REVIEW: The Versatile and Complex Enzymology of Nitric Oxide Synthase
A. C. F. Gorren* and B. Mayer
Institut für Pharmakologie und Toxikologie, Karl-Franzens-Universität Graz, Universitätsplatz 2, A-8010 Graz, Austria; fax: 43-316-380-9890; E-mail: antonius.gorren@kfunigraz.ac.at* To whom correspondence should be addressed.
Received August 8, 1997
The biogenesis of nitric oxide is catalyzed by nitric oxide synthase (NOS) which forms L-citrulline and NO from L-arginine. Here we review the enzymology of NOS. We discuss its modular structure, its prosthetic groups and cofactors, and we provide a brief account of present knowledge regarding cellular targeting and regulation of the different isoforms. The various reactions which are catalyzed by NOS are reviewed, and an inventory of different inhibitor types is given. Special attention is paid to the role of the cofactor tetrahydrobiopterin (BH4) and of the dimeric structure, and to the possibility that the main product of NOS catalysis under some conditions may not be NO. Based on a number of recent observations, we postulate that neuronal NOS with one equivalent of BH4 per dimer (a state which may be physiologically relevant) catalyzes the concerted formation of peroxynitrite.
KEY WORDS: nitric oxide synthase, enzymology, tetrahydrobiopterin, uncoupling, dimer, peroxynitrite
Abbreviations: nNOS, eNOS, and iNOS) neuronal, endothelial, and inducible nitric oxide synthase, respectively; OH-Arg) N-hydroxy-L-arginine; BH4) (6R)-5,6,7,8-tetrahydro-L-biopterin; 4-amino-BH4) (6R)-2,4-diamino-5,6,7,8-tetrahydro-6-(L-1,2-dihydroxypropyl)pteridine; sGC) soluble guanylate cyclase; cGMP) cyclic guanosine-5´-monophosphate; DPI) diphenylene iodonium; DCPIP) 2,6-dichlorophenol-indophenol; PIN) protein that inhibits NOS.
The existence of an enzyme that produces nitric oxide from L-arginine
was only surmised in the late 1980s, and purification of the enzyme
dates back no further than 1990. Yet studies involving nitric oxide
synthase (NOS) have taken such a flight in the short time that has
passed since then that currently NOS must be one the best studied
enzymes. It certainly ranks among the most reviewed ones, which is why
we will not exhaustively discuss all aspects of NOS, but focus on some
properties while glossing over others. Older more comprehensive
reviews, and recent ones on other aspects are available [1-14]. To keep this review as
short as possible, little work prior to 1994 is cited.
NITRIC OXIDE SYNTHASE AT A GLANCE
Nitric oxide synthase (EC 1.14.13.39; NOS) catalyzes the production of NO and L-citrulline from L-arginine, O2, and NADPH-derived electrons. NOS requires several cofactors and prosthetic groups for activity: thiolate-bound heme, FAD and FMN, calmodulin and Ca2+ (constitutive NOS only), and (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4). There are three distinct isoforms of the enzyme, the neuronal (nNOS), endothelial (eNOS), and inducible (iNOS) isoforms. In the active form all three are homodimers with approximate subunit molecular masses of 130 (iNOS), 135 (eNOS), and 160 kD (nNOS). NOS is a modular enzyme in which each monomer consists of several discrete domains. Starting at the C-terminus, we can discern: 1) a reductase domain, which shows extensive amino acid homology with cytochrome P-450 reductase; 2) a small calmodulin binding domain; 3) an oxygenase domain with many characteristics of cytochrome P-450 but no structural homology; 4) a N-terminal isoform-specific sequence. Catalytically, the best documented distinction between the different isoforms is that eNOS and nNOS require Ca2+ for activity, whereas calmodulin binding to iNOS is so tight that addition of Ca2+ is not necessary.
STRUCTURE AND FUNCTION
Stoichiometry and Function of Prosthetic Groups and Cofactors
NOS utilizes five different prosthetic groups and cofactors for catalysis. The reductase domain contains two flavin moieties, one FAD and one FMN. The close structural relationship between the NOS reductase domain and cytochrome P-450 reductase has allowed the putative assignment of specific functions to the two flavins by analogy. Accordingly, FAD is believed to be the primary acceptor for the electrons coming from NADPH, while FMN transfers the electrons from FAD to the heme site in the oxygenase domain.
Calmodulin is thought to convey a conformational change that is necessary for internal electron transfer. It is absolutely required for electron transfer from the reductase to the oxygenase domain, i.e., from the FMN to the heme [15], and strongly stimulates electron transfer within the reductase domain, i.e., from FAD to FMN [16]. Additionally, calmodulin is essential for proper biosynthesis of iNOS, since expression of iNOS in Escherichia coli in its absence results in synthesis of flavin- and heme-free monomers [17]; expression of eNOS in E. coli also improves by coexpression with calmodulin [18]. For eNOS there is growing evidence that calmodulin is directly involved in subcellular targeting [19, 20].
The heme pocket is the site where L-arginine and O2 are bound and where catalysis takes place. NOS is spectroscopically similar to cytochrome P-450 as both have a cysteinyl thiolate as the axial heme ligand. The cysteine providing the proximal ligand has been identified in all three isoforms by site-directed mutagenesis [21-23]. The similarities extend to both redox states, both spin states, and complexes with a whole range of external ligands. However, NOS is already largely high-spin five-coordinate as isolated, whereas, most, but not all, cytochrome P-450s become high-spin only after addition of substrate. This difference is partly due to the presence of BH4; pterin-free NOS has a considerably higher proportion of low-spin heme [24-26]. The substrate-free low-spin six-coordinate enzyme, which probably has H2O as a weak sixth ligand [27], absorbs at 418 nm [28]. As in cytochrome P-450 a shift from six-coordinate low-spin to five-coordinate high-spin (maximal absorbance at 394-397 nm) is induced by the substrates L-arginine [25, 29, 30] and N-hydroxy-L-arginine (OH-Arg) [29, 31, 32], by most substrate analogs [24, 29, 30, 33, 34], as well as by BH4 [24-26] and the BH4 analog 4-amino-BH4 [35]. Spin-shifts have been reported for a wide array of other compounds [27-30, 33, 36-39]. Comparison of the spectral effects of various ligands is starting to yield valuable information about the geometry of the heme pocket [30, 38, 39].
