binding of the folding protein to the chaperonin. For example, a twofold molar excess of GroEL chaperonin to glutamine synthetase monomers is sufficient to "arrest" further folding and assembly to form a stable GroEL--glutamine synthetase monomer complex [24]. Here, the chaperonin essentially binds and kinetically traps transiently folding glutamine synthetase monomers before they can form dimers [31].
Definition of the chaperonin capture species. Even though nucleotide-free GroEL is frequently included in various chaperonin mechanistic schemes as transient species [41, 61], this form more than likely does not exist as the primary capture complex of folding proteins in the cell. The binding interactions of nucleotides with GroEL appears to be complex [62]. By monitoring ATP hydrolysis rates with respect to ATP concentration, Yifrach and Horovitz have fit the changes in ATPase activity with a “nested cooperative” model where ATP hydrolysis displays positive cooperativity within a single heptameric GroEL ring and negative cooperativity between heptamers [63]. Within one heptameric ring of the tetradecameric GroEL, the apparent affinity for ATP is estimated to be 10 µM. For ADP the binding differences are more extensive where the first ring binds ADP with a Kd of 2-7 µM and the second ring binds ADP with a Kd of 2.3 mM. When GroES binds to GroEL, the dissociation constants for both ADP and ATP binding show a further decrease [62]. Numerous estimates of the Kd for the GroES--GroEL binding interaction give dissociation constants between 0.3 and 2 nM (reviewed in [39]). Since the steady-state concentrations of ATP and ADP in the cell are estimated to be 3 and 1 mM, respectively, at least one heptamer of the GroEL tetradecamer exists as a nucleotide bound form in vivo. In addition, given the tight binding affinity of GroES and the 2:1 molar ratio of GroES to GroEL [6], the cochaperonin can readily form complexes with GroEL. With association rates between GroES and GroEL at ~1·105 M-1·sec-1 [64] and a Kd of ~1 nM, the mean lifetime of the complex is a staggering 2.5 h at physiological concentrations of GroEL, GroES, and ADP. However, the folding polypeptides may influence the GroEL--GroES binding affinity. For example, substrates that fold while being sequestered in the GroEL--GroES cavity can still rapidly dissociate from the complex (C.-M. Low and M. T. Fisher, unpublished data). Furthermore, forming complexes between a folding protein like rhodanese with a preformed GroEL--ADP--ES asymmetric complex still results in folding and dissociation of rhodanese from this complex. These results indicate that the GroEL--GroES--ADP complex is labile in the presence of folding substrates (C.-M. Low and M. T. Fisher, unpublished data). The binding interaction between GroES and GroEL is also weakened by ATP binding at the opposite ring [43]. The nature of the steady-state capture complex is unclear but numerous schemes have suggested various nucleotide--GroES--GroEL complexes [41, 61]. It is highly unlikely that the completely unliganded form of GroEL species is the capture complex in vivo.
The nature of the initial chaperonin complex and the folding efficiency of glutamine synthetase. Both the nucleotide-free and ADP-bound GroEL can arrest glutamine synthetase folding and assembly [31]. When ATP binds to GroEL, glutamine synthetase subunits can once again continue to fold and assemble to high yields. Due to its inability to “arrest” glutamine synthetase refolding, the ATP-bound GroEL has a lower affinity for the glutamine synthetase folding intermediates than do either the nucleotide-free or ADP-bound forms of GroEL [24, 31]. These early data indicated that at least one of the roles of nucleotide binding is to simply decrease binding affinities of GroEL for folding proteins [31, 53, 54]. The structural origins for this decrease in affinity have been demonstrated in an elegant study by Helen Saibil’s group at Birbeck College. Their cryoelectron microscopy data show that binding ADP or ATP to GroEL results in a concerted conformational change that moves the multiple substrate-binding sites on GroEL away from solvent accessible orientations to differentially rotate and bury these sites at subunit interfaces [65]. These binding surfaces are further removed from interactions with folding proteins when GroES forms a complex with the nucleotide bound form of GroEL. The averaged cryoelectron images of GroEL with ADP or ATP, coupled with GroES binding can be compared in a movie format that is accessible on the World Wide Web (http://www.cryst.bbk.ac.uk/~ubcg16z/elmovies.html). The high-resolution crystal structure of the GroEL--GroES--ADP complex illustrates how these dramatic conformational shifts result in a complete sequestration of the protein-binding site upon GroES binding [66]. However, since the GroEL--GroES complex was resolved under low nucleotide concentrations (~50 µM ADP), it is still not known whether ADP or ATP is bound to the opposite, GroES-free GroEL ring under physiological conditions.
At moderate ionic strengths, the molecular interactions that primarily dictate chaperonin binding with folding proteins are predominately hydrophobic in character. Eisenstein and coworkers have preformed the first initial thermodynamic characterizations of the binding interactions between chaperonins and folding proteins. Using a folding variant of the bacterial protease subtilisin (PG9) that remains as a partially folded stable conformer in solution, they found that the binding interaction has a positive enthalpy, a hallmark of hydrophobic interactions [67]. Furthermore, the dissociation constant was temperature dependent with the highest affinity at 37°C, which is the optimal growth temperature of E. coli. At this temperature, the enthalpic contribution approaches zero indicating that the energetic contributions are mainly entropic. At lower temperatures (20°C), glutamine synthetase subunits slowly dissociate from the preformed GroEL--GS complex in the absence of nucleotide, forming a small amount of active glutamine synthetase (M. T. Fisher, unpublished data). Correspondingly, to insure optimal interactions between the chaperonins and refolding glutamine synthetase, most if not all experiments with glutamine synthetase were performed at 37°C.
