REVIEW: Chaperones in Bacteriophage T4 Assembly
E. I. Marusich1, L. P. Kurochkina1,2, and V. V. Mesyanzhinov1,2*
1Bakh Institute of Biochemistry, Russian Academy of Sciences, Leninskii pr. 33, Moscow, 117071 Russia2Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 336-6022; E-mail: vvm@ibch.siobc.ras.ru
* To whom correspondence should be addressed.

|
||
| E. I. Marusich | L. P. Kurochkina | V. V. Mesyanzhinov |
Received October 10, 1997
Protein folding in the cell is controlled at the levels of translation and post-translational modification, depends on a number of conserved proteins known as chaperones, and is catalyzed by specific enzymes, such as protein disulfide isomerase and peptidyl prolyl cis-trans isomerase. The chaperones stabilize folding intermediates and participate in assembly and disaggregation of supramolecular structures. Bacteriophage T4 is an especially convenient system for studying of protein folding mechanisms, since its genome encodes several virus-specific chaperones. In this review, the chaperones of phage T4 that take part in capsid formation (gp31 and gp40) and in folding and assembly of virion tail fibers (gp38, gp57A) have been considered. Protein encoded by gene 31 completely substitutes co-chaperonin GroES of the host cell in folding of the major capsid protein, gp23, aided by chaperonin GroEL. The product of gene 40, which is homologous to analogs of eukaryotic GroEL and peptidyl prolyl cis-trans isomerase, participates in assembly of gp20 while the formation of procapsid connector. The chaperone encoded by gene 57A is essential for folding and oligomerization of both long and short phage tail fibers. gp38, together with gp57A, participates in the formation of the distal part of the long fibers. This protein seems to represent a principally new group of chaperones that change steric structure of folded polypeptide. One phage chaperone, fibritin, encoded by gene wac (whiskers antigen control) and taking part in assembly the subunits of the long tail fibers is a constituent of the virion. Fibritin is a convenient model for studying mechanisms of folding and oligomerization of fibrous proteins due to its labile triple-stranded alpha-helical coiled-coil structure.
KEY WORDS: folding, molecular chaperone, chaperonin, bacteriophage T4, capsid, fibrous proteins
Abbreviation: gp) gene product.
The problem of the spatial protein structure formation is one of the key
questions in molecular biology and biochemistry. Details of folding of
a nascent polypeptide chain from a large number of sterically possible
states to the native conformation are still incompletely clear. The
information contained in amino acid sequence was initially suggested to
be sufficient for proper folding of a protein molecule. However, as
experimental data accumulated, particularly when molecular chaperones
were found, it became clear that protein folding in the cell is a
complex process which is controlled at the levels of translation and
post-translational modification, assisted by protein helpers called
chaperones, and catalyzed by specific enzymes, such as protein
disulfide isomerase and peptidyl prolyl cis-trans isomerase [1-5].
Chaperones take part in folding of multidomain proteins and are not commonly included in a final functional structure. The term "molecular chaperone" was firstly proposed by Laskey et al. for nucleoplasmin responsible for nucleosome assembly [6]. Recent data classify molecular chaperones more widely as proteins that recognize and stabilize partially assembled intermediates while polypeptides fold, assemble, and disaggregate.
Viruses of bacteria are particularly convenient systems to study regulation of protein folding in the cell. Investigations of them revealed much important information in molecular biology. Firstly, genetic evidences of existence of chaperones were initially obtained while bacteriophages were being studied: GroEL gene product of Escherichia coli was identified as a host factor necessary for capsid assembly of phages T4 and lambda [7, 8]. Secondly, folding mutations called tsf (temperature-sensitive folding) [9], were initially studied in detail on proteins of bacterial viruses. Tsf-mutations belonging to tss class (temperature-sensitive synthesis) destabilize folding intermediates, not altering function and thermostability of native protein that folds properly at permissive temperature.
E. coli bacteriophage T4 is a complex supramolecular machine composed of more than 50 products of various genes that form three main substructural components of a viral particle. A single DNA molecule coding more than 250 genes is packed into the capsid [10]. The contractile tail with the baseplate at its end ensures viral DNA infection into the host cell. Two kinds of tail fibers, short and long, provide specific virus recognition of cell surface receptors and irreversible adsorption. As seen from Fig. 1, phage particle morphogenesis branches on three parallel processes including, respectively, assembly of capsid, tail, and tail fibers. All viral structural proteins encoded by groups of late genes are synthesized almost simultaneously, and phage morphogenesis regulation is generally based on a mechanism of successive induction of conformational changes of interacting protein molecules [11]. Besides, because of morphogenesis process complexity, a group of protein helpers takes part in folding and assembly regulation of individual virus components. These phage-specific chaperones are products of genes 31, 38, 40, 57A, and 63. Protein encoded by gene 31 interacts with the host chaperonin GroEL and prevents aggregation of the major capsid protein, gp23 [12]. The product of gene 40 takes part in gp20 assembly and aids formation of connector that is the initiative complex of procapsid assembly [13].
