REVIEW: Role of Soluble Guanylate Cyclase in the Molecular Mechanism Underlying the Physiological Effects of Nitric Oxide
I. S. Severina
Institute of Biomedical Chemistry, Russian Academy of Medical Sciences, Pogodinskaya ul. 10, Moscow, 119832 Russia; fax: (095) 245-0857
Received June 26, 1997
In this review the molecular mechanisms underlying the antihypertensive and antiaggregatory actions of nitric oxide (NO) are discussed. It has been shown that these effects are directly connected with the activation of soluble guanylate cyclase and the accumulation of cyclic 3´,5´-guanosine monophosphate (cGMP). The mechanism of guanylate cyclase activation by NO is analyzed, especially the role and biological significance of the nitrosyl--heme complex formed as a result of interaction of guanylate cyclase heme with NO and the role of sulfhydryl groups of the enzyme in this process. Using new approaches for studying the antihypertensive and antiaggregatory actions of nitric oxide in combination with the newly obtained data on the regulatory role of guanylate cyclase in the platelet aggregation process, the most important results were obtained regarding the molecular bases providing for a directed search for and creation of new effective antihypertensive and antiaggregatory preparations. In studying the molecular mechanism for directed activation of soluble guanylate cyclase by new NO donors, a series of hitherto unknown enzyme activators generating NO and involved in the regulation of hemostasis and vascular tone were revealed.
KEY WORDS: guanylate cyclase, nitric oxide, platelet aggregation
Determination of the endogenous nature of nitric oxide (NO) was one of the most fundamental achievements in biochemistry of recent years; it allows better insight into the molecular mechanisms underlying several physiological processes in cells. Nitric oxide proved to be identical to endothelium-derived relaxing factor (EDRF) [1].
Endogenous NO is formed from L-arginine by oxidation of the guanidine amine group with catalysis by L-arginine-NO-synthase [2]. Endogenous NO is known to be involved in several important physiological processes, acting as a neurotransmitter [3], a cytotoxic agent [4], and a powerful factor in hemostasis. NO inhibits platelet aggregation [5] and is presently considered as the endogenous vasodilator. Antihypertensive and antiaggragatory properties of nitric oxide are directly associated with the functions of guanylate cyclase.
Guanylate cyclase (EC 4.6.1.2) catalyzes the biosynthesis of cyclic 3´,5´-guanosine monophosphate (cGMP) from guanosine triphosphate. cGMP is a potent regulator of cell metabolism. The hydrolytic degradation of cGMP is catalyzed by phosphodiesterase. However, the key enzyme of cGMP metabolism is guanylate cyclase, and it is due to activation of this enzyme that the increase in tissue cGMP level is brought about.
Guanylate cyclase exists in two forms: soluble and membrane bound. It has been established that these forms are not only two different proteins, but two differently regulated enzymes [6]. The present paper discusses the soluble form of guanylate cyclase because it is with this enzyme the antihypertensive and antiaggregatory action of nitric oxide is associated. Soluble guanylate cyclase is a heterodimer with two immunologically distinct subunits. One characteristic feature of the enzyme is the presence in its molecule of labile sulfhydryl groups. Oxidation of the latter stimulates the enzyme activity [7]. However, long-term exposure of the enzyme to oxidants results in the loss of activity [8]. The second characteristic feature of soluble guanylate cyclase is the presence of heme in the enzyme molecule [9]. It is known that the immediate heme precursor in vivo is protoporphyrin IX, which proved to be a potent activator of the enzyme [10]. Introduction of iron in the porphyrin ring yields ferroprotoporphyrin IX (or heme) which acts as an inhibitor of the enzyme [11]. The sequence of reactions resulting in the formation of the guanylate cyclase holoenzyme, namely, protoporphyrin IX -- iron -- heme -- guanylate cyclase, remains obscure. However, this system is essential to the endogenous regulation of guanylate cyclase.
The role of heme in the functioning of guanylate cyclase is thought to be mainly connected with enzyme activation by nitric oxide and NO-generating compounds [12, 13]. The actual activator of the enzyme is the nitrosyl--heme complex [14] which is formed on interaction of a NO group with the guanylate cyclase heme. Under these conditions the iron protrudes from the plane of the porphyrin ring; as a result the structure of the nitrosyl--heme complex formed becomes similar to that of protoporphyrin IX--one of the potent activators of the enzyme [11]. It was found that heme-deficient guanylate cyclase loses its capacity to be activated by NO [14]. On reconstruction of the heme-deficient enzyme (by hematin in the presence of dithiothreitol) to the heme-containing guanylate cyclase, the enzyme recovers its sensitivity to activation by NO and NO-generating compounds [15]. Nitrosyl--heme complexes may be formed by interactions between NO or NO-donors with heme moieties of other heme-containing proteins (hemoglobin, myoglobin, catalase, etc.). Guanylate cyclase has a high affinity to the nitrosyl--heme complex, facilitating its transfer from other heme-containing proteins to the enzyme but not in the opposite direction [16].