The role of BH4 is less clear. It binds to the oxygenase domain [40-42], probably in the immediate vicinity of the heme and the substrate L-arginine [26, 40, 43, 44]. By analogy to the functions BH4 performs in other enzymes, it is expected to be a dissociable electron carrier [45], but direct evidence for such a role is lacking. BH4 stimulates dimerization [46-48], induces a low-to-high spin shift of the heme [25, 26, 40, 49], and enhances the affinity of the enzyme for L-arginine [26, 43]. All of these effects may increase activity, but none can explain the absolute BH4 requirement for NO and L-citrulline synthesis [25, 50, 51]. For this reason the direct participation of BH4 as a redox-active agent is likely (see below).
In principle, NOS will bind one equivalent of each cofactor per monomer. The stoichiometries actually found in NOS as isolated are often considerably lower. Because of the large variations in the observed cofactor content, it is hard to assess the true stoichiometries for the in vivo situation. Most data suggest that functionally intact NOS dimers do indeed contain two equivalents of each cofactor, with the exception of BH4, for which only one equivalent per two equivalents of heme is found [46, 51, 52], unless high levels of BH4 are maintained during purification [53, 54]. The cause and possible consequences of this stoichiometry are discussed below. A schematic representation of NOS and the functions of the various cofactors is given in Fig. 1.
NOS DomainsFig. 1. Schematic representation of the NOS homodimer. The three domains that are common to all isoforms, the cofactor and substrate-binding sites, and the presumed electron pathway through the enzyme are indicated. The enzyme is drawn in the state with only one BH4. The lower panel summarizes the NOS-catalyzed reaction.
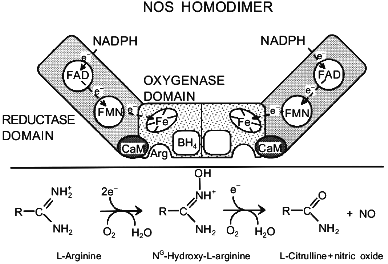
Isolated reductase domains contain the FAD and FMN moieties and are, at least in the case of iNOS, monomeric [41, 42, 55]. Provided they comprise the neighboring calmodulin binding domain as well, they are catalytically fully competent: they exhibit the same Ca2+/calmodulin-dependent electron transfer of electrons from NADPH to cytochrome c or other electron acceptors as the holoenzyme [41, 42, 56] and can also catalyze electron transfer from NADPH to the isolated oxygenase domain [42, 57]. The oxygenase domain contains the binding sites for heme, Arg, and BH4 [40-42, 55]. Isolated iNOS oxygenase domains are dimeric [41] and inactive, but activity is partially restored when supplied with NADPH and the reductase domain [42, 57].
The intermediate domain, which binds calmodulin, displays large isoform-specific variations [58-60]. The last few years have witnessed considerable progress in the search for the origin of the different Ca2+ dependence of the inducible and constitutive isoforms. Studies with chimeric NOS isoforms, in which the calmodulin-binding domains were exchanged between nNOS and iNOS [61] (or eNOS and iNOS [60]) demonstrated that the distinctions between these domains are indeed the principal cause for the different Ca2+/calmodulin binding properties, but that other sequences in the C-terminal half of iNOS contribute to the tight binding of Ca2+/calmodulin as well. An additional autoinhibitory sequence, present in nNOS and eNOS but not in iNOS, was also postulated to play a role. A detailed discussion of present ideas on this topic is given elsewhere [14].
At the N-terminus nNOS contains 300 amino acids that are peculiar to the neuronal isoform. This domain is involved in targeting nNOS towards its subcellular location [62]. It is missing in eNOS, which instead exhibits a smaller N-terminal sequence that comprises three fatty acylation sites that are important for targeting and membrane binding [63, 64]. The inducible isoform, as a soluble protein, lacks both the long sequence of nNOS and the sites for myristoylation and palmitoylation reported for eNOS.
Monomers and Dimers
NOS is only active as a dimer. Behind this deceptively simple statement hides a fundamental question which has proved very hard to answer. Since di/monomerization is always accompanied by changes in the cofactor content, it is not possible to assign a role to the dimeric structure as such. Inactive iNOS monomers that contain normal amounts of flavins and calmodulin, but no heme or BH4, are present in macrophages, and regeneration of the active dimer requires the combined presence of heme, BH4, and Arg [46]. Dimers can be dissociated in urea into inactive monomers that retain the heme but have lost BH4; re-dimerization requires both BH4 and Arg [65]. The link between BH4 and dimerization of iNOS was confirmed in several other studies [23, 50, 66]. BH4 is not essential for dimerization of nNOS and eNOS, but extremely stable dimers are formed in its presence [47, 48, 52], a phenomenon not observed with iNOS [48]. Dimerization does, however, require the heme [48, 52, 67], and all monomeric constitutive NOS is heme-free [52, 67]. The overall picture is one of eNOS and nNOS dimers being more stable than iNOS dimers, a conclusion that is supported by the observation that a greater part of the enzyme is partaking in subunit interactions in eNOS and nNOS [48]. Most importantly, for no isoform a monomer containing both essential cofactors (heme and BH4) has been reported.
As a result, hardly any property of NOS can be confidently ascribed to the dimeric structure. A study with various substrate analogs showed that many of these promoted iNOS dimerization to different extents, but no correlation was established between dimer formation capabilities of the compounds and their effects on coupled or uncoupled activity or on the heme spin state [30]. iNOS heterodimers, consisting of a wild-type monomer and an oxygenase domain, exhibited half the activity of native homodimers, and only half of the heme could be reduced by NADPH [68]. This suggests full activity of the wild-type monomer, and null activity of the oxygenase domain, although the heme that was reduced was not identified in these experiments. The data are, at most, consistent with a role for the dimer in enzyme stabilization and indicate that there is little cross-talk between the subunits. In apparent contrast with those results is a report involving iNOS heterodimers with a range of mutants, which seemed to indicate that dimerization required only one heme, but that for the enzyme to be active, both hemes had to be present [23]. This conclusion was based mainly on the observation that a heterodimer consisting of a heme-free mutant and a BH4-free mutant could be formed but was inactive. Based on their capacity to form heterodimers, it was assumed, but not determined, that mutants that were defective in heme binding still bound BH4. However, it has become increasingly clear since that NOS is unable to bind BH4 in the absence of heme [46, 51, 54, 67]. Consequently, heterodimers that were assumed to be lacking one heme and one BH4 were most likely BH4-free and inactive because of that. The crucial experiment to establish whether NOS activity requires two hemes, which would be the study of a heterodimer of a wild-type monomer and a heme-free mutant, remains to be performed.
