REVIEW: Apoptotic Cell Death and Nitric Oxide: Activating and Antagonistic Transducing Pathways
B. Brüne*, K. Sandau, and A. von Knethen
University of Erlangen-Nürnberg, Faculty of Medicine, Department of Medicine IV-Experimental Division, Loschgestrasse 8, 91054 Erlangen, Germany; fax: +49-9131-859202; E-mail: mfm423@rzmail.uni-erlangen.de* To whom correspondence should be addressed.
Received June 24, 1997
Nitric oxide (NO) is a unique diffusible molecular messenger that occupies central roles in mammalian pathophysiology. Overproduction of NO is important for nonspecific "host" defense, helping to kill tumors and intracellular pathogens. Cytotoxicity as a result of long-lasting NO generation is now established to initiate apoptosis. Apoptotic cell death defines morphological alterations and distinctive biochemical events that lead to cell demise. NO-mediated apoptosis comprises upregulation of the tumor suppressor protein p53, activation of proteases known as caspases, chromatin condensation, DNA laddering, and is associated with alterations in the expression of apoptotic associated proteins that belong to the Bcl-2 family. An active role of NO was established by blocking adverse effects by NO-synthase inhibitors. Overexpression of the classical antiapoptotic protein Bcl-2 rescued cells from apoptosis by attenuating signaling downstream of p53 and upstream of caspase activation. Accumulating evidence suggests that transducing mechanisms can intersect and therefore a cell response to a given stimulus may alter significantly. As a result, transducing pathways of NO are not only adapted to cytotoxicity but also refer to cell protection. Protection from NO-elicited apoptosis may result as a consequence of a diffusion controlled NO/O2- (superoxide) interaction. The NO/O2- interaction redirects the apoptotic initiating activity of radicals (NO or O2-) towards protection as long as reduced glutathione compensates the resultant oxidative stress. Further, NO-mediated protective principles are understood on the basis of gene transcription of protective proteins such as heat shock proteins, hemeoxygenase-1, or cyclooxygenase-2 that attenuate cell injury in a cell specific way. The crosstalk between destructive and protective principles as a result of NO formation will determine the role of NO in cell injury. The balance between pro- and anti-apoptotic signaling mechanisms, their activation or deactivation as a result of NO formation, will allow cells to cope with NO or to exit into apoptosis.
KEY WORDS: nitric oxide, apoptosis, Bcl-2, DNA fragmentation, p53 accumulation, superoxide generation, caspases, protection
Abbreviations: GSNO) S-nitrosoglutathione; IFN-gamma) murine interferon-gamma; LPS) lipopolysaccharide; NO) nitric oxide; NOS) nitric oxide synthase; ICE) interleukin-1beta converting enzyme; NMMA) NG-monomethyl-L-arginine; PARP) poly(ADP-ribose) polymerase; SNP) sodium nitroprusside.
FORMATION OF NITRIC OXIDE AND APOPTOTIC CELL DEATH
Nitric oxide formation and target interactions. Nitric oxide (NO) is endogenously generated from the terminal guanidine residue of L-arginine by a family of NO synthase (NOS) isoenzymes, with the consequent production of stoichiometric amounts of citrulline [1]. For simplistic reasons NOS isoforms are categorized by descriptive terms based on the dependence of intracellular calcium transients required for full activity and the duration of their active participation in NO production. Constitutive forms normally become activated by a transitory cytosolic calcium increase, which promotes the release of NO over several minutes. Additionally, tyrosine phosphorylation and dephosphorylation participates in the regulation of enzyme activity [2]. A cytokine-inducible NOS isoform is expressed in many cells after stimulation with immunologic or inflammatory agents, producing large amounts of NO for hours or days [3]. NOS isoform-dependent NO production allows approximation of a low versus high NO output system, as well as a transitory versus extended NO generation period. NOS inhibitors, like NG-monomethyl-L-arginine (NMMA), are commonly used to intervene pharmacologically in NO production, thus allowing to trace back individual actions to NO-signaling.
To study signal transduction of NO irrespective to NOS involvement, NO-releasing compounds are valuable tools [4]. Nitric oxide releasing compounds, generally termed nitrovasodilators or NO donors preserve NO in their molecular structure and exhibit biological activity after decomposition. These drugs show considerable variations in their chemical structure, stability, and biological activity. Different bioavailability, at least in part, arises from differences in bioactivation and enzymatic versus nonenzymatic NO-release. Examples are organic nitrates, sodium nitroprusside (SNP), 3-morpholinosydnonimine (SIN-1), S-nitrosothiols (i.e., S-nitrosoglutathione (GSNO) or S-nitrosocysteine (CysNO)), and compounds containing the N(O)NO- functional group (i.e., diethylamine--nitric oxide complex (DEA--NO) or spermine--NO). Moreover, dinitrosyl--iron complexes are valuable tools to investigate NO-dependent signaling [5, 6]. On one side, these compounds are used as NO donors, while on the other side the formation of dinitrosyl--iron complexes are used as a detection system for endogenous NO production.