The heme-dependent activation of the enzyme was proposed to be related to its sulfhydryl groups because the capacity of the enzyme to be activated by NO decreases after oxidation [17]. Another line of evidence comes from the finding that heme-deficient enzyme is still sensitive to activation by NO and NO-generating compounds, though their efficacy falls considerably [18, 19].
For the last few years (1994-1997) new data on the functional domains of soluble guanylate cyclase and the role of the cysteine residues of the enzyme in its functioning were obtained. It was shown that soluble guanylate cyclase heterodimer consists of two subunits: alpha and beta [20]. Thus the first evidence was provided that the regulatory and catalytic properties of guanylate cyclase can be attributed to different regions of the subunits and that the catalytic site is located in the COOH-terminal halves of the alpha- and beta-subunits of guanylate cyclase [21]. This catalytic site is responsible only for cGMP formation and is insensitive to NO. The NH2-terminal region of the beta-subunit is involved in activation of guanylate cyclase by nitric oxide [21, 22]. The site of heme binding is still unknown, but point mutation of His-105 to Phe in the beta-subunit disrupts heme binding with the protein and the resulting recombinant guanylate cyclase loses its ability to be activated by NO. At the same time, the basal catalytic guanylate cyclase activity is unchanged [21, 22]. Point mutation of 15 conserved cysteine residues located in the alpha- and beta-subunits of guanylate cyclase to serine yielded recombinant enzymes that were able to catalyze the synthesis of cGMP. Mutation of two cysteine residues (Cys-78 and Cys-214) located in the immediate proximity of His-105, the putative heme-binding region of the beta-subunit, yielded recombinant proteins that were insensitive to NO [23]. The role of SH-groups of soluble guanylate cyclase in enzyme activation by nitric oxide is not yet clear. It was proposed that the interaction between thiols and NO and NO-generating compounds produces S-nitrosothiols [24], which had been earlier considered the true activators of guanylate cyclase [24]. However, now S-nitrosothiols are regarded as intermediate NO donors rather than the direct stimulant of guanylate cyclase [16]. Indeed, thiols facilitate activation of guanylate cyclase by NO donors [25]. There is no consensus in the literature on the role of sulfhydryl groups of the enzyme in its activation by NO and NO donors. Apparently, the stimulatory effect may be complex and depends on the presence of thiols in the sample and on the degree of oxidation of guanylate cyclase. However, a major role is played by the guanylate cyclase heme and the nitrosyl--heme complex; the latter is the true activator of the enzyme.
MECHANISM UNDERLYING THE ANTIHYPERTENSIVE ACTION OF NITRIC
OXIDE
Apparently, the antihypertensive action of nitric oxide is directly connected with the heme dependent mechanism of guanylate cyclase activation and accumulation of cGMP. The accumulated cGMP activates cGMP-dependent protein kinase and Ca2+-ATPase, which dephosphorylate myosin light chain leading to the efflux of Ca2+ from muscle cells and ultimately to vascular relaxation [26]. The therapeutic effects of most known nitrovasodilaters (such as glycerotrinitrate, nitrosorbide, etc.) are closely related to the interaction of NO released in the course of their biotransformation with the guanylate cyclase heme (according to the above mechanism), enzyme activation, and accumulation of cGMP.
The nature of the bond between the protein and heme in guanylate cyclase has not yet been determined; however, the lability of this bond has been proved. The heme can dissociate from the protein on a decrease in pH (5.0), on storage, or purification of the enzyme, thus providing a certain degree of heme deficiency of guanylate cyclase. The tightness of heme binding to guanylate cyclase varies depending on the source of the enzyme [27, 28]. The literature contains no indication of the possible existence in tissues of soluble guanylate cyclase in the heme-deficient form. We were the first to demonstrate that rat platelet guanylate cyclase could not be activated by sodium nitroprusside [29]. More detailed investigation of this enzyme allowed us to conclude that, contrary to the generally accepted notion, heme is not a constituent part of the rat platelet guanylate cyclase molecule [30]. Therefore, rat platelets cannot be used as a model to study the effect of NO and NO-generating compounds on platelet guanylate cyclase. Naturally, the heme-deficiency of guanylate cyclase impairs the endogenous regulation of the enzyme, decreases the efficacy of nitrovasodilaters, and ultimately leads to disorders of vascular tone.