The T4 tail fibers are complex structures and they are likely to have a novel fold. Folding and oligomerization of these fibrous proteins are aided by several phage-specific protein-chaperones. Functions of phage fibers, steps of their assembly and attachment to viral particle are largely clear. However, the action mechanism of phage-specific chaperones remains obscure. gp38 participates in formation of the distal part of the long tail fibers [14]. In addition to gp38 action, another phage chaperone encoded by gene 57A is necessary for folding and oligomerization of both the proximal part of long tail fibers and short tail fibers [15]. gp63, a bifunctional protein with RNA-ligase activity, accelerates connection of the tail fibers to the baseplate [16].Fig. 1. Pathway of bacteriophage T4 morphogenesis. Genes participating in assembly are numbered. Chaperones that are not part of the final structure are presented in parentheses.
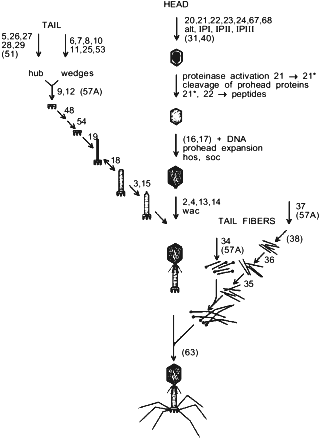
Here we consider chaperones participating in capsid formation (gp31 and gp40) as well as in tail fibers assembly (gp38, gp57A). Moreover, one more phage chaperone is known, which is the product of gene wac (whisker antigen control), or fibritin, being a structural component of phage particles. Fibritin accelerates assembly of the distal and proximal halves of long fibers and attachment of assembled fibers to the baseplate. Recently we have completed determination of spatial structure of fibritin [17], which explained the features of this chaperone functioning. At the same time, fibritin itself proved to be an attractive model to study folding and oligomerization mechanisms of a large class of fibrous proteins [18].
PHAGE T4 CAPSID FORMATION
Interaction of cell chaperonin GroEL and phage T4 gp31. As seen from Fig. 1, not less than 25 genes products take part in phage T4 capsid morphogenesis. Capsid formation is initiated from connector (gp20) assembly on the cell membrane, with procapsid core including main protein (gp22), a number of minor proteins (gp67, gp68, gpalt, IPI, IPII, IPIII), and virus-specific proteinase (gp21) polymerizing on the connector. On the core surface gp23 assembles the major procapsid protein. Its folding is aided by E. coli GroEL chaperonin and phage co-chaperonin encoded by gene 31.
Folding-active GroEL is an oligomer containing 14 identical subunits (with molecular weight of ~57 kD) that form two rings with 7-fold axis disposed with the diadic symmetry relatively to each other. GroEL-assisted protein folding proceeds while interacting with GroES-co-chaperonin (97 amino acid residues, molecular weight of 10 kD), whose subunits form a dome-shaped heptamer [4]. Chaperonin-assisted protein folding is supposed to proceed according to the Anfinsen model ("Anfinsen cage") [19] and to be carried out within a sequestered compartment forming when GroEL and GroES associate. They interact at the cost of the GroEL flexible loop consisting of 16 amino acid residues [12, 20]. According to the NMR data, this loop is disarrayed in the free heptamer but structured in the GroES/GroEL complex [21]. Basic contacts with GroEL are provided by a cluster of hydrophobic residues Ile-25--Val-26--Leu-27.