The only property which does appear to be directly affected by the dimeric structure is the binding of BH4. Key observations are: 1) a stoichiometry of approximately one equivalent of BH4 per dimer in the enzyme as isolated [46, 51, 52, 69]; 2) a strict correlation of 1:2 for the BH4/heme content [51]; 3) the ability of NOS to bind a second equivalent as judged from the increase in activity in the presence of exogenous BH4; 4) the inhibition of this stimulation, but not of the basal activity, by pteridine antagonists [35, 43, 70]. Based on these observations and on binding studies with radiolabeled BH4, it was postulated that NOS dimers contain two identical, but highly anticooperative BH4 binding sites [26]. As a result, unless exceedingly high or low levels of BH4 are present, nNOS contains one equivalent of BH4 per dimer. This appears to be the only major point of interaction between the catalytic sites, which otherwise function independently. The case for two independent subunits, one with and one without BH4, was strengthened by studies of the effect of thiols on the spectral and catalytic properties of nNOS [28], and, more recently, by comparing the pH dependence of the absorbance spectra of BH4-free and native nNOS (Gorren, Schrammel, Schmidt, and Mayer, unpublished results).
Function, Localization, and Regulation of NOS
We do not intend to give a full account of all that is known about the physiological functions, the (sub)cellular localizations, and the various mechanisms of regulation of NOS, which deserves a better treatment than we can give in these few pages.
Although its presence can be demonstrated in a great variety of cell types, iNOS is associated mainly with cells like macrophages, that partake in the immune system. Its normal concentration is exceedingly low, but it is induced to high levels by cytokines and a wide range of other agents. Since it does not require Ca2+, iNOS, once synthesized, will maintain a high activity over a period of days. The nitric oxide produced during this period is thought to be one of the ingredients of the mix of aggressive compounds used in defense against invading organisms. In how far NO itself is involved is a matter of debate, since it is unlikely to survive long under the oxidative conditions created during the respiratory burst. Particularly if O2- is simultaneously produced, rapid formation of ONOO- is to be expected.
The other two isoforms are constitutively expressed, and their activity is regulated by the Ca2+ concentration. They occur not only in endothelial and neuronal cells, but in various other cell types, and in some cells both isoforms may be present. They are thought to be part of various signalling pathways, in which their role is the transduction of an increase in Ca2+ levels into the production of cGMP, which is triggered by the binding of NO to its main target, the soluble guanylate cyclase (sGC). This is particularly true for eNOS, the physiological role of which is well-documented, whereas the function of nNOS is not exactly known.
The endothelial enzyme is myristoylated [63, 71] and palmitoylated [64, 72], which determines its subcellular localization and, indirectly, its activity. Fatty acylation is required for targeting eNOS to the caveolae and for anchoring it to the membrane [63, 64, 71-74]. In addition, eNOS is regulated by phosphorylation. Ser-phosphorylation results in dissociation from the membrane with concomitant loss of activity [75]. There is evidence for stimulation of eNOS activity by Tyr-phosphorylation [76]. On the other hand, Tyr-phosphorylation was reported to stimulate binding of eNOS to the caveolar protein caveolin-1 and to decrease its activity [77]. An indirect effect was observed after stimulation with bradykinin, via Tyr-phosphorylation of a regulatory protein (ENAP-1) that binds tightly and specifically to eNOS, and targets eNOS to the cytoskeletal subcellular compartment when phosphorylated [78]. Phosphorylation, accompanied by changes in activity, has also been reported for nNOS and iNOS; in the latter case Tyr-phosphorylation decreased the activity [79].
The neuronal enzyme is not fatty-acylated, but is nonetheless membrane-associated due to the interaction of the N-terminal PDZ/GLGF-domain with proteins like PSD-95, PSD-93, and the dystrophin-associated protein syntrophin [62]. nNOS activity may also be regulated in vivo by the newly discovered protein PIN [80]. This small protein inhibits NOS, presumably by preventing dimer formation. It is specific for the neuronal isoform, but appears not to be NOS-specific. It occurs in organisms ranging from plants to insects to mammals, and its sequence has been highly conserved.
The Isoforms
As discussed above, the inducible, endothelial, and neuronal isoforms perform different functions, which are reflected in different regulatory mechanisms and different subcellular locations. As far as the enzymological, spectroscopic, and structural properties are concerned, the enzymes are quite similar, with one exception, and in this case nNOS appears to be the odd one. It has been known for some time that nNOS, but not iNOS, readily shifts to the production of O2- instead of NO under conditions of limited Arg or BH4 availibility. Recent results [32, 52, 81] unexpectedly indicated that eNOS groups with iNOS in this respect and that facile uncoupling of NADPH and Arg oxidation is unique for the neuronal isoform. This finding may have important consequences, as it suggests that only the neuronal enzyme will yield significant amounts of a product different from NO under certain conditions (see below).
There are no other well-characterized NOS isoforms. Somewhat different is the NOS found in the salivary glands of the blood-sucking insect Rhodnius prolixus [82]. This enzyme resembles the constitutive isoforms in being Ca2+-dependent, but it is soluble like iNOS, and shows only moderate sequence homology (<50%) to any mammalian enzyme. Fascinating is the recent purification from rat cerebellum of a constitutive NOS that can efficiently utilize both bradykinin and L-arginine as substrates, with a preference for the former [83]. It has a lower apparent molecular weight (105 kD per subunit) than any other NOS, and NO formation from bradykinin, but not from L-arginine is calmodulin-independent. Confirmation of these results would certainly introduce a new member to the NOS family.
MECHANISM
The Reaction
It is surprising that the unambiguous identification of NO as a reaction product has remained one of the more problematic aspects of NOS catalysis. The main characteristics of the reaction are as follows: 1) NOS catalysis requires L-arginine as a substrate, with only a couple of other compounds (L-homoarginine, NG-methyl-L-arginine) that serve as poor alternative substrates; 2) the product of the reaction is L-citrulline; 3) the reaction takes place in two discrete steps with N-hydroxy-L-arginine (OH-Arg) being formed as the intermediate; 4) OH-Arg, which is the only good NOS substrate besides Arg, hardly accumulates during catalysis; 5) both steps require O2 as a co-substrate; 6) both steps require NADPH-derived electrons; 7) the NADP+-to-L-citrulline stoichiometry is 1.5, indicating that three electrons are consumed to produce one molecule of L-citrulline; 8) NO formation is mostly inferred by indirect methods, from the oxidation products nitrite and nitrate, from the scavenging by oxyhemoglobin, or from the stimulation of cGMP production; 9) the nitrogen in the product NO derives from the Arg guanidino group, and 10) the oxygen molecules from O2 are split between NO and L-citrulline. With this information and borrowing from known cytochrome P-450 chemistry, several reaction schemes have been proposed. For an excellent review of the postulated mechanisms, we refer to Griffith and Stuehr [7]. It is generally assumed that the first reaction step, the oxidation of Arg to OH-Arg, represents typical cytochrome P-450 chemistry, whereas the second step is more unusual. Curiously, no cytochrome P-450 is known to catalyze the first step, whereas the facile transformation by cytochrome P-450 of OH-Arg into L-citrulline and NO has been reported. The overall reaction for the two steps is schematically shown in the bottom half of Fig. 1. Recently, products other than nitrite and nitrate (hydroxylamine, nitrous oxide, nitrosothiols) were reported to be formed by NOS in low yields [84].