This confronts us with the problem of measuring heme saturation of guanylate cyclase. The problem was settled with the use of carnosine. Carnosine (beta-alanyl-L-histidine) is a water-soluble antioxidant, capable of forming chelate complexes. Because of its antioxidant properties the substance is widely used in treatment of inflammation, in wound healing, and cataract treatment [31, 32]. In studying the effect of carnosine on human platelet guanylate cyclase we have first shown [33] that carnosine, at a concentration producing no effect on the basal activity of enzyme, strongly (by ~70%) inhibited the stimulatory effect of sodium nitroprusside. However, carnosine did not affect the slight activation by sodium nitroprusside of a heme-deficient guanylate cyclase preparation, obtained by ion-exchange chromatography [33]. Carnosine did not inhibit the stimulatory effect of protoporphyrin IX on guanylate cyclase, which is heme-independent [33]. Data obtained have demonstrated that stimulation of guanylate cyclase by nitric oxide is a combined result of: 1) a slight nonspecific and heme-independent increase in activity (probably caused by oxidation of labile sulfhydryl groups of the enzyme), and 2) stimulation of activity due to formation of nitrosyl--heme complex. Carnosine inhibits the latter stimulation. Since further experiments have shown that carnosine interacts with the guanylate cyclase heme, it was postulated that carnosine specifically inhibits heme-dependent NO-stimulation of guanylate cyclase and this phenomenon can be used to estimate the degree of heme saturation of the enzyme [33]. With the use of carnosine additional data were obtained on the mechanism of vasodilatory action of sodium nitroprusside and other nitroso complexes of some transition metals differing in the character of NO oxidation. It is known that an important factor in the vasodilatory action of sodium nitroprusside is the degree of oxidation of the NO group and the readiness of its release from the sodium nitroprusside molecule. It was shown earlier that the NO group in sodium nitroprusside Na2[FeNO+(CN)5] carries a positive charge due to the nitrosonium cation [34]. Other nitroso complexes of transition metals (structure analogs of sodium nitroprusside) are poorly investigated. There is only one report [35] mentioning the absence of a pharmacological (antihypertensive) effect of the anion [Mn2+NO(CN)5]3- in which the NO group is electrically neutral. Therefore, it seemed to us to be worthwhile to study two analogs of sodium nitroprusside, the nitroso complexes of transition metals (Cr and Co): K3[CrNO(CN)5] and [CoNO(NH3)5]SO4, the NO groups of which are neutral. Effects of these compounds on guanylate cyclase activity from human platelets (heme-containing [36]) and rat platelets (heme-deficient [36]) were compared with the effect of sodium nitroprusside on these same enzymes. As seen from the data presented in the table, sodium nitroprusside is the most potent (16.2-fold) activator of human platelet guanylate cyclase. NO activation is due to the interaction between the sodium nitroprusside NO group and the guanylate cyclase heme. This fact explains why sodium nitroprusside cannot stimulate rat platelet guanylate cyclase, which is initially heme-deficient. The table also shows that the Cr and Co nitroso complexes only slightly stimulate guanylate cyclase activity, the effects on the heme-containing (human platelet) and heme-deficient (rat platelet) soluble guanylate cyclases are virtually equal in magnitude. The data suggest that the mechanism of guanylate cyclase activation by Cr and Co nitroso complexes is not heme-dependent. Further experiments have confirmed this suggestion. It was shown [37] that carnosine, a specific inhibitor of a heme-dependent stimulatory effect on guanylate cyclase, reduces sodium nitroprusside-elicited enzyme activation by 66 ± 4% without affecting the degree of the enzyme activation by Cr and Co nitroso complexes [37]. In the case of heme-deficient human platelet guanylate cyclase preparation obtained by ion-exchange chromatography, its capacity to be activated by sodium nitroprusside was decreased by 90% but the magnitude of the stimulatory effects of Cr and Co nitroso complexes remained virtually unchanged [37]. It should be noted that nitroso complexes of Cr and Co (in contrast to sodium nitroprusside) possessed no hypertensive action. Data obtained allow us to conclude that in order for the hypotensive effect to be materialized the activation of soluble guanylate cyclase must proceed by the heme-dependent mechanism.
Effect of sodium nitroprusside (SNP),
K3[CrNO(CN)5] (I) and
[CoNO(NH3)5]SO4 (II) on guanylate
cyclase activity in 105,000g supernatants of human and rat
platelets
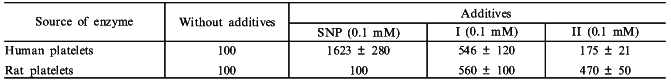
Note: The table shows the percent changes of guanylate cyclase
specific activities in the presence of SNP and compounds I or II. The
basal activity in the presence of Mg2+ was taken as 100%.