T4 protein encoded by gene 31, gp31 (111 amino acid residues, molecular weight of ~12 kD), completely replaces E. coli co-chaperonin GroES during bacteriophages lambda and T5 morphogenesis as well as chaperonin-assisted folding of ribulose bisphosphate decarboxylase in vivo and in vitro [22]. Like GroES, gp31 forms a stable complex with GroEL chaperonin in the presence of Mg-ATP and inhibits GroEL ATPase activity in vitro [22]. gp31, together with E. coli GroEL chaperonin, is absolutely necessary for folding in vivo of phage T4 major capsid protein, gp23 [23]. With mutations in both gene 31 and gene groEL amorphous aggregates of gp23 accumulate onto the cell membrane [13]. Moreover, mutations in gene groEL may be suppressed by additional mutations in gene 31 [24, 25].
gp31 and GroES are similar in molecular weight; both are acidic proteins (pI 4.88 for gp31 and 4.71 for GroES) and have anomalous electrophoretic mobility in denaturing polyacrylamide gel (Mr ~16,000 for gp31 and ~15,000 for GroES). Although the amino acid sequences of the two proteins do not have pronounced homology, gp31 also includes a highly flexible loop (Gln-24--Val-45) that can adopt the same beta-hairpin conformation when interacting with GroEL as the loop of GroES [22]. gp31 binding to GroEL proceeds at the cost of that loop, which is confirmed by the following facts: a) addition of GroEL and Mg-ATP results in proteolysis site protection in the gp31 loop [22]; b) all mutations in gene 31 that recover GroEL-assisted folding of gp23 with defected GroEL are situated in the sequence of gp31 flexible loop [23].
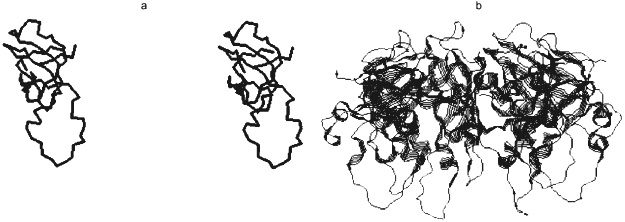
Crystallographic 2.3 Å resolution data showed that in spite of weak homology of gp31 and GroES amino acid sequences (only 14% identity), the spatial structures of these proteins were principally similar (Fig. 2) [26]. Active gp31 also is a heptamer and has toroidal shape; however, the opening diameter in gp31 heptamer is more (26 Å) than in GroES (16 Å). Amino acid residues forming the opening of the toroid are different in their nature: preferentially hydrophobic in GroES and hydrophilic in gp31. The last property may cause immediate participation of gp31 in gp23 folding. More extended high-flexible loop of gp31 compared to GroES results in cavity height increase in the GroEL/gp31 chaperonin complex, which is crucial for the folding of capsid protein gp23 (molecular weight of 56 kD) that, probably, cannot be accommodated into the smaller size cavity of GroEL/GroES chaperonin.
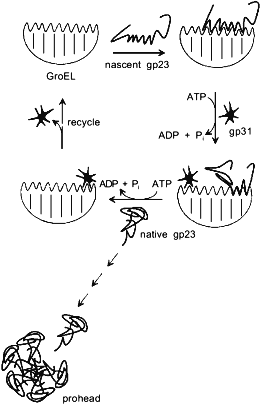
By analogy with functioning of the GroEL/GroES chaperonin complex, a model of GroEL/gp31-assisted folding of gp23 has been proposed (Fig. 3) [23]. According to this model, GroEL interacts with nascent chain of gp23, preventing its inappropriate aggregation. One of the functions of gp31 seems to be effective release of gp23 from the chaperonin by means of ATP hydrolysis. It is also possible that the GroEL/gp31 complex contributes to the formation of gp23 dimers [27] and properly folded polypeptide of gp23 forms procapsid lattice at the core surface. Since single procapsid assembly requires 960 copies of gp23 and as high as 200 phage particles are generated into one infected cell, then, assuming a 50% efficiency of assembly, between 2·105 and 4·105 molecules of gp23 must be synthesized per cell. The average number of GroEL monomers per cell at 37°C is about 104 [28], which results in formation of 700 GroEL decatetramer structures. Therefore, during gp23 folding, GroEL effectively recycles.Fig. 2. X-Ray structure of gp31 [26]. a) Stereopair of gp31 monomer. Disposition of C-alpha atoms is shown. The mobile loop is in the lower part of the structure. b) gp31 heptamer projection.
The main question why it is gp31 that is required for gp23 folding rather than GroES co-chaperonin is still unsolved. Several phage T4 gene 23 mutants have been selected that do not required gp31 co-chaperonin for proper folding of gp23 [29]. Analysis of mutations points out of availability of a region within gp23 whose deletion results in less effective GroEL/gp31-aided folding of gp23 both in vivo and in vitro. Previously we proposed that gp23 folding anomalies may be related to a translational pause during gp23 synthesis [5]. However, extended site-directed mutagenesis recently performed by L. Black's laboratory has not found such a pause [29]. Since gp31 does not participate in gp23 binding to GroEL and GroES is not required for gp23 folding, co-chaperonin gp31 may form local supersecondary structure which interacts with GroEL [29]. Although the spatial structures of GroEL, GroES, and gp31 chaperonins are resolved, principal difficulties of interpretation of the gp31 action mechanism are related to the absence of detailed information on the structure of the major capsid protein--gp23.Fig. 3. Putative model of gp23 folding aided by gp31 and GroEL [23].