Glutamine synthetase has been classified as a protein that is not a “true” chaperonin substrate because its efficient folding can occur under less stringent conditions [68] (i.e., without GroES). The high-affinity nucleotide-free form of GroEL was initially used to bind folding intermediates. When ATP was added to the GroEL--glutamine synthetase complex, high yields of active glutamine synthetase were achieved [24]. However, our laboratory has now shown that the efficient refolding of glutamine synthetase previously defined with the high-affinity nucleotide-free chaperonin complex (GroEL alone) is substantially diminished if the ATP--GroEL--GroES or ADP--GroEL--GroES "low affinity" chaperonin complexes are used to capture folding intermediates of glutamine synthetase. When these physiologically more relevant "low affinity" chaperonin complexes are used, the folding efficiency decreases to 40-50% of the original activity compared to the 70-80% yields generated with the “high affinity” nucleotide-free chaperonin form (P. A. Voziyan and M. T. Fisher, unpublished data). It is only when the ratio of chaperonin to folding protein increases that the folding efficiency of these activated "low affinity" complexes approaches optimal levels (~60-70%). This is a rather interesting observation because glutamine synthetase has relaxed requirements for the chaperonin system. Even more surprising is the observation that over the same concentration range of folding protein, the “stringent” substrates such as mammalian mitochondrial rhodanese or mitochondrial malate dehydrogenase do not show any dramatic differences in yields or rates from either the “high affinity” (GroEL alone) or “low affinity” GroE (ATP--GroEL--GroES) complexes (P. A. Voziyan and M. T. Fisher, unpublished data).
There are at least three major factors that may contribute to the observed decline in recoverable glutamine synthetase activity with the “low affinity” chaperonin complexes. First, this complex has a decreased binding affinity for the folding intermediate(s) [53]. Second, there is a loss of one substrate-binding site in the “low affinity” complex when GroES binds to GroEL compared to the “high-affinity” GroEL tetradecamer that has two potential binding sites for folding proteins and thus an increased probability for binding and arresting the folding of glutamine synthetase subunits. These two factors alone can largely account for the inherent decrease in effective binding of transient folding intermediates by the “low affinity” form. The third and final factor is dependent on the folding properties of the isolated glutamine synthetase subunit itself. For most oligomeric proteins, the assembly competent conformation has a defined lifetime. The decay rate of this conformer is often directly proportional to the concentration of the folding protein [19, 20]. Using methodologies developed by Lorimer and Clarke, the rates of irreversible misfolding can be determined by initiating spontaneous folding and then adding the chaperonins and ATP to the folding reaction at various delay times [35, 57]. At a 0.1 µM subunit concentration, the assembly competence of the glutamine synthetase subunits declined more rapidly in the presence of the “low affinity” complex compared to the “high affinity complex” (rate constants 1.7 and 0.38 sec-1, respectively; P. A. Voziyan and M. T. Fisher, unpublished data). No differences in the misfolding kinetics were observed when rhodanese refolding was observed with “high” and “low” affinity complexes (~0.6 sec-1). The data show that the glutamine synthetase folding intermediates are more sensitive to changes in the parameters of chaperonin--substrate binding.
In experiments particularly germane to the properties of a “low affinity” chaperonin complex, elegant studies by Horwich and coworkers have shown that the most efficient chaperonin-assisted folding occurs when GroES binds to the GroEL-binding site that is occupied by the substrates to form the cis-complex [69]. In their studies, when ornithine transcarbamylase subunits bound to GroEL at the binding site opposite to the GroES-binding site to form a trans-complex, a decline in the folding rate was observed. However, the final folding yields remained the same for both cis- and trans-complexes. Since the folding of glutamine synthetase was less efficient with preformed GroEL--GroES complexes compared to GroEL alone, substrate folding variability with the chaperonin proteins seems to depend, in part, on the properties of the folding intermediates that rapidly accumulate prior to their collision and binding to GroEL.
It has recently been reported that an equal mixture of cis and trans ADP--GroEL--GroES--rhodanese complexes are formed when ADP and GroES are added to preformed unfolded rhodanese--GroEL complexes [69]. This observation is surprising. It seems to indicate that the bound substrate has little influence on the GroEL--GroES complex formation. However, since GroES influences substrate binding, thermodynamic arguments would suggest that the substrate would reciprocally influence the GroES binding. Indeed, the distribution of cis- and trans-complexes could vary for different substrates. If this is the case, the affinities of substrate binding may influence the partitioning of GroES to the same or opposite side occupied by substrate. It is clear that more systematic studies are needed to determine whether changes in binding affinities of substrates to the chaperonin will influence the chaperonin mechanism directly.
Variability of the release requirements of glutamine synthetase monomers from GroEL. Although chaperonins have been implicated in facilitating assembly of oligomers, experimental data indicates that chaperonins are indirectly involved in the actual assembly process. During assisted assembly reactions, it has been repeatedly shown that the chaperonins are capable of binding and releasing monomeric subunits prior to the assembly of the oligomer [31, 70-72]. For dodecameric glutamine synthetase, optimal release of its monomers from GroEL--substrate complexes occurs when ATP and GroES are present. These released monomers then assemble into the glutamine synthetase dodecamer. Although the release of assembly-competent monomers from GroEL can be accomplished with a variety of chaperonin species, the rates are often very different from conditions with the complete chaperonin system present. To understand the details of the chaperonin-assisted glutamine synthetase assembly reaction, one must define: 1) the conditions where the release of assembly-competent monomers can occur; 2) the physical properties of the released subunits and; 3) the factors that may control the lifetime of the chaperonin--glutamine synthetase subunit complex.
Numerous studies of chaperonin-assisted protein folding show there are different requirements for GroES and nucleotides during polypeptide folding. That is, some proteins can fold in the presence of ATP (or ADP) and GroEL alone while others absolutely require ATP and groES. These results indicate that the detailed mechanism for protein folding by chaperonins may depend on the nature of the protein substrate.
Lorimer and coworkers have proposed requirements for GroES in chaperonin-assisted folding depends on the propensity of the protein to attain its final fold once it is released from the chaperonin [68]. This hypothesis was tested using three different protein substrates. By lowering the temperature of folding reactions to conditions where the protein could fold to an active form without the aid of the chaperonin system (“folding permissive” conditions), it was found that GroES was no longer required for successful folding from GroEL. From this limited data, Lorimer and coworkers suggest that the nature of the folding environment will dictate the GroES requirement during folding. However, from the point of view of chaperonin--substrate interactions, the changes in the folding environment would directly influence the binding affinities of chaperonin for the folding intermediates. At lower temperatures, these interactions, defined primarily by hydrophobic forces, would be weakened, thus allowing for less stringent chaperonin requirements. Alternatively, under “non-permissive” folding conditions, when the temperature is elevated to physiological levels, folding intermediates would have more exposed hydrophobic regions and exhibit stronger binding to GroEL. In order to release these more tightly bound intermediates, GroES would be required in the chaperonin mechanism. In a slightly different set of experiments, Lorimer and coworkers have also observed that the GroES requirements for rubisco shift from “non-permissive” to “permissive” conditions at constant temperature if a low molecular weight additive (in this case chloride ions) is added in the renaturation mixture [68]. Since the Cl- concentrations used were as high as 0.5 M, it is possible that the binding affinities between rubisco and GroEL were also significantly perturbed. In these examples, the GroES requirements would be directly linked to chaperonin--substrate interactions rather than being indirectly related to the nature of the folding environment.