MECHANISM UNDERLYING THE ANTIAGGREGATORY ACTION OF NITRIC
OXIDE
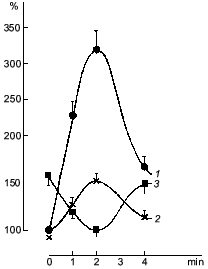
It was known that nitric oxide and sodium nitroprusside both inhibit platelet aggregation. This inhibitory effect is associated with the ability of these compounds to activate soluble guanylate cyclase [38]. The antiaggregatory action of nitric oxide was cleared up in detail after investigation of the role of guanylate cyclase in regulation of platelet aggregation [39, 40]. When studying the dynamics of changes in functioning of human platelet guanylate cyclase during ADP-induced reversible aggregation in vitro, we found that immediately after ADP addition the guanylate cyclase response to sodium nitroprusside begins to increase with the concurrent increasing intraplatelet cGMP level (Fig. 1, curves 1 and 2, respectively). Curve 3 (Fig. 1) demonstrates the effect of hemoglobin on guanylate cyclase activation by sodium nitroprusside. It can be seen that in intact platelets, before ADP addition hemoglobin increases the stimulatory effect of sodium nitroprusside. This was interpreted as being due to the additional formation of the nitrosyl--heme complex (at the expense of hemoglobin) and its transfer to guanylate cyclase. Thus it would seem that in natural conditions the guanylate cyclase is partly heme-deficient. However, in the course of aggregation the stimulatory effect of hemoglobin decreased and ultimately disappeared at the time point corresponding to the maximum of the nitroprusside-induced activation of the enzyme and to the highest cGMP level (see Fig. 1, curves 1-3). On reaching the aggregation maximum (following by disaggregation) all the values reverted to the baseline. Dynamics of the hemoglobin effect magnitude on the nitroprusside activation (see Fig. 1, curve 3) reflects the gradually increasing heme saturation of the enzyme throughout the aggregation process in accord with the enhanced capacity of the enzyme to be activated by sodium nitroprusside. It should be noted that dynamics of changes in guanylate cyclase parameters is the same at all aggregation levels and is independent of the degree of aggregation. However, the proportionality between the time of achievement of the aggregation peak and the maximum of the nitroprusside-produced activation was retained only with 45-50% aggregation (see Fig. 2). In other words, these changes occurred at the earliest stages of aggregation within the first minutes of the aggregation process (see Fig. 2). This is best demonstrated in the case of irreversible aggregation (Fig. 2d). At this stage the regulatory role of guanylate cyclase is no longer revealed.
Fig. 1. Dynamics of changes in the cGMP level (1), guanylate cyclase activation by sodium nitroprusside (2), and guanylate cyclase heme saturation (3) at 45-50% reversible platelet aggregation induced by 4-6 µM ADP. Abscissa, time (min) after ADP addition. Ordinate, changes in the measured parameters (% of initial value) (curves 1 and 2); changes in the effect of hemoglobin on the nitroprusside-induced guanylate cyclase activation (%) (curve 3). The mean values of five experiments of the same type are given.
If sodium nitroprusside was introduced to platelets before the addition of the aggregation inducer no increase in guanylate cyclase activation was observed [41]. Thus, guanylate cyclase provides a defense against aggregation. Moreover, sodium nitroprusside not only prevents aggregation but also facilitates disaggregation. Figure 3 shows that adding sodium nitroprusside at the peak of aggregation initiates disaggregation, which explains the inhibitory effect of nitroprusside on aggregation. Thus, guanylate cyclase effects negative control over platelet aggregation: initiation of aggregation is accompanied by increasing the enzyme activation and accumulation of cGMP; the latter mediates a signal that induces disaggregation. Therefore, the functioning of platelet guanylate cyclase and the ability of platelets for aggregation are interrelated. Indeed in comparing the functioning of platelet guanylate cyclase in platelets of diabetes mellitus patients (types I and II) characterized by increased aggregability and in normal platelets of healthy donors we have found a decrease in the basal guanylate cyclase activity and a reduced capacity of the enzyme to activation in platelets of diabetes mellitus patients [42]. Moreover, the higher was the platelet aggregability, the lower was the basal guanylate cyclase activity and, accordingly, its capacity for activation [42]. It should be noted that the degree in guanylate cyclase parameters was not associated with the aetiology of diabetes mellitus but solely with hemostases system disturbances. Thus, guanylate cyclase may be considered as a protective mechanism blocking the development of aggregation. In this connection the directed activation of guanylate cyclase by nitric oxide and NO-generating compounds may be used to normalize pathologically increased platelet aggregability. Since the regulatory role of cGMP is manifested at the earliest stages of aggregation (see Fig. 2) new activators of guanylate cyclase will be capable not only to reduce platelet aggregability but also prevent their spontaneous aggregation and hence the appearance and development of vascular complications.Fig. 2. Dynamics of changes in degree of guanylate cyclase activation by sodium nitroprusside (1) at 20% (a), 50% (b), and 70% (c) reversible and 70% irreversible (d) ADP-induced human platelet aggregation with platelet concentration 2.5·108 (a-c) and 4.5·108 (d) per ml of plasma. Aggregograms (curves 2). Ordinate, changes in the measured parameters (% of the initial values). The results of a typical experiment are given. Arrows indicate time of addition of ADP.