If Arg or BH4 are not present, nNOS can still oxidize NADPH with similar rates, but in that case O2 is reduced to O2-. The other two isoforms display strongly reduced NADPH oxidation rates under those conditions [15, 52], but NADPH oxidation can be increased by certain (not all) substrate analogs [30, 33, 52]. NOS-catalyzed electron transfer from NADPH to O2 is designated the uncoupled reaction because of the uncoupling of O2 reduction from NO production. It should not be confused with the peroxide-shunt known from several members of the cytochrome P-450 family, which is also sometimes called uncoupled. In that reaction product formation is uncoupled from reduction of the heme by cytochrome P-450 reductase, because H2O2 serves as the source of both oxygen and electrons. The latter reaction is also catalyzed, with low efficiency, by nNOS and iNOS with OH-Arg as the substrate [13, 85].
In addition, the reductase domain, isolated or within the holoenzyme, catalyzes the oxidation of NADPH by a range of electron acceptors, either independently of Ca2+/calmodulin (nitroblue tetrazolium), or Ca2+/calmodulin-stimulated (cytochrome c, ferricyanide, DCPIP).
In Support of a Redox-Active Role of BH4
A special problem with respect to the reaction mechanism is posed by the role of BH4. As already noted, the well-documented structural and allosteric effects of BH4 on NOS cannot explain the absolute BH4 requirement for formation of L-citrulline and NO, suggesting that BH4 acts as a redox-active cofactor in NOS catalysis. Although a direct demonstration of the participation of BH4 as an electron donor is still lacking, there is some evidence in favor of such a function. The regeneration of BH4 from quinonoid-BH2, a necessary step if BH4 is indeed oxidized during catalysis, was recently found to be catalyzed by NOS itself at an unspecified site on the reductase domain [86]. The application of redox-inactive BH4 analogs has provided additional arguments for a redox function of BH4 [43]. Especially the recent observation that 4-amino-BH4 emulates all the structural effects of BH4 (increased substrate affinity, stabilization of the dimer, low-to-high spin shift of the heme), but does not sustain catalysis, strongly suggests the participation of BH4 as a redox-active agent [35, 70].
It would be of interest to know whether BH4 participates in both reaction steps or if its involvement is restricted to the first or second one. In this respect it may be relevant that, in limited turnover experiments, conversion of one equivalent of Arg into OH-Arg required O2, BH4, and Ca2+/calmodulin, but no NADPH, whereas the further conversion of OH-Arg into L-citrulline was strictly NADPH-dependent [87]. These results suggest that BH4 may serve as an electron source for the first but not for the second step. In a recent review [11] it was stated that these data are in conflict with the established requirement for NADPH in both steps, and with the observation that NOS catalyzes H2O2-supported oxidation of OH-Arg but not of Arg [85]. However, the latter reaction, which has H2O2 present as an alternative electron source, cannot be easily equated with the O2-supported reaction, in which all electrons ultimately derive from NADPH. Furthermore, it should be noted that these were single turnover experiments in which oxidation of endogenous BH4 would yield sufficient electrons to account for product formation, whereas sustained catalysis requires NADPH. It remains to be established whether BH4 performs different functions in the two catalytic steps.
Inhibitors
There is a vast and still expanding literature on NOS inhibitors [2, 3, 7, 8, 12]. The inhibitors can be classified in several groups depending on where they interfere with the catalytic mechanism. Since some of them have great significance as tools for the elucidation of NOS structure and mechanism, we give a short overview on the various classes of inhibitors.
Substrate analogs. This group comprises the well-known typical NOS inhibitors, such as NG-methyl-L-arginine (NMA), NG-nitro-L-arginine (NNA), and NG-amino-L-arginine (NAA). They are by nature Arg-competitive and reversible, but on longer exposure some of them induce irreversible or slowly reversible inactivation of NOS. It is this component that endows some of the substrate analogs with moderate isoform specificity. A recent addition to the list is the compound 1400W (N-(3-(aminomethyl)benzyl)acetamidine), which shows considerable specificity towards the inducible isoform, especially in comparison to eNOS [88].
BH4 analogs. This small group consists of pteridines (BH2, 4-amino-BH4) that compete with BH4 for its binding site, but do not support catalysis. Their action is by nature reversible and BH4-competitive. As inhibitors they are not particularly potent: they only inhibit the 2-fold stimulation by exogenous BH4, since they seem not to be able to expel the tightly bound endogenous BH4 from its site [43, 70]. This is true even for the novel inhibitor 4-amino-BH4, which appears to have a higher affinity for NOS than BH4 itself. At present their main use is in the elucidation of the functions of BH4.
Heme ligands. This group includes small diatomic molecules (CO, CN-, NO), the imidazoles [37], and many other compounds. Mechanistically they are a mixed bag. They are expected to be reversible inhibitors, that are either non-competitive, if they prevent heme reduction, or O2-competitive, if they do not. Regarding this issue, however, few have been studied. Since Arg and, probably, BH4 bind close to the heme, members of this class may also be Arg- and/or BH4-competitive. The best studied example is imidazole. Although originally categorized by two groups as Arg-non-competitive [29, 89], there is now a wealth of data identifying it as a reversible Arg-competitive inhibitor of all three isoforms [37, 38, 42, 53, 90, 91]. The slight competition with BH4 [37, 90] can confidently be ascribed to an indirect effect, caused by the cooperativity between Arg and BH4 binding [25, 26, 43, 52, 91]. The weak inhibitor dithiothreitol, on the other hand, is clearly BH4-competitive [28], whereas Zn2+ cations were reported to be Arg- and BH4-non-competitive [36]. The affinity of the members of this class is low in comparison to the other inhibitors. Since most heme ligands bring about a high-to-low spin-shift, binding is accompanied by spectroscopic changes that allow easy determination of their binding constant. A notable exception is 7-nitro-indazole, which has been labeled a heme ligand because of its imidazole-like structure, but may be misclassified, as it produces a low-to-high spin shift (Gorren and Mayer, unpublished observations; [39]) and exhibits atypically high affinity [92, 93].