Physiological and pathophysiological activity of NO are classified by cGMP-dependent and cGMP-independent transducing pathways [7]. NO is a key transducer of the vasodilator message from the endothelium to vascular cells, is a constituent in central and peripheral neuronal transmission, and participates in the nonspecific immune defense. In terms of physiology, activation of guanylyl cyclase, formation of cGMP, and concomitant protein phosphorylation is considered of utmost importance [8]. For understanding the cytostatic or cytotoxic signals one has to consider the reactions with oxygen, superoxide, and transition metals. Reaction products (NOx, peroxynitrite (ONOO-), and metal--NO adducts, respectively) support additional reactions through their interactions with targets via redox and additive chemistry [9]. Examples for toxic actions of NO are major neurodegenerative diseases or pancreatic beta-cell destruction linked to type I diabetes mellitus. Mechanistically, the diffusion limited reaction of NO with superoxide known to generate ONOO-, inhibition of FeS-enzymes like the Krebs cycle aconitase, complex I and II of the mitochondrial respiratory chain, or ribonucleotide reductase, deregulation of poly-ADP-ribosyltransferase, interactions with thiols, and energy depletion are discussed as a likely scenario for cell death (for references, see [10, 11]).
Apoptosis and necrosis: two modes of cell death. Cell death is believed to occur by either necrotic or apoptotic mechanisms [12, 13]. These are two distinct forms of cell death which have different defining morphological and molecular features and implications for the surrounding tissue. Necrosis includes swelling of the cytoplasm and organelles, especially the mitochondria, with only minor changes in the nucleus, ultimately followed by cell dissolution. Intense mitochondrial damage is associated with rapid energy loss and disruption of internal homeostasis. Thus, intraorganelle and cytoplasmic contents leak out into the extracellular space as a result of direct membrane damage. DNA is exposed to lysosomal nucleases, causing DNA degradation, with fragments displaying a continuous spectrum of sizes.
The term "apoptosis" was introduced by Kerr and coworkers in 1972 to describe an active process of cell destruction characterized by cell shrinkage, chromatin aggregation with extensive genomic fragmentation and nuclear pyknosis [14]. The purpose of this process is to kill unwanted host cells in situations like development and homeostasis, defense, and aging. Initiation of programmed cell death is achieved by external signals delivered through surface receptors or originates inside the cell from the action of a drug or toxin. The active form of programmed cell death comprises several distinctive steps. First, morphological alterations of nuclear and cytoplasmic condensation are observed when the cell forms localized protrusions of the cell surface, which separate into multiple membrane-bound bodies also known as "apoptotic bodies" containing nuclear remnants and intact organelles. In a second stage these cell fragments are phagocytosed and rapidly degraded by neighboring cells. Apoptosis in contrast to necrosis usually occurs in isolated, single cells. This ensures that inflammatory mediators are not released, thereby preventing a secondary inflammatory response in the surrounding tissue. Although morphological criteria still remain an important standard for documentation of apoptosis, endonuclease-mediated DNA degradation is an established marker as well. DNA fragmentation starts early in the death process, appearing several hours before cell viability starts to decline. Analytically, fragmented DNA is separated by agarose gel electrophoresis into a "ladder" of fragments, generated by an endonuclease that cleaves the linker regions of DNA into 180-200 bp fragments and multiples of it. However, DNA ladder formation is not ultimately required or causatively linked to the death process.
For apoptotic stimuli propagation, initiating signals or metabolic states have to be sensed and transduced to the death effector machinery, which is highly regulated, in part, by a complex interplay between regulatory proteins. The prototypic regulator of mammalian cell death is the protooncogene bcl-2 [15]. The bcl-2 gene was first discovered because of its involvement in t(14:18) chromosomal translocation found in follicular B-cell lymphomas where it contributes to neoplastic expansion of germinal center B-cells [16, 17]. Constitutive expression of Bcl-2 protein by transfection experiments has proven that Bcl-2 can protect many cell types exposed to a variety of adverse conditions. This suggests that the protein controls a distal step in a signaling pathway leading to apoptotic cell death. Although the ultimate function of Bcl-2 remains elusive, a model for the regulation of cell survival by Bcl-2 family proteins has been suggested in which the ratio between Bax and Bcl-2 either promotes survival (excess of Bcl-2 or Bcl-xL) or augments death (excess Bax) [18]. Additionally, regulation by phosphorylation is most likely.