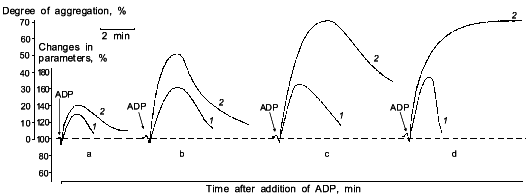
Clearly, the molecular mechanism of antiaggregatory action of nitric oxide and NO donors is connected with activation of soluble guanylate cyclase and accumulation of cGMP. However, the molecular mechanism of cGMP participation in the aggregation process is yet incompletely understood.Fig. 3. Effect of sodium nitroprusside (0 (1), 10-4 (2), 10-5 (3), 10-6 (4), 10-7 M (5)) on ADP-induced human platelet aggregation. Abscissa, time (min) after ADP addition; ordinate, platelet aggregation (%). The arrows indicate the time of addition of ADP and sodium nitroprusside (SNP). The results of five experiments of the same type are presented.
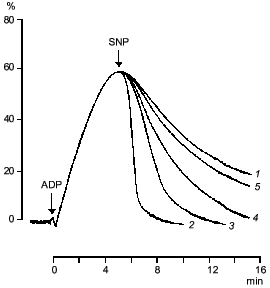
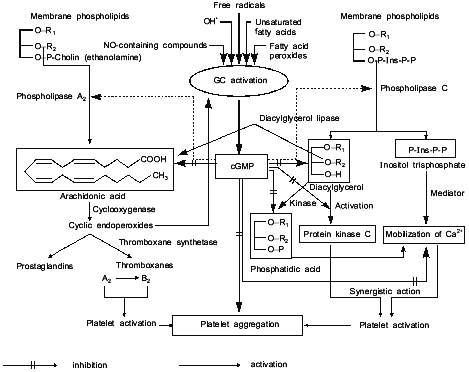
Figure 4 represents our hypothetical scheme of likely sites of action of cGMP as a regulator of platelet aggregation. Figure 4 shows that cGMP inhibits the liberation of arachidonic acid, thus preventing the activation of phospholipase A2; however, cGMP does not affect the subsequent steps of arachidonic acid breakdown mediated by cyclooxygenase and thromboxane synthetase, both of which stimulate the accumulation of Ca2+ and cause platelet activation and aggregation. cGMP impedes the formation of 1,2-diacylglycerol and inositol trisphosphate, through the inhibition of phospholipase C activation. 1,2-Diacylglycerol is a potent activator of protein kinase C which phosphorylates platelet proteins (20 and 40 kD), causes platelet activation and aggregation. cGMP prevents the formation of inositol trisphosphate, suppresses the accumulation of Ca2+ thereby inhibiting the platelet activation and aggregation. cGMP also inhibits the formation of phosphatidic acid, which is readily formed in platelets from 1,2-diacylglycerol under the action of 1,2-diacylglycerol phospholipase; phosphatidic acid acts as an ionophore, facilitating the release of intracellular Ca2+ and causing activation and aggregation of platelets. In other words, cGMP prevents phospholipid breakdown (including inositol phospholipids) and inhibits platelet aggregation through a common mechanism inhibiting Ca2+ accumulation [43].
Thus, antihypertensive and antiaggregatory properties of nitric oxide are due to soluble guanylate cyclase activation and accumulation of cGMP.Fig. 4. Model summarizing the possible role of platelet guanylate cyclase (GC) and cGMP in regulation of platelet aggregation.
In connection with the above-reviewed data, the synthesis of new NO donors and revealing among them possible guanylate cyclase activators seems to be promising and pertinent for solution of one the most fundamental problems of modern biological and medical chemistry, the directed search for and synthesis of new effective antihypertensive and antiaggregatory preparations based on the investigation of the effects of NO-generating compounds on soluble guanylate cyclase.