The role of gp40 in phage T4 capsid assembly. gp40 is required for assembly of procapsid initiative complex--connector formed by gp20 [30, 31] (Fig. 1). It is interesting that gp40 is mapped into the group of genes responsible for replication of phage T4 DNA and it is situated at a great distance away from the main genes controlling capsid morphogenesis.
Capsid connector is a gp20 oligomer (molecular weight of the monomer is 65 kD). Twelve subunits of gp20 assemble into the ordered structure (diameter of 18 nm) with a 12-fold rotational symmetry, forming the central opening of 3 nm in diameter [32]. Two such disks associate with each other and form double ring structure. That complex is compatible to phage tail that has the 6-fold symmetry.
The connector has two functions. First, assembly of the procapsid core is initiated from that, then the major capsid protein, gp23, polymerizes onto the core. Second, the connector situated in the portal vertex of procapsid is a channel through which viral DNA is packed into the capsid. After finishing DNA packing, other structural proteins of phage (gp2, gp4, gp13, gp14, gp50, gp64, gp65) attach to the connector. Then the tail attaches to the complicated connector (phage neck).
In spite of available genetic evidence of interaction of gp40 with gp20, the action mechanism of gp40 is still unknown [33]. gp40 is necessary for phage reproduction only at high temperature; however, at all temperatures it is a supplementary factor for gp20 effective attachment to the membrane [34, 35]. gp40 is a small protein with molecular weight of 13.3 kD which might form oligomer complexes. When amino acid sequence of gp40 was analyzed, homologous regions with two eukaryotic chaperones are found that are cyclofilin C (an analog of peptidyl prolyl cis-trans isomerase) and CPN 1 protein (an analog of GroEL) (Mesyanzhinov, unpublished data). Moreover, gp40 is a pleiotropic protein and it has suppressive effect on mutations in gene e coding for phage T4 lysozyme [36].
TAIL FIBERS FOLDING AND ASSEMBLY
Phage T4 infection process in E. coli cells is initiated from recognition of receptors onto the host cell surface by carboxyterminal region of the distal part of the long tail fibers encoded by gene 37 [37-40]. Then, with the aid of the short tail fibers encoded by gene 12, irreversible virus adsorption occurs [41]. Complex application of techniques of electron microscopy, image processing, and computer analysis revealed features of structural organization of tail fibers [42]. Bacteriophage T4 long fibers consist of two halves, proximal and distal, each having a length about 75 nm. Three copies of gp34 (140 kD) form the proximal half attached to the baseplate and three copies of gp37 (109 kD), one molecule of gp35 (30 kD), and two copies of gp36 (23 kD) form the distal half [42]. Long fibers have domain structure and contain not less than 17 domains. The globular domain of gp34 which is responsible for the connection of the fiber proximal half to the baseplate is followed by a relatively plane region that seems to contain a cluster of 7 quasi-repeats, each of 34-39 amino acid residues. The proximal half is ended by three globular domains. The fiber distal part includes 10 globular domains, various in size and distance between them, and is ended by a rigid tip.
Three identical chains of gp34 and gp37 within each of fiber halves are packed in parallel and seem to form a novel structure type--beta-helix, like structure of P22 phage adhesin [43]. Phage T4 carboxyterminal region of gp37 molecule forms the tip of the fiber distal part that interacts with cell receptors [44, 45]. Assembly and trimerization of both distal and proximal halves of fibers are aided by chaperones encoded by genes 57A and 38.
Short phage fibers, which we name adhesin, also are trimers of packed in parallel gp12 subunits. Besides the main function of irreversible attachment of phage to E. coli cell, short fibers take part in formation of a specific channel through the host cell membrane and, while added to E. coli in purified state, they kill the cell. Short fibers, which are 38 nm in length, as well as long fibers, have domain structure [46]. Globular domain (10 nm in length, 6 nm in diameter) is followed by a plane shaft with a length of 24 nm and 3.8 nm in diameter and a short rigid tip at the end. The sequence of gp12 (526 amino acid residues) contains, as well as gp34 sequence, a tandem of quasi-repeats of about 40 amino acids in each that are situated between 48 and 320 amino acid residues [46].