Chaperonin-assisted folding of glutamine synthetase provides useful information with respect to possible mechanisms of GroES requirements. As mentioned previously, glutamine synthetase was not considered to be a stringent substrate because: 1) it can fold spontaneously, and 2) it does not require GroES to fold under “permissive” conditions [68]. Curiously, our laboratory has found that the general rule of “permissive” versus “non-permissive” folding dictating GroES requirements does not hold for chaperonin-assisted folding of glutamine synthetase. At a constant temperature of 37°C, the conditions of spontaneous reactivation of glutamine synthetase can also be shifted from “permissive” (10-30% reactivation) to “non-permissive” (0% reactivation) just by lowering the concentration of Mg2+ ions present in the renaturation solution from 5 to 3 mM. Under these more stringent folding conditions, ATP can still reactivate glutamine synthetase from the arrested GroEL--glutamine synthetase complex without the aid of GroES (M. T. Fisher, unpublished data). Since the solution conditions for glutamine synthetase refolding are largely unperturbed, it is safe to assume that the folding intermediate conformations and the binding affinities between GroEL and glutamine synthetase would also remain relatively unchanged. Thus, the GroES stringency for facilitated folding appears to be dependent on the nature of the interactions between glutamine synthetase and GroEL rather than on the propensity for the glutamine synthetase to fold by itself.
With regard to increased substrate-binding affinities, earlier experiments by Buchner and coworkers had suggested that the degree of protein unfolding may dictate the GroES requirement. Using reduced (disulfide bonds are reduced) and oxidized (disulfide bonds are intact) antibody Fab fragments, it was demonstrated that the chaperonin requirements seem to depend on the stability of the substrate--chaperonin complex [55]. Whether the reduced Fab fragment intermediate binds tighter to the chaperonin or simply folds slower (i.e., has an extended intermediate lifetime) is not known.
“True” chaperonin substrates (albeit in vitro) have been defined as those substrates that have stringent requirements for the chaperonin system [41, 68]. According to this standard, the substrates such as mammalian mitochondrial rhodanese, porcine mitochondrial malate dehydrogenase, bacterial ribulose diphosphate decarboxylase, and porcine mitochondrial citrate synthase belong in this category. However, it is interesting to note that rhodanese, the most often used model substrate for chaperonin-assisted folding, can still fold spontaneously to 25% of its original activity at low protein concentrations [73]. Even though some spontaneous folding can occur, no rhodanese refolding is observed if ATP is added to a GroEL--rhodanese complex without GroES. For rhodanese, the stringency for the GroES requirement remains even under conditions when a portion of the population can fold by itself. This result again suggests that the GroES requirement is not entirely dictated by the folding propensity of the protein substrate. Clearly, the linkage between stringent chaperonin folding requirements and binding strengths between substrates and GroEL need to be explored in more detail.
Influence of Glutamine Synthetase on the Stability of the GroEL--GroES Complex
In addition to the variability with GroES requirements, chaperonin-assisted folding and assembly of glutamine synthetase can occur with non-hydrolyzable ATP analogs like ATP-gamma-S or AMP-PNP [24, 31]. These results show once again that the GroEL affinity for folding substrates is modulated by binding interactions between nucleotides and GroEL. Although ADP does not induce glutamine synthetase reactivation from GroEL--glutamine synthetase complexes, adding GroES to a ADP--GroEL--glutamine synthetase complex results in reactivation rates that are the same as those observed with ATP [31]. With both ATP or ADP, the addition of groES greatly accelerates glutamine synthetase monomer release and consequently, glutamine synthetase dodecamer formation [24, 31]. Given that GroES accelerates chaperonin-assisted folding of glutamine synthetase with non-hydrolyzable ATP analogs and ADP, it is logical to conclude that it is the binding free energy between GroES and the nucleotide-bound GroEL that provides the additional energy source to facilitate the conformational changes within GroEL which is necessary to decrease GroEL affinity for glutamine synthetase monomers. Although the chaperonin-assisted assembly rates of glutamine synthetase with AMP-PNP are significantly slower, the final reactivation yield is equal to what is observed with ATP and ADP. A slower assembly rate is most likely due to a slower dissociation of glutamine synthetase from the GroEL--ES--AMP-PNP complex. As an interesting comparison, this same AMP-PNP--chaperonin complex effectively traps the substrate rhodanese inside the enclosed GroEL--GroES cavity ([74]; C.-M. Low and M. T. Fisher, unpublished data), emphasizing the importance of specific substrate--chaperonin interactions in the overall mechanism.
It has been suggested that ATP hydrolysis is necessary to “prime” GroES dissociation from the chaperonin complex [43, 75]. This may explain why the GroEL--GroES--AMP-PNP complex is so stable. Although the molecular details still remain obscure, it is logical to assume that this “priming” interaction is probably linked to an overall decrease in the binding affinity between GroEL and GroES as ATP is hydrolyzed to ADP. Since glutamine synthetase monomers must be released prior to assembly, the variable release rates observed with the various nucleotides may reflect the different binding strengths and interactions of the bound nucleotide with the GroEL--GroES--protein complex. It is still unclear whether the substrate itself has a significant influence on the stability of the GroEL--GroES complex. However, given the inherent reciprocity in polypeptide and GroES binding [64], it is not unreasonable to assume that different substrates will affect the stabilities of the GroEL--GroES complex.