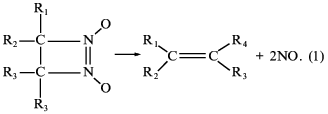
Thus, we have first studied a new class of newly synthesized compounds, derivatives of 1,2-diazetine-1,2-di-N-oxides capable of non-enzymatic generation of nitric oxide by a principally new mechanism of nitric oxide splitting at physiological pH values and without the participation of thiols [44, 45] according to Eq. (1):
Four of the seven derivatives tested exhibited a distinct correlation between the ability of being decomposed with nitric oxide formation, activation of soluble guanylate cyclase [45], inhibition of platelet aggregation, and acceleration of platelet disaggregation [46]. Furthermore, studies of the spasmolytic activity of compounds on isolated rings of aorta and their hypotensive effect on urethane-narcoticized spontaneously hypertensive rats (carried out at the Chemical Pharmaceutical Research Institute, Moscow) have shown that with each of the four derivatives of diazetine-di-N-oxides there is a full correlation between the ability of being decomposed with NO formation, soluble guanylate cyclase activation, and manifestation of the above-mentioned physiological effects [47]. Among the compounds tested, 3-brom-4-methyl-3,4-tetramethylene-diazetine-di-N-oxide has proved to be most effective, its spasmolytic effect being commensurate with glycerotrinitrate activity [47].
The next class of new guanylate cyclase activators capable of nitric oxide generation were derivatives of guanidine thiols. Guanidine thiols contain both the guanidine and SH groups which act, respectively, as donor and acceptor of nitric oxide. It was shown that beta-mercaptoethylguanidine (HS-CH2-CH2-NH-C=NH(NH2)) proved to be a much more potent activator of soluble guanylate cyclase than was L-arginine [48]. In our view, this is explained by formation of unstable intramolecular nitrosothiols promoting NO transfer to the guanylate cyclase heme and hence the enzyme activation. To ascertain the role of guanidine thiols SH groups in increasing their stimulatory effect on guanylate cyclase, three compounds were tested: beta-mercaptoethylguanidine (MEG), mercaptoethylguanidine disulfide (MEG-disulfide), and the SH-group methylated MEG derivative S-methyl-mercaptoethylguanidine (S-methyl-MEG) with the following formula:
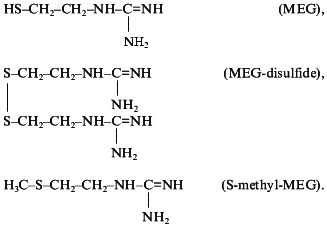
All these compounds proved to be guanylate cyclase activators. The stimulatory effect of these compounds was blocked by NG-monomethyl-L-arginine, a well-known inhibitor of NO-synthase [49]. The degree of guanylate cyclase activation by MEG and MEG-disulfide was, respectively, two and four times higher than that of L-arginine [49]. The stimulatory effect of S-methyl-MEG was of the same order as that of L-arginine [49]. Thus, the important role of S-acceptor group of guanidine thiols in the intensification of guanylate cyclase activation was demonstrated providing a plausible explanation for different intensities of guanylate cyclase activation by the compounds tested [49]. In full accordance with the intensity of the stimulatory effect of these compounds on guanylate cyclase activity are their antiaggregatory properties [50] and their hypotensive effect on intravenous injection to spontaneously hypertensive rats [51].
Of interest is the dependence between the chemical structure of a compound as well as its ability to generate NO and to activate human platelet guanylate cyclase revealed in studies yet another class of compounds--derivatives of oximes of quinuclidin-3-one--generating NO by oxidation [52].
A group of compounds with different stereochemistry identified on the basis of H1-NMR-spectroscopy was obtained [52]. It was shown that the most active compound in this line, based on electrochemical data and on stimulatory effect on guanylate cyclase, is the compound containing the ortho-hydroxy group in the aryl ring. Consequently, the oxime and phenolic fragments are in near proximity to each other; it may be suggested, therefore, that the phenolic hydroxy group possesses NO acceptor properties allowing it to promote NO transfer onto guanylate cyclase heme and consequently, the activation of the enzyme. The methylation of phenolic hydroxyl blocks its NO acceptor properties and decrease its stimulatory effect on guanylate cyclase [52]. An analogous dependence of the guanylate cyclase activation intensity on the presence of free phenolic hydroxyl in the oxime molecule was revealed with some other oximes--derivatives of p-hydroxy- and p-methoxybenzaldehydes. The former compound (OH-C6H4-CH=NOH) (0.1 mM) was able to activate soluble guanylate cyclase (3.5 ± 0.41)-fold, the latter (H3C-O-C6H4-CH=NOH) (0.1 mM), (1.4 ± 0.17)-fold. Thus, the acceptor properties of phenolic hydroxyls were first demonstrated.