Interaction of chaperone gp57A with the tail fibers. Processes of assembly of tail fibers distal and proximal parts occur in vivo simultaneously. Assembly of the distal part is the most complex. First, three copies of gp37 form its precursor, then two molecules of gp36 are attached to the N-terminus of the gp37 trimer, extending the distal part by 20%. Then one molecule of gp35 is attached to gp36, providing following interaction with the proximal part.
Chaperone gp57A takes part in assembly of both halves of the long fibers and the short tail fibers. Phage T4 DNA sequence of the region of gene 57 includes two open reading frames coding for proteins gp57A and gp57B with molecular weight of 6 and 18 kD, respectively [47]. However, only gp57A is essential for phage growth. Active in fiber assembly protein seems to be a tetramer with molecular weight of 33.9 kD [48]. gp57A amino acid sequence is unusual in its own way unique in the prokaryotic world. gp57A polypeptide chain containing only 79 amino acid residues has the excess of nine negatively charged amino acids that make it very acidic. Moreover, it lacks Pro, Cys, His, and aromatic residues Phe, Trp, and Tyr. Analysis of secondary structure by circular dichroism technique showed that gp57A was alpha-helical protein. When expressed from plasmid, the protein forms unusual extended crystal-like structures and is toxic for cells.
Some attempts to study interaction of gp57A with gp12 during short fiber formation have been undertaken. In the absence of gp57A, gp12 accumulates onto the membrane in the form of inclusion bodies (Marusich, unpublished data). However, when gene 12 and gene 57A were co-expressed, recombinant gp12 assembled into trimers, though remaining associated with the cell membrane. EDTA treatment released gp12 readily from the membranes [48].
Participation of gp38 in tail fiber oligomerization and assembly. gp38 is necessary for assembly of the distal part of the long fibers. Gene 38 of phage T4 codes for a protein containing 183 amino acid residues with molecular weight of 22.3 kD [49]. Together with genes 36 and 37, whose products are structural proteins of the fiber distal part, gene 38 forms one transcription unit. Analysis of gene 38 sequences of other T-even phages--T2, K3, Ox2, and M1 [50]--showed that they coded for protein strongly differing from the protein of phage T4, both structurally and functionally. Moreover, it turned out that gp38 of T2, K3, Ox2, and M1 phages was a structural a component of the phage particle; it was attached immediately to the distal ends of the long fibers and responsible for phage association with cell receptors. However, gp38 of phage T4 is a chaperone and it is not included into the phage particle [51]. Phages K3, Ox2, and M1 use membrane protein of E. coli OmpA as a receptor, phage T2--proteins OmpF and Ttr, and phage T4--porin OmpC.
Within the sequence of gp38 of all bacteriophages related to T4, three domains are distinguished. First 120 N-terminal amino acids and 25 C-terminal amino acids are conservative regions. The sequence between them is highly variable with many amino acid substitutions as well as insertions and deletions. Conservative regions of the polypeptide chain are rich in Gly residues that seem to be essential for recognition of cell receptors. There is significant homology between gene 37 C-end of phages T2, K3, Ox2, and M1 and gene 38 of phage T4, this region of gene 37 missing in phage T4. Therefore, phage T4 is considered to borrow gene 38 from C-terminal part of gene 37 of T2-like bacteriophage [50].
The carboxyterminal domain of gp37 of phages TuIa and TuIb, which are also related to phage T4, recognizes the same E. coli cell receptors--porin OmpC--as phage T4. Mutants of phages TuIa and TuIb have been obtained that can recognize other receptors, not OmpC [44]. Since all amino acid substitutions of these mutants are located within the variable region of gp37, the authors concluded that this region was directly responsible for association with cell receptors. Moreover, conditionally lethal mutants with lowered ability to adsorb have been obtained that, under non-permissive conditions (42°C), form phage with normal morphology. All substitutions of these mutants are located in conservative regions of the C-terminal domain. One of the mutants (L43) has completely lacked the ability to use traditional for phage T4 porin OmpC as a receptor. To explain these peculiarities, the hypothesis of "snap-back" conformation of ts-mutants tail fiber has been proposed: C-terminal region of gp37 may fold back.