PROPERTIES OF GLUTAMINE SYNTHETASE SUBUNITS RELEASED FROM THE GroE
CHAPERONINS
The glutamine synthetase subunits released from GroEL have a reduced tendency to misfold and aggregate. As mentioned before, an increase in the concentration of the folding protein often leads to an increased flux toward misfolded and/or aggregated off-pathway protein conformers. Although the GroE chaperonins enhance renaturation yields of glutamine synthetase at optimal substrate concentrations of 0.5 µM, initial experiments showed that these renaturation yields still declined with the increase of concentration regardless of whether the GroE chaperonins were present or not [25]. Other chaperonin-assisted folding reactions showed similar trends [57, 58]. The origin of this decline in activity is primarily due to competing kinetic partitioning reactions that lead to misfolded forms. It must be remembered that there are at least three probable folding pathways that can occur with chaperonins present. For example, the glutamine synthetase monomers can: 1) misfold and aggregate; 2) fold and form native glutamine synthetase; or 3) interact and bind to the GroEL chaperonin forming a stable GroEL--glutamine synthetase complex. As the concentration of folding glutamine synthetase monomers increases, off-pathway interactions between misfolded conformers become more frequent. This leads to an increase in the kinetics of irreversible misfolding and aggregation. However, if the stable GroEL--glutamine synthetase complexes are preformed under optimal renaturation concentrations and are concentrated prior to activation with ATP and GroES, the yields of assembled and activated glutamine synthetase remain at their highest levels without any concentration-dependent decline [25]. These results suggest that the formation of the initial GroEL--glutamine synthetase complex is crucial for insuring optimal folding and assembly. Once again, this observation emphasizes the importance of the interactions between initially folded intermediates and the chaperonins. In vivo, the proper folding yields of some overexpressed proteins can be improved when the relative chaperonin concentration is simultaneously increased [52]. Thus, the subtle balance between chaperonin and initial folding intermediates concentrations can prevent the folding protein from kinetically partitioning into misfolded or aggregated forms. Once a complex with GroEL is formed with glutamine synthetase, the released subunits show little tendency to misfold and aggregate even at subunit concentrations as high as 2 mg/ml. Under optimal unfolding and refolding conditions, other substrates such as rhodanese also avoid aggregation if the folding is initiated from preformed GroEL--rhodanese complexes [45].
To test whether the prevention of misfolding at high concentrations results from the encapsulation of glutamine synthetases monomers by the GroEL--GroES complex, the following experiment was performed. When ATP alone is added to the concentrated GroEL--glutamine synthetase complex (2 mg/ml glutamine synthetase, final concentration) without GroES present, the folding yields still remained as high as those observed under optimal mixing conditions (M. T. Fisher, unpublished data). Thus, at least for glutamine synthetase, the critical steps appear to be the initial association reaction and the ATP-induced conformational change of GroEL. This result implies that glutamine synthetase folding yields are not dependent on the encapsulation of the folding glutamine synthetase monomers in a GroEL--GroES complex. For glutamine synthetase assembly, the advantage of GroES inclusion in the chaperonin mechanism is to enhance the release of monomers from the complex.
The commitment rate of folding glutamine synthetase to a native protein conformation. Although the glutamine synthetase monomers released from GroEL are less susceptible to misfolding and aggregation, this observation does not address the question of how released glutamine synthetase monomers remain in an assembly competent state while avoiding misfolding and aggregation. The released monomers appear to remain assembly competent for a long period of time while the initially observed stable GroEL--glutamine synthetase complexes rapidly disappear after groES and ATP addition [31]. It was reasoned that released monomers could either attain a conformation that is sufficiently folded such that they would no longer need to interact with the chaperonins or the monomers could maintain a steady state assembly competence through repeated cycles of release and rebinding reactions with the GroE chaperonins. An experimental protocol was designed to determine when monomers attain an assembly competent conformation that no longer interacts with the chaperonin [76]. For these experiments, GroEL was removed by rapid immunoprecipitation at various times during the chaperonin-assisted refolding of glutamine synthetase. The released monomers that remained in solution were allowed to fold to a native state. The maximum yields were then determined after two hours. The kinetics of assembly of released monomers into active glutamine synthetase were represented as an activity amplitude at a specific GroEL removal time. Surprisingly, the kinetics at which glutamine synthetase monomers commit to an assembly competent monomer at 37°C was extremely rapid (t1/2 = 10 sec; see table) compared to the kinetics of the appearance of glutamine synthetase activity (t1/2 = 4 min) [24, 31]. Although ADP and AMP-PNP can replace ATP in facilitating glutamine synthetase refolding from the chaperonins, their measured commitment rates were substantially slower than those rates obtained with ATP (see table). The slower commitment rates observed with AMP-PNP results from a slower dissociation rate of glutamine synthetase monomers from the GroEL--GroES--AMP-PNP complex [31]. Since GroES dissociation is a prerequisite for substrate dissociation, this seems to indicates that GroES dissociation from the complex is slow. Additional experimental evidence supports this hypothesis ([74]; C.-M. Low and M. T. Fisher, unpublished data). Interestingly, other substrates such as the mitochondrial dimeric malate dehydrogenase cannot fold without ATP and monomeric rhodanese can become active but remains trapped in the GroEL--GroES complex. It is not known whether these drastic differences in release rates are indications of the influence of glutamine synthetase monomers themselves on the stability and thus the affinity of GroEL--GroES complex. Here again, the degree with which the polypeptide-binding reaction itself influences the chaperonin mechanism has yet to be determined.
Commitment rates for glutamine synthetase with various nucleotides
(37°C)*
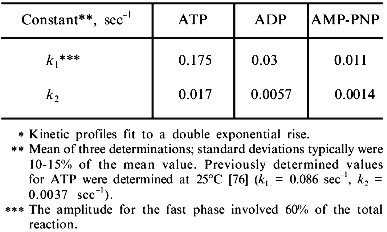
Release of glutamine synthetase subunits from nucleotide-free chaperonin complexes. Glutamine synthetase monomers commit to the native state at a much faster rate than other chaperonin substrates such as rhodanese and malate dehydrogenase [35, 76]. Thus, the lifetime of the GroE--glutamine synthetase complex must be short. Although this interaction is transient, the nucleotide-induced conformational changes within the GroE chaperonins may have a dramatic effect on the folding yields of glutamine synthetase. At present, it is still not clear precisely how the chaperonin actually enhances the folding yield of this protein.