The basic structure of the executive cell death machinery has evolutionarily been conserved, thus allowing a diverse class of initiating agents to converge in the final death promoting signaling steps [19, 20]. This includes the catalytic processing of proteases that are activated during apoptosis, as well as their negative and positive regulators (for references, see [21, 22]). Our knowledge stems from genetic analysis of programmed cell death in the nematode Caenorhabditis elegans that identified ced-3 as a key regulator gene that encodes a cysteine protease homologous to the interleukin-1beta converting enzyme (ICE) [23, 24]. In mammalian cells multiple cDNAs encode ICE-related proteases that are termed “caspases” based on the catalytic properties of these enzymes [25], reflecting a cysteine protease mechanism, and their ability to cleave after aspartic acid. Caspases are broadly divided into two subdivisions based on sequence homology. There are proteins resembling ICE (caspase-1), such as TX/ICH-2/ICErel-II and ICErel-III, and enzymes resembling more closely Ced-3, such as CPP32/Yama/apopain (caspase-3), Mch2, and CMH-1. Ich-1 may represent a separate category (for review, see [22, 26]). All members of the ICE/Ced-3 family are found as inactive proenzymes that are activated by proteolytic cleavage to the active dimeric or tetrameric species. The functional relationship between family members is not clear but it is possible that proteases act in sequence or parallel [27]. Peptide based inhibitors that mimic the active site of caspases are pharmacological tools that block the apoptotic process by caspase inhibition. Caspase substrates include poly(ADP-ribose) polymerase (PARP), D4-GDI (GDP dissociation inhibitor for the rho family), sterol regulatory element-binding proteins, 70 kD component of U1 small nuclear ribonucleoprotein, catalytic subunit of DNA protein kinase, laminin A, DNA topoisomerase, and others [21, 22, 28]. Substrate cleavage in its causative relation to apoptotic cell death is not fully understood but the degradation of some proteins may ensure the inevitability of the death process.
Both, apoptotic and necrotic signaling pathways have been coupled to PARP. In many cell types apoptosis is accompanied by the proteolytic cleavage of PARP from a 116 kD polypeptide to a 31 kD fragment containing the N-terminal DNA binding domain and a 85 kD polypeptide containing the automodification site and the NAD+-binding domain of the protein [29]. Thus, proteolytic digestion of PARP results in functional attenuation. In contrast, it is believed that DNA damage elicits a rapid stress response in mammalian cells, which involves attachment of PARP to strand breaks and extensive synthesis of short-lived polymers by the bound enzyme. Although PARP has no direct role in DNA excision repair, the enzyme binds tightly to DNA strand breaks and sometimes interferes with repair if poly(ADP-ribose) synthesis is prevented. However, massive PARP activation following extensive DNA damage leads to NAD+, the ADP-ribose donor, depletion. In effort to resynthesize NAD+, ATP becomes depleted which ultimately results in cell death due to energy deprivation. With regard to NO signaling, PARP digestion and PARP activation followed by energy depletion has been reported [30-32].
The following sections deal with NO-mediated apoptosis. Basic observations and regulatory components such as p53 accumulation, caspase activation, and regulation by Bcl-2 overexpression are discussed. Furthermore, we will touch on a mechanism that modulates NO-dependent apoptosis, thereby regulating cellular susceptibility towards this potential cytotoxic agent.
INITIATION OF APOPTOSIS BY NITRIC OXIDE
NO donors and an active inducible NOS promote apoptotic cell death. Since the beginning of the nineties it has been realized that NO, derived from exogenously supplied NO donors or generated from an active NOS causes cellular toxicity. The table gives a selected reference source on important findings related to this area. Due to space limitations not all available articles are listed. Restrictions especially apply to a continuously increasing number of more recently published reports. Moreover, the table does not consider some initial and crucial observations on the bacteriostatic and parasitic activity of NO ([33]; for references, see [1]).