An unusual gene 38 point mutant has been obtained that increases infectivity of phage T4 gene 37 ts-mutants. This observation points out that gp38 can change the conformation of receptor-recognizing domain of gp37 [44]. It is known that gene tfa of phage lambda complements phage T4 gene 38 amber-mutants, whose sequences have 40% homology. It turned out that gptfa could restore the ability of phage T4 L43 mutant to recognize porin OmpC. Consequently, one of functions of chaperones gp38 and gptfa may be altering steric conformation of the final polypeptide.
Phage T4 mutant has been isolated containing a region with duplicated sequence of gp37 amino acid residues from 797 to 805 that interacts with gp38 [52]. In that mutant, assembly of the distal part of the long tail fibers is violated. This fact also confirms that gp38 acts as a specific chaperone during assembly rather than covalently modifies gp37.
BACTERIOPHAGE T4 FIBRITIN AS A CHAPERONE
One of the objects studied in our laboratory, phage T4 fibritin, is a chaperone encoded by late gene wac (whisker antigen control). The singularity of this protein is that, providing assembly of the halves of the long tail fibers, it is a structural component of the viral particle. Fibritin forms thin strings--“whiskers” with a length of about 50 nm joined to the lower part of the capsid (neck) [18]. Fibritin accelerates assembly of the distal and proximal parts of the long fibers and seems to act as a bicomplementary matrix, increasing the number of interacting sites. Fibritin also is a primitive molecular sensor. Under unfavorable for phage growth conditions (at low temperature or pH) it holds the long fibers pressed to the tail and capsid. Such phage particles are not infectious because the long fibers in the lifted and fixed state cannot bind to the receptor.
Analysis of fibritin amino acid sequence of 486 residues has found extended heptad repeats that are typical for "coiled coil" alpha-helical proteins [53]. Fibritin sequence consists of three distinguished modules: the long central region and small N- and C-terminal domains. The central module includes heptad periodicity of hydrophobic residues typical for coiled coil superhelical proteins. However, a singularity of fibritin is that its coiled coil part is segmented. Short fragments of the polypeptide chain situated between 12 regular "coiled coil" regions are rich in Gly and Pro residues and resemble loops of globular proteins. N- and C-terminal domains of the protein (47 and 29 residues, respectively) do not contain heptad repeats. CD-spectroscopy showed that fibritin included about 90% alpha-helices. Data of equilibrium centrifugation and reassociation of mutant proteins in vitro pointed out that fibritin was a trimer and calorimetric studies showed that the protein contained not less than 5 domains melting at different temperatures [53].
Recently we have obtained crystals of two mutants: fibritin E containing the last 119 C-terminal amino acid residues of the monomer and fibritin M (respectively, 75 residues), which are suitable for X-ray analysis [17]. At present, crystallographic studies of the two proteins are completed in collaboration with professor M. Rossmann's laboratory (Purdue University, USA) with support of a grant of Howard Hughes Medical Institute.
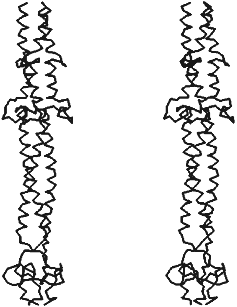
X-Ray analysis has shown that fibritin does belong to the class of proteins constructed of three parallel alpha-helices like fibrinogen, laminin, influenza virus hemoagglutinin, and macrophage scavenger receptor protein. Three identical subunits of fibritin E associate into alpha-helical "coiled coil" along the 3-fold crystallographic axis (Fig. 4). Our predictions about segmented structure of fibritin also were true. Fibritin E includes two "coiled coil" segments and two loops, L11 and L12, each of these begins and ends by Gly residues, which provides flexibility to the molecule in loop inclusion sites. However, the sequence of the three last "coiled coil" segments of fibritin in arrangement of hydrophobic residues in a and d positions is closer to a dimer protein. Therefore, superhelices packing in fibritin is less tight (4.42 Å) compared to other trimer "coiled coil" as, for example, isoleucine zipper (3.95 Å). Analysis of parameters and geometry of packing of the molecule [17] has shown that fibritin "coiled coil" part, as in the case of influenza virus hemagglutinin, is labile. Such lability of fibritin seems to ensure flexibility of the molecule and its local conformational changes, which is necessary for its functioning as a chaperone while interacting with the long fibers.