Two predominant hypotheses have been advanced to explain why higher folding yields are frequently observed with chaperonins. The first hypothesis suggests that the GroE chaperonins insure that the protein can fold to a more native-like conformation through sequestered folding inside the GroEL--GroES complex. The chaperonins simply provide an enclosed environment where folding to the native state can occur unperturbed by off-pathway aggregation events. Similar in vitro situations are found when proteins are allowed to fold on immobilized supports and after completion, the newly folded proteins are simply removed from the support [77, 78].
The second hypothesis stresses the importance of the initial interactions between the chaperonins and folding proteins. Here, the chaperonins would bind and partially unfold misfolded intermediates that would initially accumulate and essentially initiate the refolding reaction from a more unfolded state [35, 42]. By unfolding the initially misfolded protein, the chaperonins are envisioned to raise the energy levels of locally trapped off-pathway kinetic intermediates and allow the protein to kinetically partition once again to its native state. The closest analogy of this mechanism to an in vitro folding situation would be a procedure where one would take a previously misfolded protein and subject it to repeated unfolding and refolding cycles, removing the native protein formed and continuing the cycle until all the protein folds to an active conformation. Although the accumulating experimental evidence suggests that both mechanisms may be operational [39], it appears that the final yields are ultimately dictated by the initial binding interactions. This important aspect in chaperonin assisted folding is illustrated by the following observation. If active rhodanese was unfolded in the absence of thiosulfate (a ligand for folded rhodanese) and GroEL--rhodanese complexes were prepared and concentrated as described before [25], the released rhodanese underwent substantial misfolding and aggregation upon release from the GroE chaperonins [45]. This result was not observed when rhodanese was unfolded with thiosulfate present. Thus, it appears that the initial interaction conformation of the substrate which binds to the chaperonin can directly influence the folding yields of the GroEL released protein.
Presently, it is hard to determine how much folding to native structure occurs in the sequestered environment with the complete chaperonin system. If the substrate could fold unobstructed in the GroEL--GroES cavity, the formation of active protein would depend on the intrinsic folding rates of the protein substrates, as well as the release of this protein from the cavity. It has been repeatedly demonstrated that proteins can acquire their native folds while sequestered inside stable GroEL--GroES complexes that are: 1) stabilized by non-hydrolyzable nucleotides, or 2) generated using genetically engineered single ring-chaperonin complexes [69, 74]. However, since a significant amount of non-native proteins is always released from wild-type chaperonin complexes in vitro and in vivo, folding yields can only be increased if the misfolded proteins are allowed to rebind and undergo another round of partial unfolding and refolding.
Because glutamine synthetase interacts briefly with the chaperonin system, it was hypothesized that a simple binding and release reaction was all that was needed to achieve efficient folding of this oligomer. If true, one of the predictions of this hypothesis would be the following. After forming a complex between glutamine synthetase monomers and GroEL, a simple competitive displacement of the bound glutamine synthetase with another protein substrate in the absence of nucleotide would result in the release of functional glutamine synthetase. However, contrary to these expectations, it was found that simply competing off GroEL-bound glutamine synthetase with a vast excess of a metastable molten globule protein alphaa1-casein at 37°C results in just a relatively small increase in refolding yield over what is observed in the absence of chaperonins (L. Jadhav and M. T. Fisher, unpublished data). In contrast, the addition of casein to stable GroEL--glutamine synthetase--ADP complexes results in efficient dissociation and folding of glutamine synthetase to levels seen with the complete system. This suggests that simple binding to and release from the chaperonin is not enough to insure that a majority of the released glutamine synthetase subunits acquire a folding-competent form. The higher folding yields observed with nucleotide suggest that the resulting conformational change in GroEL is crucial for insuring that the bound glutamine synthetase subunits attain on-pathway folding-competent states. Based on the high resolution X-ray crystallographic analysis of the GroEL--GroES--ADP complex [65] and cryoelectron microscopic imaging [66], Lorimer has proposed a mechanical model wherein the apical domains would bind the protein and would physically unfold the protein when these binding domains move following nucleotide binding [79].
In the casein competition experiments, the disappearance of the GroEL--glutamine synthetase complexes was more rapid with nucleotide (ADP) present. In the absence of nucleotides, GroEL--glutamine synthetase complexes were observed up to 4 h after casein addition but eventually all of the glutamine synthetase subunits dissociated from GroEL. In summary, these results suggest that the nucleotide-induced movements of the chaperonin domains and increased dissociation rates may be important for proper folding of glutamine synthetase once this protein interacts with GroEL.
THE NEXT STAGE IN CHAPERONIN RESEARCH: FROM “MODEL” TO
“AUTHENTIC” SUBSTRATES
It is becoming very clear that attempts to explain the mechanism of chaperonin action with a limited number of substrates may not be adequate. Furthermore, using substrates that are not even isolated from the same organism where the molecular chaperones reside may miss important adaptive factors governing intrinsic protein--molecular chaperone interactions. Once the protein is fully synthesized and is released from the ribosome, the overall folding efficiency of that protein depends on its rate of folding to the active conformation(s) and its ability to avoid off-pathway folding reactions. Ironically, a majority of the stringent protein substrates that are used for in vitro studies of chaperonin mechanism are mitochondrial proteins. Since these proteins are synthesized in the cytoplasm and are then transported into the mitochondria, they do not fold rapidly to a native conformation because rapid folding in the cytoplasm would prevent transport.
Currently, one of the major debates in the chaperonin field relates to the question: What is the most representative protein substrate for the chaperonin system? The answer may be as varied as the number of proteins that can be folded by the chaperonins. Most “good” or “optimal” substrates are defined as those that cannot acquire an active fold in the absence of the chaperonins. Thus, the overall folding reaction with chaperonins is viewed as that of the “all or none” variety. Another caveat is that “representative” substrates can only fold with the complete chaperonin system present. However, these substrates often (but not necessarily) interact with the chaperonins for extended periods of time because they must undergo multiple rebinding and release reactions with the cycling chaperonin system. Fortunately, these substrates are “good” or “optimal” for research into the mechanism because, for the most part, their interaction times are extended, and trapped species can be easily isolated. Conversely, with regards to the organism itself, authentic “good” or “optimal” protein substrates in E. coli may only require transient interactions with their natural chaperonin system (GroE). Because of the rapid generation times of E. coli (~23-30 min at 37°C with enriched nutrient broth) and the limited number of chaperonins in the cell, such interactions may also be very rapid.