Study of the dependence of the degree of guanylate cyclase activation on the structure of NO generating compounds has shown that directed introduction of NO acceptor groups (such as SH groups and phenolic hydroxyl) into molecules of new NO donors increases the stimulatory effect of these compounds on the enzyme activity.
Thus, synthesis of new NO donors and study of their effects on soluble guanylate cyclase--to reveal among them the most active enzyme stimulators for pharmacological uses--serve as a molecular basis for directed search for and creation of new effective antihypertensive and antiaggregatory preparations.
Indeed, in studying a great number of newly synthesized NO donors belonging to different classes of compounds we have clearly demonstrated that, inside each class, the more active was a stimulator of guanylate cyclase activity the more pronounced were its antihypertensive and antiaggregatory effects. It appears that, based on the intensity of guanylate cyclase stimulation by a given compound, the pharmacological efficiency of each particular newly synthesized NO donor may be predicted.
This work received financial support from the Russian Foundation for Basic Research.
REFERENCES
1.Palmer, R. M. J., Ferrige, A. G., and Moncada, S.
(1987) Nature, 327, 524-526.
2.Palmer, R. M. J., Ashton, D. S., and Moncada, S.
(1988) Nature, 333, 664-666.
3.Knowles, R. J., Palacios, M., Palmer, R. M. J., and
Moncada, S. (1989) Proc. Natl. Acad. Sci. USA, 86,
5159-5162.
4.Hibs, G. B., Tailor, R. E., and Vavrin, Z. (1987)
Science, 235, 473-476.
5.Busse, R., Luchoff, A., and Bassenge, E. (1987)
Naunyn-Schmiedebergs Arch. Pharmacol., 336, 566-571.
6.Tremblay, G., Gerzer, R., and Hamet, P. (1988)
Adv. Second Messenger and Phosphoprotein Res., 22,
319-385.
7.Braughler, J. M. (1980) Biochim. Biophys.
Acta, 616, 94-104.
8.Braughler, J. M., Mittal, S. K., and Murad, F.
(1979) J. Biol. Chem. Acta, 254, 12450-12454.
9.Gerzer, R., Bohme, E., Hofmann F., and Schultz, G.
(1981) FEBS Lett., 132, 71-74.
10.Ignarro, L. J., Wood, K, S., and Wolin, M. S.
(1982) Proc. Natl. Acad. Sci. USA, 79, 2870-2873.
11.Ignarro, L. J., Wood, K. S., and Wolin, M. S.
(1984) Adv. Cycl. Nucl. Protein Phosphor. Res., 17,
267-274.
12.Ignarro, L. J., and Wood, K. S. (1987)
Biochim. Biophys. Acta, 928, 160-170.
13.Gerzer, R., and Garbers, D. L. (1982) Fed.
Proc., 41, 1410.
14.Graven, P., and De Rubertis, F. (1983)
Biochim. Biophys. Acta, 745, 310-321.
15.Ignarro, L. J., Degnan, J. N., Baricos, W. H.,
Kadowitz, P. J., and Wolin, M. S. (1982) Biochim. Biophys. Acta,
718, 49-59.
16.Ignarro, L. J., Adams, J. B., Horwitz, P. M., and
Wood, K. S. (1986) J. Biol. Chem., 261, 4997-5002.
17.Braughler, J. M. (1983) Biochem.
Pharmacol., 32, 811-818.
18.Edwards, J. C., Barry, B. K., Grutter, D. Y.,
Ohlstein, E. H., Baricos, W. H., and Ignarro, L. J. (1981) Biochem.
Pharmacol., 30, 2531-2538.
19.Bussygina, O. G., and Severina, I. S. (1990)
Biokhimiya, 55, 1812-1818.
20.Garbers, D. L., and Lowe, D. G. (1994) J.
Biol. Chem., 269, 30741-30744.
21.Wedel, B., Harteneck, C., Forster, J., Friebe,
A., Schultz, G., and Koesling, D. (1995) J. Biol. Chem.,
270, 24871-24875.
22.Stone, J. R., and Marletta, M. A. (1995)
Biochemistry, 34, 14668-14674.
23.Friebe, A., Wedel, B., Harteneck, C., Foerster,
J., Friebe, A., Schultz, G., and Koesling, D. (1997)
Biochemistry, 36, 1194-1198.