The carboxyterminal domain of the molecule contains two short antiparallel beta-strands in each chain of the trimer. In contrast to the labile "coiled coil" part, three symmetrical beta-hairpins, each of those has five H-bonds in the main chain of the monomer, form stable structure like a beta-propeller. When expressed in the cell, the carboxyterminal domain determines proper folding of fibritin into the native structure, ensuring, obviously, alignment of the monomers and their following trimerization and "coiled coil" formation. We name that domain in fibritin monomer “foldon”, whose stable secondary structure should form before the rest of the molecule. As the results of site-directed mutagenesis have showed, point mutations in foldon results in violation of protein folding and formation of aggregates like inclusion bodies which are often formed when improper protein folding in the cell [5, 53]. In the structure of the C-terminal domain, interactions of side-chains of aromatic residues, in particular Trp-476, are essential. Substitutions of Trp-476 by Leu or Ser residues completely blocks fibritin trimerization. The principle of initiation of fibritin folding and oligomerization at the cost of the stable carboxyterminal foldon seems to be carried out with other fibrous proteins too.Fig. 4. Stereopair of X-ray structure of fibritin E [17]. Disposition of C-alpha atoms is shown.
We are grateful to the Russian Foundation for Basic Research (grants 96-04-48035 and 97-04-49708), Protein Engineering Council and Genetics Council of the Russian Federation Science Department as well as Howard Hughes Medical Institute for support of studies of our laboratory. We thank Dr. J. Hunt for kindly provided coordinates of gp31 atoms.
REFERENCES
1.Kim, P. S., and Baldwin, R. L. (1990) Annu. Rev.
Biochem., 59, 631-652.
2.Gething, M.-J., and Sambrook, J. (1992)
Nature, 355, 33-45.
3.Hartl, F. U. (1996) Nature, 381,
571-580.
4.Martin, J. (1998) Biochemistry (Moscow),
63, 374-381.
5. Kurochkina, L. P., and Mesyanzhinov, V. V. (1996)
Uspekhi Biol. Khim., 36, 49-86.
6.Laskey, R. A., Honda, B. M., Mills, A. D., and
Finch, J. T. (1978) Nature, 275, 416-420.
7.Georgopoulos, C. P., Hendrix, R. W., Casjens, S.
R., and Kaiser, A. D. (1973) J. Mol. Biol., 76,
45-60.
8.Sternberg, N. (1973) J. Mol. Biol.,
67, 1-24.
9.King, J., Haase, C., and Yu, M.-H. (1994) in
Protein Engineering (Oxender, D. L., and Fox, C. F., eds.) Alan
R. Liss, New York, pp. 109-118.
10.Kutter, E., Stidham, T., Guttman, B., Kutter, E.,
Batts, D., Peterson, S., Djavakhishvili, T., Arisaka, F., Mesyanzhinov,
V., Ruger, W., and Mosig, G. (1994) in Molecular Biology of
Bacteriophage T4 (Karam, J. D., ed.) American Society for
Microbiology, Washington, DC, pp. 491-518.
11.Kellenberger, E. (1990) Eur. J.
Biochem., 190, 233-248.
12.Zeilstra-Ryalls, J., Fayet, O., and Georgopoulos,
C. P. (1991) Annu. Rev. Microbiol., 45, 301-325.
13.Laemmli, U. K., Beguin, F., and
Kellenberger-Gujer, K. G. (1970) J. Mol. Biol., 47,
69-85.
14.Ward, S., and Dickson, R. C. (1971) J. Mol
Biol., 62, 479-492.
15.Dickson, R. C. (1973) J. Mol. Biol.,
79, 633-647.
16.Wood, W. B., Conley, M. P., Lyle, H. L., and
Dickson, R. C. (1978) J. Mol. Biol., 253, 2437-2445.
17.Tao, Y., Strelkov, S. V., Mesyanzhinov, V. V.,
and Rossmann, M. G. (1997) Structure, 5, 789-798.
18.Mesyanzhinov, V. V. (1997) Mol. Biol.
(Moscow), 31, 389-397.
19.Ellis, R. J. (1994) Curr. Biol., 4,
633-635.
20.Xu, Z., Horwich, A. L., and Sigler, P. B. (1997)
Nature, 388, 741-750.
21.Zeilstra-Ryalls, J., Fayet, O., and Georgopoulos,
C. P. (1994) J. Bacteriol., 176, 6558-6565.
22.Landry, S. J., Zeilstra-Ryalls, J., Fayet, O.,
Georgopoulos, C. P., and Gierasch, L. M. (1993) Nature,
364, 255-258.
23.Van der Vies, S. M., Gatenby, A. A., and
Georgopoulos, C. P. (1994) Nature, 368, 654-656.
24.Georgopoulos, C. P., Hendrix, R. W., Kaiser, A.
D., and Wood, W. B. (1972) Nature (London) New Biol.,
239, 38-41.