Calmodulin binding antagonists. Compounds that interfere with calmodulin--protein interactions, from the rather trivial Ca2+ chelators, like EGTA and EDTA, to more sophisticated compounds that occupy the protein-interaction regions on calmodulin, or the calmodulin-binding domain on NOS, fall into this category [2, 3]. They are reversible, Ca2+- or calmodulin-competitive inhibitors, and usually specific for the constitutive isoforms. Unlike the members of the previous three groups, they inhibit all calmodulin-dependent reactions.
NADPH analogs. Diphenylene iodonium (DPI) is a potent NADPH-competitive and irreversible inhibitor of cytochrome P-450 reductases, including NOS. Since it interferes with binding of NADPH to the reductase domain, it blocks all NADPH-dependent reactions.
RECENT DEVELOPMENTS
NO or Not to Know
The compound that provided the main impetus for NOS research and gave the enzyme its name, more often than not eludes detection. Identification of NO as the reaction product has relied heavily on two methods. The method of choice in cells and in vivo is the Griess-assay for NO2- and NO3-, the stable oxidation products of NO under aerobic conditions. With the purified enzyme good use has been made of the NO-scavenging properties of oxyhemoglobin. Unfortunately, neither method is specific for NO: notably, they cannot distinguish NO from ONOO- [94, 95]. When NO is measured more directly, by chemiluminescence or electrochemically, no formation of NO is observed unless high concentrations of superoxide dismutase are added [84, 96]. As discussed in an earlier review [97], NO may never accumulate, at least in the case of nNOS, since at low BH4 levels, the partly uncoupled enzyme produces NO and O2- simultaneously [98], while at high BH4 concentrations NOS does produce NO, but then O2- is formed from the autooxidation of the excess BH4 [96, 99]. Thus, under all conditions the rapid reaction between NO and O2*- would result in the formation of ONOO- [100].
The inability to detect NO unless special precautions are taken has left lingering doubts whether NO synthase produces NO at all. A model by which NOS does not form NO, but the nitroxyl anion NO-, was advanced as one of several alternative mechanisms for NOS catalysis early on [101]. The idea received incidental support since then [102, 103], and recently resurfaced [84]. Crucial to the model in its most recent guise is the notion that the generally accepted NADPH-to-L-citrulline ratio is overestimated because of the rapid oxidation of NADPH by ONOO- in the absence of thiols. It is unclear to us how an overestimated stoichiometry could cause NO- production to be masked as NO production, since the former requires one more electron than the latter. Moreover, unpublished observations in our laboratory indicate that 1.5 is the minimum value for the stoichiometry in the presence of 2.4 mM 2-mercaptoethanol over a wide pH range, including at pH 9.0 and above, conditions under which peroxynitrite is fairly unreactive, and which favor sulfhydryl oxidation (Gorren, Schrammel, and Mayer, unpublished observations). Although the ephemeral nature of NO- makes it hard to exclude the possibility that it is transiently formed, we believe that the case for NOS really being a NO- synthase is weak.
An indication that NOS nonetheless may not form NO, at least not exclusively, lies hidden in the explanation Abu-Soud et al. provide for the auto-inhibition of NOS by NO [104]. In their model a large proportion of the NO originally produced remains tightly bound to the NOS heme, only to be released after reaction with O2. Although not expressly stated, this would imply that a considerable part (40-50%) of NOS catalysis results in nitrate production rather than NO formation.
Earlier, it was demonstrated that ONOO- stimulated cGMP production in the presence of glutathione, but with a potency several orders of magnitude less than authentic NO [105], which all but ruled out ONOO- as an intermediate in the cascade leading up to sGC activation. Nonetheless, the likelihood that ONOO- is indeed produced by nNOS under many conditions, keeps our interest in this topic alive, and research into the nature of the NOS reaction products is ongoing in our laboratory.
The nNOS Dimer: a Peroxynitrite Synthase?
In this section we shall merge some of the more intriguing properties of NOS, highlighted in this review, into a hypothetical, but coherent model of how we believe nNOS may function.
The only structural aspect in which dimers are potentially different from monomers is in their ability to regulate the BH4 content, due to the strong negative cooperativity between the two BH4 moieties [26]. One BH4 molecule will be tightly bound at very low external BH4 levels, since its dissociation constant is probably smaller than 1 nM; the affinity of the second molecule will be much lower, with a dissociation constant greater than 1 µM. The intracellular BH4 level is hard to estimate, but a concentration in between these two Kd values seems reasonable. Consequently, the dimer with one bound BH4 may well be the form of NOS that is present in the cell.
The two hemes within the dimer appear to function independently from each other [28, 68]. Therefore the NOS dimer with one BH4 can, in a way, be regarded as an assembly of two different enzymes, one of which produces NO, whereas the other catalyzes the uncoupled reaction and forms O2-. Consequently, not only the rate of catalytic turnover, but also the identity of the reaction products may be regulated by the BH4 concentration (Fig. 2). At very low BH4 levels NOS would generate superoxide; at very high levels nitric oxide would be formed. In the wide range in between, though, the simultaneous production of O2- and NO at virtually the same site may render the nNOS dimer an efficient peroxynitrite synthase.
The relative yields of the products would depend on the rates at which O2- and NO are formed. If NADPH is oxidized equally fast by both subunits, three equivalents of O2- are produced for each NO. However, there is some evidence suggesting that L-arginine raises the heme reduction potential [27, 30, 33], as is well documented for cytochrome P-450 [106]. Corresponding data for BH4 are lacking, but the ability of BH4 to shift the heme towards high-spin [26, 40], suggests that BH4, too, may enhance the reduction potential. In that case the enzyme could be seriously tilted towards producing NO. If, indeed, the BH4-containing subunit yields NO much faster than O2- is formed by the other subunit, rapid formation of the autoinhibitory ferrous NO complex might ensue [104]. This complex may then react not with O2, as suggested by Abu-Soud et al., but with the O2- produced by the second subunit, which would represent a truly concerted mechanism for peroxynitrite production by nNOS.Fig. 2. Putative modulation by BH4 of nNOS-catalyzed O2-, NO, and "ONOO-" production. The anticooperative binding of BH4 to the dimer generates a wide BH4 concentration range (10-9 M <= [BH4] <= 10-6 M), in which the main reaction product of nNOS may be peroxynitrite, rather than NO. The dimeric structure of NOS is crucial to the postulated mechanism.