In E. coli, the folding reactions aided by chaperone proteins may not necessarily be “all or none” folding reactions. The following example illustrates a situation where obtaining a moderate recovery of folded protein would still be detrimental to the growth of E. coli. Prior to cell division, it has been shown that the first molecular step is the formation of a large macromolecular structure called the Z-ring. The Z-ring is composed of a tubulin-like protein called FtsZ, and it determines the position of the division plane and septum formation. It has been shown that a critical intracellular concentration of FtsZ is required to form such a large structure [80]. If folding of FtsZ is diminished, a decrease in the final concentration of the FtsZ protein disrupts cell division. It appears that this process depends on an acquisition of a sufficient concentration of properly folded FtsZ to initiate cell division and is not consistent with “all or none” folding. It is interesting to note that E coli carrying temperature sensitive mutants of DnaK (hsp70) and GroEL are defective in septum formation and cell division [80].
In addition, slight inefficiencies in folding may have serious ramifications with respect to generation time. Selection pressures may select out those strains that do not efficiently convert substrate sources into biomass. Therefore, E. coli, with its rapid generation times, may favor transient interactions between folding proteins and chaperonins. It is of interest to note that while mammalian dihydrofolate reductase (DHFR) can interact and bind quite efficiently with the GroE chaperonins, the E. coli DHFR interactions are only transient under the same conditions [81]. Curiously, these two forms of DHFR have virtually identical tertiary structures. Here, the differences in the folding kinetics between mammalian DHFR and E. coli DHFR may dictate different interaction times with GroEL. It is unclear how much one can extrapolate in vitro studies into in vivo processes using heterogeneous substrate--chaperonin interactions when the homologous substrates do not show similar interactions in the host chaperonins. With this in mind, the finding that (1) assembly competent E. coli glutamine synthetase monomers are bound to newly isolated GroEL, and (2) the dodecamer formation of glutamine synthetase is substantially aided by E. coli chaperonins emphasizes a particular importance of using this and other authentic in vivo substrates of GroE for understanding of the chaperonin mechanism.
Both folding and misfolding are dramatically influenced by molecular chaperones. With in vivo evidence for interaction between glutamine synthetase and the GroE chaperonins, it is useful to examine these specific interactions in detail. If the turnover rates of chaperonins with glutamine synthetase in vitro are representative of the cellular turnover rates, then using the turnover estimations outlined by Lorimer [82] reveals that the chaperonin could potentially interact with only 20 to 30% of the total E. coli cellular proteins. These estimates are in good agreement with the estimates obtained by Horwich and coworkers [83]. However, one must be cautious with such estimates of chaperonin turnover numbers. Since chaperonin proteins can interact with a large array of proteins that probably have very different folding rates, binding interactions, and different concentrations, obtaining a “turnover number” that would represent general protein--chaperonin interactions in vivo becomes difficult. A partial solution would be to use authentic E. coli substrates for such estimates of global turnover numbers.
Recent estimates of the rate of release for a wide range of newly synthesized E. coli proteins do indicate that a majority of them are released from the chaperonin with half times of 10 to 30 sec [84]. Although these experiments cannot tell whether the released proteins have folded correctly, the reported interaction times are very similar to the interaction times estimated for glutamine synthetase (see table). These estimates, however, were obtained at 30°C while the optimal growth temperature of E. coli is 37°C. Although the glutamine synthetase commitment rate does not dramatically increase as the temperature increases from 25 to 37°C (from 5.13 to 10.5 min-1) for the fastest rate constant (see table), the number of proteins that could interact with chaperonins increases from 13.5 to 27.5% over this same temperature range. In addition, the amount of chaperonins increases by ~0.5% of the total cell protein [7]. Thus, the 30°C measurement may underestimate both the mean resident time and the amount of protein that can potentially interact with the chaperonins under optimal growth conditions.
For the reaction between E. coli chaperonin and folding of E. coli glutamine synthetase subunits, some insights into the efficiency of both systems have come to light. For example, in contrast to other more commonly used exogenous substrates, glutamine synthetase subunits interact briefly with the chaperonin system. Furthermore, the transient interactions between folding glutamine synthetase and the activated GroE chaperonins leads to a folding efficiency that is much more sensitive to changes in chaperonin concentrations. It appears that the lifetime and frequency of the initial interactions between the folding subunits of glutamine synthetase and GroEL are critical for insuring successful folding. Because glutamine synthetase is more prone to misfolding when folding occurs with the physiologically relevant “lower affinity” complexes of GroEL--GroES, the influence of other prominent molecular chaperone systems such as the Hsp70, Hsp90, or Hsp25 must be explored along with the chaperonin interactions. Transient interactions may enable the limited number of chaperonins in the cell to interact productivity with more folding proteins. In contrast, a large population of proteins interacting with the chaperonins for extended periods of time, may be highly detrimental to the organism. Thus, from a kinetic point of view, normal growth conditions in E. coli would favor transient interactions and long term interactions would be avoided [85]. Using authentic E. coli substrates with E. coli chaperonins will reveal whether the commonly observed long interaction times and multiple rebinding reactions observed with non-E. coli substrates are characteristic for E. coli substrates.
In relation to interaction times, it is also important to determine whether the properties of the substrate can influence the chaperonin mechanism itself. For example, binding properties of the substrates interacting with the GroE chaperonins may directly or indirectly influence interactions times by altering GroEL--GroES interactions. Although these interactions would certainly affect the chaperonin mechanism, very little quantitative data is currently available that examines how polypeptide binding affects the GroEL--GroES binding equilibrium [64]. However, some tantalizing evidence for reciprocity between GroES and the polypeptide in the form of shifts in allosteric states of GroEL has come out of the recent work by Horowitz and coworkers [86, 87]. Surprisingly, one of the most important challenges for chaperonin research is the characterization of the molecular interactions between authentic E. coli protein substrates and the GroE chaperonin system [39, 41]. Because the important steps for substrate recognition depend on the properties of the folding intermediate(s), these interactions are substantially defined even before the folding intermediate encounters the chaperonin. Thus, the efficiency of the chaperonin mechanism may still remain incomplete until the other molecular chaperone systems are included in folding experiments.