25.Keppel, F., Lipinska, B., Ang, D., and
Georgopoulos, C. P. (1990) Gene, 86, 19-25.
26.Hunt, J. F., van der Vies, S. M., Henry, L., and
Deisenhofer, J. (1997) Cell, 90, 361-371.
27.Muller, M., Mesyanzhinov, V. V., and Aebi, U.
(1994) J. Struct. Biol., 112, 199-215.
28.Neidhardt, F. C., VanBogelen, R. A., and Vaughn,
V. (1984) Annu. Rev. Genet., 18, 295-329.
29.Simon, L. D., and Randolf, B. (1984) J.
Virol., 51, 321-328.
30.Eiserling, F. A., Geiduschek, P., and Metter, E.
J. (1970) J. Mol. Biol., 6, 865-876.
31.Marusich, E. I., and Mesyanzhinov, V. V. (1989)
Nucleic Acids Res., 17, 7514.
32.Black, L. W., and Showe, M. K. (1983) in
Bacteriophage T4 (Mathew, C. K., Kutter, E., Mosig, G., and
Berget, P. B., eds.) American Society for Microbiology, Washington, pp.
219-245.
33.Michaud, C., Zahary, A., Rao, V. B., and Black,
L. W. (1989) J. Mol. Biol., 209, 667-681.
34.Hsiao, C. L., and Black, L. W. (1978)
Virology, 91, 1-14.
35.Hsiao, C. L., and Black, L. W. (1978)
Virology, 91, 15-25.
36.Brown, S. M., and Eiserling, F. A. (1979)
Virology, 97, 68-76.
37.Wood, W. B., and Revel, H. R. (1976)
Bacteriol. Rev., 40, 847-868.
38.Kellenberger, E., Bolle, A., Boy de la Tour, E.
B., Epstein, R. H, Franklin, N. C., Jerge, N. K., Reale-Scafati, A.,
Sechaud, J., Berget, I., Goldstein, D., and Lauffier, M. A. (1965)
Virology, 26, 419-440.
39.Beckendorf, S. K., Kim, J. S., and Leilausis, I.
(1973) J. Mol. Biol., 73, 17-35.
40.Wood, W. B., Eiserling, F. A., and Crowther, R.
A. (1994) in Molecular Biology of Bacteriophage T4 (Karam, J.
D., ed.) American Society for Microbiology, Washington, pp.
282-290.
41.Kells, S. S., and Haselkorn, R. (1974) J. Mol.
Biol., 83, 473-485.
42.Cerritelli, M. E., Wall, J. S., Simon, M. N.,
Conway, J. F., and Steven, A. C. (1996) J. Mol. Biol.,
260, 767-780.
43.Steinbacher, S., Secler, R., Miller, S., Steipe,
B., Huber, R., and Reinemer, P. (1994), Science, 265,
383-386.
44.Hashemolhosseini, S., Montag, D., Kramer, L., and
Henning, U. (1994) J. Mol. Biol., 142, 524-533.
45.Montag, D., Hashimolhosseini, S., and Henning, U.
(1990) J. Mol. Biol., 216, 327-334.
46.Makhov, A. M., Trus, B. L., Conway, J. F., Simon,
M. N., Zurabishvili, T. G., Mesyanzhinov, V. V., and Steven, A. C.
(1993) Virology, 194, 117-127.
47.Herrmann, R. (1982) Nucleic Acids Res.,
10, 1105-1115.
48.Matsui, T., Griniuviene, B., Goldberg, A.,
Tsugita, A., Tanaka, N., and Arisaka, F. (1997) J.
Bacteriol., 179, 1846-1851.
49.Oliver, D. B., and Crowther, R. A. (1981) J.
Mol. Biol., 153, 545-568.
50.Montag, D., Riede, I., Eschbach, M.-L., Degen,
M., and Henning, U. (1987) J. Mol. Biol., 196,
165-174.
51.Riede, I., Drexler, K., Schwarz, H., and Henning,
U. (1987) J. Mol. Biol., 194, 23-30.
52.Tetard, F., Repoila, F., Monod, C., and Krisch,
H. M. (1996) J. Mol. Biol., 258, 726-731.
53.Efimov, V. P., Nepluev, I. V., Sobolev, B. N.,
Zurabishvili, T. G., Schulthess, T., Lustig, A., Engel, J., Haener, M.,
Aebi, U., Venyaminov, S. Yu., Potekhin, S. A., and Mesyanzhinov, V. V.
(1994) J. Mol. Biol., 242, 470-486.