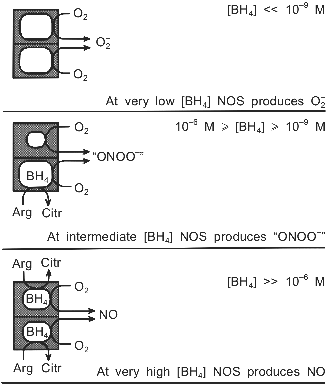
An open question is whether this mechanism is isoform-specific. Most observations discussed in this section were made with nNOS, and it seems that substantial uncoupling only occurs with the neuronal isoform [15, 32, 52, 98]. If confirmed, the capacity of ONOO- production by nNOS but not by eNOS, may explain the otherwise puzzling observation that nNOS is implicated more often than eNOS in pathophysiological settings.
Outlook
The picture we have drawn of the latest developments in NOS research is hopelessly incomplete. By focusing on just a few aspects of NOS enzymology we have necessarily neglected progress in other areas. Within the restricted context of NOS enzymology, however, we think the most exciting questions are the ones discussed in the final part of this review. What role does BH4 play in catalysis? Which are the products of NOS catalysis in vivo? What part, if any, is reserved for the dimeric structure? Are there fundamental enzymological differences between the three isoforms? We are confident that the next few years will yield answers to some of these questions, and we expect to encounter some more surprises along the way.
Work in the authors' laboratory was supported by the Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich.
REFERENCES
1.Nathan, C. (1992) FASEB J., 6,
3051-3064.
2.Marletta, M. A. (1994) J. Med. Chem.,
37, 1899-1907.
3.Knowles, R. G., and Moncada, S. (1994) Biochem.
J., 298, 249-258.
4.Bredt, D. S., and Snyder, S. H. (1994) Annu.
Rev. Biochem., 63, 175-195.
5.Masters, B. S. S. (1994) Annu. Rev. Nutr.,
14, 131-145.
6.Nathan, C., and Xie, Q. (1994) J. Biol.
Chem., 269, 13725-13728.
7.Griffith, O. W., and Stuehr, D. J. (1995) Annu.
Rev. Physiol., 57, 707-736.
8.Mayer, B. (1995) in Nitric Oxide in the Nervous
System (Vincent, S. R., ed.), Neuroscience Perspectives
(Jenner, P., ed.) Academic Press, London, pp. 21-42.
9.Förstermann, U. (1995) in Nitric Oxide.
Biochemistry, Molecular Biology, and Therapeutic Implications
(Ignarro, L., and Murad, F., eds.), Advances in Pharmacology,
Vol. 34 (August, J. T., Anders, M. W., Murad, F., and Coyle, J. T.,
eds.) Academic Press, San Diego, pp. 171-186.
10.Wang, Y., and Marsden, P. A. (1995) in Nitric
Oxide. Biochemistry, Molecular Biology, and Therapeutic
Implications (Ignarro, L., and Murad, F., eds.), Advances in
Pharmacology, Vol. 34 (August, J. T., Anders, M. W., Murad, F., and
Coyle, J. T., eds.) Academic Press, San Diego, pp. 71-90.
11.Masters, B. S. S. K. M., Sheta, E. A., Nishimura,
J. S., Roman, L. J., and Martasek, P. (1996) FASEB J.,
10, 552-558.
12.Griffith, O. W., and Kilbourn, R. G. (1996)
Meth. Enzymol., 268, 375-392.
13.Stuehr, D. J. (1997) Annu. Rev. Pharmacol.
Toxicol., 37, 339-359.
14.Hemmens, B., and Mayer, B. (1997) in Nitric
Oxide Protocols (Titheradge, M. A., ed.) Methods Mol. Biol.,
Vol. 100, Humana Press, Totowa, in press.
15.Abu-Soud, H. M., and Stuehr, D. J. (1993)
Proc. Natl. Acad. Sci. USA, 90, 10769-10772.
16.Abu-Soud, H. M., Yoho, L. L., and Stuehr, D. J.
(1994) J. Biol. Chem., 269, 32047-32050.
17.Wu, C., Zhang, J., Abu-Soud, H., Ghosh, D. K.,
and Stuehr, D. J. (1996) Biochem. Biophys. Res. Commun.,
222, 439-444.
18.Rodríguez-Crespo, I., and Ortiz de
Montellano, P. R. (1996) Arch. Biochem. Biophys., 336,
151-156.
19.Matsubara, M., Titani, K., and Taniguchi, H.
(1996) Biochemistry, 35, 14651-14658.
20.Michel, J. B., Feron, O., Sacks, D., and Michel,
T. (1997) J. Biol. Chem., 272, 15583-15586.
21.Richards, M. K., and Marletta, M. A. (1994)
Biochemistry, 33, 14723-14732.
22.Chen, P.-F., Tsai, A.-L., and Wu, K. K. (1994)
J. Biol. Chem., 269, 25062-25066.
23.Xie, Q., Leung, M., Fuortes, M., Sassa, S., and
Nathan, C. (1996) Proc. Natl. Acad. Sci. USA, 93,
4891-4896.
24.Salerno, J. C., Martasek, P., Roman, L. J., and
Masters, B. S. S. (1996) Biochemistry, 35, 7626-7630.
25.Rodríguez-Crespo, I., Gerber, N. C., and
Ortiz de Montellano, P. R. (1996) J. Biol. Chem., 271,
11462-11467.
26.Gorren, A. C. F., List, B. M., Schrammel, A.,
Pitters, E., Hemmens, B., Werner, E. R., Schmidt, K., and Mayer, B.
(1996) Biochemistry, 35, 16735-16745.
27.Matsuoka, A., Stuehr, D. J., Olson, J. S., Clark,
P., and Ikeda-Saito, M. (1994) J. Biol. Chem., 269,
20335-20339.
28.Gorren, A. C. F., Schrammel, A., Schmidt, K., and
Mayer, B. (1997) Biochemistry, 36, 4360-4366.
29.McMillan, K., and Masters, B. S. S. (1993)
Biochemistry, 32, 9875-9880.
30.Sennequier, N., and Stuehr, D. J. (1996)
Biochemistry, 35, 5883-5892.