The author is grateful to Paul Voziyan and Bryan Tieman for helpful discussions. This research is supported by the National Institutes of Health, Grant GM49309.
REFERENCES
1.Ellis, R. J. (1990) Seminars in Cell
Biology, 1, 1-10.
2.Sternberg, N. (1973) J. Mol. Biol.,
67, 1-24.
3.Georgopoulos, C., Hendrix, R. W., Casjens, S. R.,
and Kaiser, A. D. (1973) J. Mol. Biol., 76, 45-60.
4.Hohn, T., Hohn, B., Engel, A., Wortz, M., and
Smith, P. R. (1979) J. Mol. Biol., 129, 359-373.
5.Hendrix, R. W. (1979) J. Mol. Biol.,
129, 375-392.
6.Hemmingsen, S. M., Woolford, C., van der Vies, S.
M., Tilly, K., Dennis, D. T., Georgopoulos, C. P., Hendrix, R. W., and
Ellis, R. J. (1988) Nature, 333, 330-334.
7.Niehardt, F. C., Phillips, T. A., VanBogelen, R.
A., Smith, M. W., Georgalis, Y., and Subramanian, A. P. (1981) J.
Bacteriol., 145, 513-520.
8.Fayet, O., Ziegelhoffer, T., and Georgopoulos, C.
P. (1989) J. Bacteriol., 171, 1379-1387.
9.Georgopoulos, C., Ang, D., Liberek, K., and Zylicz,
M. (1990) in Stress Proteins in Biology and Medicine (Morimoto,
R. I., Tissieres, A., and Georgopoulos, C., eds.) Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, N. Y., pp. 191-278.
10.Bennett, M. J., Schlunegger, M. P., and
Eisenberg, D. (1995) Protein Sci., 4, 2455-2468.
11.Shapiro, B. M., Kingdon, H. S., and Stadtman, E.
R. (1967) Proc. Natl. Acad. Sci. USA, 58, 642-649.
12.Holzer, H., Mecke, D., Wulff, K., Liess, K., and
Heilmeyer, L., Jr. (1967) Adv. Enzyme Regul., 5,
211-225.
13.Valentine, R. C., Shapiro, B. M., and Stadtman,
E. R. (1968) Biochemistry, 7, 2143-2152.
14.Frey, T. G., Eisenberg, D., and Eiserling, F. A.
(1975) Proc. Nalt. Acad. Sci. USA, 72, 3402-3406.
15.Almassy, R. J., Janson, C. A., Hamlin, R., Xuong,
N-H., and Eisenberg, D. (1986) Nature, 323, 304-309.
16.Yamashita, M. Y., Almassey, R. J., Jenson, C. A.,
Cascio, D., and Eisenberg, D. (1989) J. Biol. Chem., 264,
17681-17690.
17.Maurizi, M. R., and Ginsburg, A. (1982) J.
Biol. Chem.,257, 7246-7251.
18.Haschmeyer, R. H., Wall, J. S., Hainfield, J.,
and Maurizi, M. R. (1982) J. Biol. Chem., 257,
7252-7253.
19.Jaenicke, R., and Rudolph, R. (1986) Meth.
Enzymol., 131, 218-250.
20.Rudolph, R., and Lilie, H. (1996) FASEB
J., 10, 49-56.
21.Ciardi, J. E., Cimino, F., and Stadtman, E. R.
(1973) Biochemistry, 12, 4321-4329.
22.Shen, C. L., and Murphy, R. M. (1995) Biophys.
J., 69, 640-651.
23.Fisher, M. T., and Stadtman, E. R. (1992) J.
Biol. Chem., 267, 1872-1880.
24.Fisher, M. T. (1992) Biochemistry,
31, 3955-3963.
25.Fisher, M. T. (1993) J. Biol. Chem.,
268, 13777-13779.
26.Zettlmeissl, G., Rudolph, R., and Jaenicke, R.
(1979) Biochemistry, 18, 5567-5571.
27.Goldberg, M. E., Rudolph, R., and Jaenicke, R.
(1991) Biochemistry, 30, 2790-2797.
28.Zhi, W., Landry, S., Gierasch, L. M., and Srere,
P. A. (1992) Protein Sci., 1, 522-529.
29.Woolfolk, C. A., Shapiro, B., and Stadtman, E. R.
(1966) Arch. Biochem. Biophys., 163, 155-162.
30.Hunt, J. B., and Ginsburg, A. (1972)
Biochemistry, 11, 3723-3735.
31.Fisher, M. T. (1994) J. Biol. Chem.,
269, 13629-13636.
32.McCoy, L. F., Rowe, E. S., and Wong, K. P. (1980)
Biochemistry, 19, 4738-4743.
33.Kiefhaber, T., Grunert, H. P., Hahn, U., and
Schmid, F. X. (1992) Proteins. Struct. Funct. Genet., 12,
171-179.
34.Camacho, C. J., and Thirumalai, D. (1996)
Protein Sci., 5, 1826-1832.
35.Ranson, N. A., Burston, S. G., and Clarke, A. R.
(1995) J. Mol. Biol., 250, 581-586.
36.Dill, K. A., and Chan, H. S. (1997) Nat.
Struct. Biol., 4, 10-19.
37.Rudiger, S., Germeroth, L., Schneider-Mergener,
J., and Bukau, B. (1997) EMBO J., 16, 1501-1507.
38.Horowitz, J. (1992) Proc. Natl. Acad. Sci.
USA, 89, 10449-10453.
39.Fenton, W. A., and Horwich, A. L. (1997)
Protein. Sci., 6, 743-760.
40.Gross, M., Robinson, C. V., Mayhew, M., Hartl,
F.-U., and Radford, S. E. (1996) Protein Sci., 5,
2506-2513.
41.Martin, J., and Hartl, F.-U. (1997) Curr.
Opin. Struct. Biol., 7, 41-52.
42.Todd, M. J., Lorimer, G. H., and Thirumalai, D.
(1996) Proc. Natl. Acad. Sci. USA, 93, 4030-4035.
43.Rye, H. S., Burston, S. G., Fenton, W. A.,
Beecham, J. M., Xu, Z., and Horwich, A. L. (1997) Nature,
388, 792-798.
44.Wiessman, J. S., Kashi, Y., Fenton, W. A., and
Horwich, A. L. (1994) Cell, 78, 693-702.