24.Ignarro, L. J., Barry, B. K., Gruetter, D. Y.,
Edwards, J. C., Ohlstein, E. H., Gruetter, C. A., and Baricos, W. H.
(1980) Biochem. Biophys. Res. Commun., 94, 93-100.
25.Chirkov, Yu. Yu., Tyshchuk, I. A., and Severina,
I. S. (1988) Biokhimiya, 53, 1520-1527.
26.Ignarro, L. J. (1990) Pharmacol. Toxicol.,
67, 1-7.
27.Gerzer, R., Hofmann, F., and Schultz, G. (1981)
Eur. J. Biochem., 116, 479-486.
28.Tsai, S., Adamic, R. A., Manganiello, V., and
Vaughan, M. (1988) Biochem. J., 215, 447-455.
29.Severina, I. S. (1991) Biochem. Int.,
17, 265-278.
30.Bussygina, O. G., and Severina, I. S. (1991)
Biokhimiya, 56, 487-493.
31.Nagai, K., Suda, T., Kawasaki, K., and
Matsuchara, S. (1986) Surgery, 100, 815-821.
32.Boldyrev, A. A., Dupin, A. M., Babizhaev, M. A.,
and Severin, S. E. (1987) Biochem. Int., 15,
1105-1113.
33.Severina, I. S., and Bussygina, O. G. (1990)
Biochem. Int., 22, 455-465.
34.Swinehart, J. H. (1967) Coord. Chem. Rev.,
2, 385-402.
35.Markwardt, F., Glusa, E., Strebecher, J., Jehn,
W., and Kaizer, B. (1978) Acta Biol. Med. Germ., 37,
469-478.
36.Severina, I. S., and Bussygina, O. G. (1991)
Biochem. Int., 23, 1143-1154.
37.Severina, I. S., Bussygina, O. G., and Grigoryev,
N. B. (1992) Biochem. Int., 26, 695-705.
38.Mellion, B. T., Ignarro, L. J., Ohlstein, E. H.,
Pontecorvo, E. G., Hyman, A. L., and Kadowitz, P. J. (1981)
Blood, 57, 946-955.
39.Chirkov, Yu. Yu., Belushkina, N. N., Tyshchuk, I.
A., and Severina, I. S. (1991) Vestnik Akad. Med. Nauk SSSR,
10, 51-54.
40.Severina, I. S., and Belushkina, N. N. (1992)
Biochem. Int., 28, 621-631.
41.Chirkov, Yu. Yu., Belushkina, N. N., Tyshchuk, I.
A., and Severina, I. S. (1991) Byul. Eksp. Biol. Med., No. 2,
152-154.
42.Chirkov, Yu. Yu., Tyshchuk, I. A., and Severina,
I. S. (1990) Experientia, 46, 697-699.
43.Severina, I. S. (1992) Adv. Enzyme Regul.,
32, 35-56.
44.Volodarsky, L. B., and Tikhonova, L. A. (1985)
Khim. Geterotsikl. Soedin., No. 6, 748-752.
45.Ryaposova, I. K., Grigoryev, N. B., and Severina,
I. S. (1994) Biochemistry (Moscow), 59, 389-392.
46.Severina, I. S., Belushkina, N. N., and
Grigoryev, N. B. (1994) Biochem. Mol. Biol. Int., 33,
957-967.
47.Severina, I. S., Ryaposova, I. K., Volodarsky, L.
B., Mazhukin, D. G., Tichonov, A. Ya., Schwartz, G. Ya., Granik, V. G.,
Grigoryev, D. A., and Grigoryev, N. B. (1993) Biochem. Mol. Biol.
Int., 30, 357-366.
48.Severina, I. S., Bussygina, O. G., Belushkina, N.
N., and Grigoryev, N. B. (1995) Biochem. Mol. Biol. Int.,
36, 913-925.
49.Severina, I. S., Bussygina, O. G., Vinograd, L.
H., and Grigoryev, N. B. (1996) Biochem. Mol. Biol. Int.,
38, 509-518.
50.Belushkina, N. N., and Severina, I. S. (1996)
Biochemistry (Moscow), 61, 2140-2146.
51.Granik, V. G., Grigoryev, N. B., Vinograd, L. H.,
Severina, I. S., Mashkovskyi, M. D., Nikitin, V. B., Engalycheva, G.
N., Kalinina, M. A., and Bussygina, O. G. (1996) Mendeleev
Commun., 161-163.
52.Koikov, L. N., Alexeeva, N. V., Grigoryev, N. B.,
Levina, V. I., Turchin, K. F., Fillipenko, T. Ya., Severina, I. S.,
Ryaposova, I. K., and Granik, V. G. (1996) Mendeleev Commun.,
94-96.