31.Pufahl, R. A., and Marletta, M. A. (1993)
Biochem. Biophys. Res. Commun., 193, 963-970.
32.Presta, A., Liu, J., Sessa, W. C., and Stuehr, D.
J. (1997) Nitric Oxide, 1, 74-87.
33.Abu-Soud, H. M., Feldman, P. L., Clark, P., and
Stuehr, D. J. (1994) J. Biol. Chem., 269,
32318-32326.
34.Salerno, J. C., McMillan, K., and Masters, B. S.
S. (1996) Biochemistry, 35, 11839-11845.
35.Mayer, B., Wu, C., Gorren, A. C. F., Pfeiffer,
S., Schmidt, K., Clark, P., Stuehr, D. J., and Werner, E. R. (1997)
Biochemistry, 36, 8422-8427.
36.Persechini, A., McMillan, K., and Masters, B. S.
S. (1995) Biochemistry, 34, 15091-15095.
37.Chabin, R. M., McCauley, E., Calaycay, J. R.,
Kelly, T. M., MacNaul, K. L., Wolfe, G. C., Hutchinson, N. I.,
Madhusudanaraju, S., Schmidt, J. A., Kozarich, J. W., and Wong, K. K.
(1996) Biochemistry, 35, 9567-9575.
38.Berka, V., Chen, P.-F., and Tsai, A.-L. (1996)
J. Biol. Chem., 271, 33293-33300.
39.Tsai, A.-L., Berka, V., Chen, P.-F., and Palmer,
G. (1996) J. Biol. Chem., 271, 32563-32571.
40.McMillan, K., and Masters, B. S. S. (1995)
Biochemistry, 34, 3686-3693.
41.Ghosh, D. K., and Stuehr, D. J. (1995)
Biochemistry, 34, 801-807.
42.Chen, P.-F., Tsai, A.-L., Berka, V., and Wu, K.
K. (1996) J. Biol. Chem., 271, 14631-14635.
43.Klatt, P., Schmid, M., Leopold, E., Schmidt, K.,
Werner, E. R., and Mayer, B. (1994) J. Biol. Chem., 269,
13861-13866.
44.Gerber, N. C., Rodriguez-Crespo, I., Nishida, C.
R., and Ortiz de Montellano, P. R. (1997) J. Biol. Chem.,
272, 6285-6290.
45.Kaufman, S. (1993) Annu. Rev. Nutr.,
13, 261-286.
46.Baek, K. J., Thiel, B. A., Lucas, S., and Stuehr,
D. J. (1993) J. Biol. Chem., 268, 21120-21129.
47.Klatt, P., Schmidt, K., Lehner, D., Glatter, O.,
Bächinger, H. P., and Mayer, B. (1995) EMBO J., 14,
3687-3695.
48.Venema, R. C., Ju, H., Zou, R., Ryan, J. W., and
Venema, V. J. (1997) J. Biol. Chem., 272, 1276-1282.
49.Ghosh, D. K., Abu-Soud, H. M., and Stuehr, D. J.
(1996) Biochemistry, 35, 1444-1449.
50.Tzeng, E., Billiar, T. R., Robbins, P. D.,
Loftus, M., and Stuehr, D. J. (1995) Proc. Natl. Acad. Sci. USA,
92, 11771-11775.
51.List, B. M., Klatt, P., Werner, E. R., Schmidt,
K., and Mayer, B. (1996) Biochem. J., 315, 57-63.
52.List, B. M., Klösch, B., Völker, C.,
Gorren, A. C. F., Sessa, W. C., Werner, E. R., Kukovetz, W. R.,
Schmidt, K., and Mayer, B. (1997) Biochem. J., 323,
159-165.
53.Calaycay, J. R., Kelly, T. M., MacNaul, K. L.,
McCauley, E. D., Qi, H., Grant, S. K., Griffin, P. R., Klatt, T., Raju,
S. M., Nussler, A. K., Shah, S., Weidner, J. R., Williams, H. R.,
Wolfe, G. C., Geller, D. A., Billiar, T. R., MacCoss, M., Mumford, R.
A., Tocci, M. J., Schmidt, J. A., Wong, K. K., and Hutchinson, N. I.
(1996) J. Biol. Chem., 271, 28212-28219.
54.Richards, M. K., Clague, M. J., and Marletta, M.
A. (1996) Biochemistry, 35, 7772-7780.
55.Sheta, E. A., McMillan, K., and Masters, B. S. S.
(1994) J. Biol. Chem., 269, 15147-15153.
56.Gachhui, R., Presta, A., Bentley, D. F.,
Abu-Soud, H. M., McArthur, R., Brudvig, G., Ghosh, D. K., and Stuehr,
D. J. (1996) J. Biol. Chem., 271, 20594-20602.
57.Ghosh, D. K., Abu-Soud, H. M., and Stuehr, D. J.
(1995) Biochemistry, 34, 11316-11320.
58.Vorherr, T., Knöpfel, L., Hofmann, F.,
Mollner, S., Pfeuffer, T., and Carafoli, E. (1993) Biochemistry,
32, 6081-6088.
59.Anagli, J., Hofmann, F., Quadroni, M., Vorherr,
T., and Carafoli, E. (1995) Eur. J. Biochem., 233,
701-708.
60.Venema, R. C., Sayegh, H. S., Kent, J. D., and
Harrison, D. G. (1996) J. Biol. Chem., 271,
6435-6440.
61.Ruan, J., Xie, Q., Hutchinson, N., Cho, H.,
Wolfe, G. C., and Nathan, C. (1996) J. Biol. Chem., 271,
22679-22686.
62.Brenman, J. E., Chao, D. S., Gee, S. H., McGee,
A. W., Craven, S. E., Santillano, D. R., Wu, Z., Huang, F., Xia, H.,
Peters, M. F., Froehner, S. C., and Bredt, D. S. (1996) Cell,
84, 757-767.
63.Busconi, L., and Michel, T. (1993) J. Biol.
Chem., 268, 8410-8413.
64.Garcia-Cardena, G., Oh, P., Liu, J., Schnitzer,
J. S., and Sessa, W. C. (1996) Proc. Natl. Acad. Sci. USA,
93, 6448-6453.
65.Abu-Soud, H. M., Loftus, M., and Stuehr, D. J.
(1995) Biochemistry, 34, 11167-11175.
66.Cho, H. J., Martin, E., Xie, Q., Sassa, S., and
Nathan, C. (1995) Proc. Natl. Acad. Sci. USA, 92,
11514-11518.