45.Smith, K. E., and Fisher, M. T. (1995) J.
Biol. Chem., 270, 21517-21523.
46.Burston, S. G., Wiessman, J. S., Farr, G. W.,
Fenton, W. A., and Horwich, A. L. (1996) Nature, 383,
96-99.
47.Buchberger, A., Schroder, H., Hesterkamp, T.,
Schonfeld, H. J., and Bukau, B. (1996) J. Mol. Biol.,
261, 328-333.
48.Lubben, T. H., Donaldson, G. K., Viitanen, P. V.,
and Gatenby, A. A. (1989) Plant Cell, 1, 1223-12231.
49.Phadtare, S., Fisher, M. T., and Yarbrough, L. R.
(1994) Biochim. Biophys. Acta, 1208, 189-192.
50.Shrake, A., Fisher, M. T., McFarland, P. J., and
Ginsburg, A. (1989) Biochemistry, 28, 6281-6294.
51.Musgrove, J. E., and Ellis, R. J. (1986)
Philos. Trans. R. Soc. Lond., 313, 419-423.
52.Goloubinoff, P., Gatenby, A. A., and Lorimer, G.
H. (1989) Nature, 337, 44-47.
53.Staniforth, R. A., Burston, S. G., Atkinson, T.,
and Clarke, A. R. (1994) Biochem. J., 300, 651-658.
54.Badcoe, I. G., Smith, C. J., Wood, S., Halsall,
D. J., Holbrook, J. J., Lund, P., and Clarke, A. R. (1991)
Biochemistry, 30, 9195-9200.
55.Schmidt, M., Bucheler, U., Kaluza, B., and
Buchner, J. (1994) J. Biol. Chem., 269, 27964-27972.
56.Lilie, H., and Buchner, J. (1995) Proc. Natl.
Acad. Sci. USA, 92, 8100-8104.
57.Goloubinoff, P., Christeller, J. R., Gatenby, A.
A., and Lorimer, G. H. (1989) Nature, 342, 884-889.
58.Buchner, J., Schmidt, M., Fuchs, M., Jaenicke,
R., Rudolph, R., Schmid, F. X., and Kiefhaber, T. (1991)
Biochemistry, 30, 1586-1591.
59.Martin, J., Langer, T., Boteva, R., Schramel, A.,
Horwich, A. L., and Hartl, F.-U. (1991) Nature, 352,
36-42.
60.Lin, Z., and Eisenstein, E. (1996) Proc. Natl.
Acad. Sci. USA, 93, 1977-1981.
61.Corrales, F. J., and Fersht, A. R. (1996)
Proc. Natl. Acad. Sci. USA, 93, 4509-4512.
62.Jackson, G. S., Staniforth, R. A., Halsall, D.
J., Atkinson, T., Holbrook, J. J., Clarke, A. R., and Burston, S. G.
(1993) Biochemistry, 32, 2554-2563.
63.Yifrach, O., and Horovitz, A. (1995)
Biochemistry, 34, 5303-5308.
64.Hayer-Hartl, M., Martin, J., and Hartl, F.-U.
(1995) Science, 269, 836-841.
65.Roseman, A., Chen, S., White, H., Braig, K., and
Saibil, H. (1996) Cell, 87, 241-251.
66.Xu, Z., Horwich, A. L., and Sigler, P. B.
(1997) Nature, 388, 741-750.
67.Lin, Z., Schwarz, F. P., and Eisenstein, E.
(1995) J. Biol. Chem., 270, 1011-1014.
68.Schmidt, M., Buchner, J., Todd, M. J., Lorimer,
G. H., and Viitanen, P. V. (1994) J. Biol. Chem., 269,
10304-10311.
69.Wiessman, J. S., Hohl, C. M., Kovalenko, O.,
Kashi, Y., Chen, S., Braig, K., Saibil, H. R., Fenton, W. A., and
Horwich, A. L. (1995) Cell, 83, 577-588.
70.Zheng, X., Rosenberg, L. E., Kalousek, K., and
Fenton, W. A. (1993) J. Biol. Chem., 268, 7489-7493.
71.Miller, A. D., Maghlaoui, K., Albanese, G.,
Kleinjan, D. A., and Smith, C. (1993) Biochem. J., 291,
139-144.
72.Wynn, R. M., Davie, J. R., Cox, R. P., and
Chuang, D. T. (1992) J. Biol. Chem., 267,
12400-12403.
73.Mendoza, J. A., Rogers, E., Lorimer, G. H., and
Horowitz, P. M. (1991) J. Biol. Chem., 266,
13044-13049.
74.Hayer-Hartl, M. J., Weber, F., and Hartl, F.-U.
(1996) EMBO J., 75, 6111-6121.
75.Burston, S. G., Ranson, N. A., and Clarke, A. R.
(1995) J. Mol. Biol., 249, 138-152.
76.Fisher, M. T., and Yuan, X. (1994) J. Biol.
Chem., 269, 29598-29601.
77.Epstein, C. J., and Anfinsen, C. B. (1962) J.
Biol. Chem., 237, 2175-2179.
78.Creighton, T. E. (1985) in Protein Structure,
Folding and Design (Oxender, D. L., ed.) Alan R. Liss, N. Y., pp.
249-257.
79.Lorimer, G. (1997) Nature, 338,
720-722.
80.Addinall, S. G., and Lutkenhaus, J. (1997)
Annu. Rev. Biochem., 66, 93-116.
81.Clark, A. C., Hugo, E., and Frieden, C. (1996)
Biochemistry, 35, 5893-5901.
82.Lorimer, G. H. (1996) FASEB J., 10,
5-9.
83.Horwich, A. L., Low, K. B., Fenton, W. A.,
Hirschfield, I. N., and Furtak, K. (1993) Cell, 74,
909-917.
84.Ewalt, K. L., Hendrick, J. P., Houry, W. A., and
Hartl, F.-U. (1997) Cell, 90, 491-500.
85.Frieden, C., and Clark, C. A. (1997) Proc.
Natl. Acad. Sci. USA, 94, 5535-5538.
86.Yifrach, O., and Horovitz, A. (1996) J. Mol.
Biol., 255, 356-361.
87.Inbar, E., and Horovitz, A. (1997)
Biochemistry, 36, 12276-12281.