67.Klatt, P., Pfeiffer, S., List, B. M., Lehner, D.,
Glatter, O., Bächinger, H. P., Werner, E. R., Schmidt, K., and
Mayer, B. (1996) J. Biol. Chem., 271, 7336-7342.
68.Siddhanta, U., Wu, C., Abu-Soud, H. M., Zhang,
J., Ghosh, D. K., and Stuehr, D. J. (1996) J. Biol. Chem.,
271, 7309-7312.
69.Harteneck, C., Klatt, P., Schmidt, K., and Mayer,
B. (1994) Biochem. J., 304, 683-686.
70.Werner, E. R., Pitters, E., Schmidt, K., Wachter,
H., Werner-Felmayer, G., and Mayer, B. (1996) Biochem. J.,
320, 193-196.
71.Sessa, W. C., Barber, C. M., and Lynch, K. R.
(1993) Circ. Res., 72, 921-924.
72.Liu, J. W., Garcia-Cardena, W., and Sessa, W. C.
(1995) Biochemistry, 34, 12333-12340.
73.Robinson, L. J., and Michel, T. (1995) Proc.
Natl. Acad. Sci. USA, 92, 11776-11780.
74.Liu, J. L., García-Cardeña, G., and
Sessa, W. C. (1996) Biochemistry, 35, 13277-13281.
75.Michel, T., Li, G. K., and Busconi, L. (1993)
Proc. Natl. Acad. Sci. USA, 90, 6252-6256.
76.Ayajiki, K., Kindermann, H., Hecker, M., Fleming,
I., and Busse, R. (1996) Circ. Res., 78, 750-758.
77.Garcia-Cardeña, G., Fan, R., Stern, D. F.,
Liu, J., and Sessa, W. C. (1996) J. Biol. Chem., 271,
27237-27240.
78.Venema, V. J., Marrero, M. B., and Venema, R. C.
(1996) Biochem. Biophys. Res. Commun., 226, 703-710.
79.Pan, J., Burgher, K. L., Sczcepanik, A. M., and
Ringheim, G. E. (1996) Biochem. J., 314, 889-894.
80.Jaffrey, S. R., and Snyder, S. H. (1996)
Science, 274, 774-777.
81.Martasek, P., Liu, Q., Liu, J., Roman, L. J.,
Gross, S. S., Sessa, W. C., and Masters, B. S. S. (1996) Biochem.
Biophys. Res. Commun., 219, 359-365.
82.Yuda, M., Hirai, M., Miura, K., Matsumura, H.,
Ando, K., and Chinzei, Y. (1996) Eur. J. Biochem., 242,
807-812.
83.Chen, Y., and Rosazza, J. P. N. (1996)
Biochem. Biophys. Res. Commun., 224, 303-308.
84.Schmidt, H. H. H. W., Hofmann, H., Schindler, U.,
Shutenko, Z. S., Cunningham, D. D., and Feelisch, M. (1996) Proc.
Natl. Acad. Sci. USA, 93, 14492-14497.
85.Pufahl, R. A., Wishnok, J. S., and Marletta, M.
A. (1995) Biochemistry, 34, 1930-1941.
86.Witteveen, C. F. B., Giovanelli, J., and Kaufman,
S. (1996) J. Biol. Chem., 271, 4143-4147.
87.Campos, K. L., Giovanelli, J., and Kaufman, S.
(1995) J. Biol. Chem., 270, 1721-1728.
88.Garvey, E. P., Oplinger, J. A., Furfine, E. S.,
Kiff, R. J., Laszlo, F., Whittle, B. J. R., and Knowles, R. G. (1997)
J. Biol. Chem., 272, 4959-4963.
89.Wolff, D. J., Datto, G. A., Samatovicz, R. A.,
and Tempsick, R. A. (1993) J. Biol. Chem., 268,
9425-9429.
90.Mayer, B., Klatt, P., Werner, E. R., and Schmidt,
K. (1994) FEBS Lett., 350, 199-202.
91.Roman, L. J., Sheta, E. A., Martasek, P., Gross,
S. S., Liu, Q., and Masters, B. S. S. (1995) Proc. Natl. Acad. Sci.
USA, 92, 8428-8432.
92.Mayer, B., Klatt, P., Werner, E. R., and Schmidt,
K. (1994) Neuropharmacology, 33, 1253-1259.
93.Wolff, D. J., and Gribin, B. J. (1994) Arch.
Biochem. Biophys., 311, 300-306.
94.Schmidt, K., Klatt, P., and Mayer, B. (1994)
Biochem. J., 301, 645-647.
95.Pfeiffer, S., Gorren, A. C. F., Schmidt, K.,
Werner, E. R., Hansert, B., Bohle, D. S., and Mayer, B. (1997) J.
Biol. Chem., 272, 3465-3470.
96.Mayer, B., Klatt, P., Werner, E. R., and Schmidt,
K. (1995) J. Biol. Chem., 270, 655-659.
97.Mayer, B., and Werner, E. R. (1995)
Naunyn-Schmiedeberg's Arch. Pharmacol., 351, 453-463.
98.Heinzel, B., John, M., Klatt, P., Böhme, E.,
and Mayer, B. (1992) Biochem. J., 281, 627-630.
99.Fisher, D. B., and Kaufman, S. (1973) J. Biol.
Chem., 248, 4300-4304.
100.Huie, R. E., and Padmaja, S. (1993) Free
Rad. Res. Commun., 18, 195-199.
101.Stuehr, D. J., and Griffith, O. W. (1992)
Adv. Enzymol., 65, 287-346.
102.Fukuto, J. M., Stuehr, D. J., Feldman, P. L.,
Bova, M. P., and Wong, P. (1993) J. Med. Chem., 36,
2666-2670.
103.Hobbs, A. J., Fukuto, J. M., and Ignarro, L. J.
(1994) Proc. Natl. Acad. Sci. USA, 91, 10992-10996.
104.Abu-Soud, H. M., Wang, J., Rousseau, D. L.,
Fukuto, J. M., Ignarro, L. J., and Stuehr, D. J. (1995) J. Biol.
Chem., 270, 22997-23006.
105.Mayer, B., Schrammel, A., Klatt, P., Koesling,
D., and Schmidt, K. (1995) J. Biol. Chem., 270,
17355-17360.
106.Sligar, S. G., and Murray, R. I. (1986) in
Cytochrome P-450: Structure, Mechanism, and Biochemistry (Ortiz
de Montellano, P. R., ed.) Plenum Publishing Corp., New York, pp.
429-503.