NO-modulated toxicity has been described in the following systems
(selected reading)
| Cellular systems | Remarks and crucial observations | References |
|---|---|---|
| I. NO-mediated toxicity | ||
| Melanoma cells | cell lysis via iNOS (tr) in tumor and bystander cells | K. P. Xie et al; J. Natl. Cancer Inst., 89, 421-427, 1997 |
| Macrophages (J774) | cell lysis by donors; protection by glucose and glutathione | R. Zamora et al; Europ. J. Pharmacol., 321, 87-96, 1997 |
| ESbL T lymphoma cells | cell lysis by NO which is derived from EC | V. Umansky et al; Int. J. Oncology, 10, 465-571, 1997 |
| RAW 264.7 macrophages | apoptosis; role of superoxide -- nitric oxide, p53 | B. Brüne et al; J. Biol. Chem., 272, 7253-7258, 1997 |
| RAW 264.7 macrophages | apoptosis blocked by phorbol ester; role of p53 and Bcl-2 | U. K. Meßmer and B. Brüne; Br. J. Pharmacol., in press |
| T lymphocytes (from spleen) | apoptosis by donors | Y. Okuda et al; Immunol. Lett., 52, 135-138, 1996 |
| Cerebellar granule cells | apoptosis by donors; cytoskeletal breakdown | E. Bonfoco et al; J. Neurochem., 67, 2484-2493, 1996 |
| Cancer cells (COLO205, HepG2) | apoptosis by donors in its relation to p53 accumulation | Y. S. Ho et al; Mol. Carcinog., 16, 20-31, 1996 |
| Thymocytes | apoptosis by donors; modulation by the cellular redox-state | K. Sandau and B. Brüne; Cell. Signal., 8, 173-177, 1996 |
| Mesangial cells | apoptosis by donors, p53 accumulation; protection by IL-1beta | H. Mühl et al; Europ. J. Pharmacol., 317, 137-149, 1996 |
| RAW 264.7 macrophages | apoptosis by donors; time- and concentration dependency | U. K. Meßmer and B. Brüne; Arch. Biochem. Biophys., 327, 1-10, 1996 |
| RAW 264.7 macrophages | apoptosis; p53-dependent and -independent pathways | U. K. Meßmer and B. Brüne; Biochem. J., 319, 299-305, 1996 |
| RAW 264.7 macrophages | apoptosis; initial report of PARP-cleavage, protection by Bcl-2 | U. K. Meßmer et al; FEBS Lett., 348, 162-166, 1996 |
| Smooth muscle cells | cell lysis via iNOS-induction | K. Fukuo et al; J. Clin. Invest., 95, 669-676, 1995 |
| Cortical neurons | apoptosis and necrosis by donors, ONOO- killing | E. Bonfoco et al; Proc. Natl. Acad. Sci., 92, 7162-7166, 1995 |
| HL-60/EC | apoptosis by ONOO- in HL-60 cells but not in EC | K. T. Lin et al; J. Biol. Chem., 270, 16487-16490, 1995 |
| PC12 cells | apoptosis by ONOO-; modulation by neurotrophic factors | A. G. Estevez et al; J. Neurochem., 65, 1543-1550, 1995 |
| Cerebellar granule cells | cell lysis via NOS-induction | P. G. Gunasekar et al; J. Neurochem., 65, 2016-2021, 1995 |
| Thymocytes | apoptosis by ONOO-; inhibition by trolox | M. G. Salgo et al; Arch. Biochem. Biophys., 322, 500-505, 1995 |
| Myocytes | lysis via iNOS | D. J. Pinsky et al; J. Clin. Invest., 95, 677-685, 1995 |
| Thymocytes | apoptosis by donors, p53 mRNA, protection by NO preexposure | K. Fehsel et al; J. Immunol., 155, 2858-2865, 1995 |
| RAW 264.7 macrophages | apoptosis by donors and iNOS, p53 protein, role of kinases | U. K. Meßmer et al; Mol. Pharmacol., 47, 757-765, 1995 |
| RAW 264.7 macrophages | apoptosis via iNOS, initial observation on p53 accumulation | U. K. Meßmer et al; FEBS Lett., 355, 23-26, 1994 |
| Hepatocytes | cell lysis by donors; role of mitochondrial calcium | C. Richter et al; Biochem. Biophys. Res. Commun., 205, 1143-1150, 1994 |
| Insulinoma RINm5F cells | apoptosis via iNOS | W. L. Suarez-Pinzon et al; Endocrnology, 134, 1006-1010, 1994 |
| P815 mastocytoma cells | apoptosis via iNOS | I. Kitajima et al; Biochem. Biophys. Res. Commun., 204, 244-251, 1994 |
| Hepatoma cells | cell lysis via iNOS | K. Aono et al; Biochem. Biophys. Res. Commun., 201, 1175-1181, 1994 |
| Fibroblasts (L929) | apoptosis via iNOS | S. Cui et al; Cancer Res., 54, 2462-2467, 1994 |
| Insulinoma RINm5F cells | apoptosis by donors and via iNOS, NMMA inhibition | M. Ankarcrona et al; Exp. Cell Res., 213, 172-177, 1994 |
| P815 mastocytoma cells | lysis via iNOS-induction in RAW 264.7 macrophages | R. B. Lorsbach et al; J. Biol. Chem., 268, 1908-1913, 1993 |
| Hepatoma cells (Fu5) | cell lysis by donors; cooperative action with H2O2 | I. Ioannidis and H. deGroot; Biochem. J., 296, 341-345, 1993 |
| Macrophages (peritoneum) | apoptosis via iNOS; initial report on apoptosis | J. E. Albina et al; J. Immunol., 150, 5080-5085, 1993 |
| Macrophages (peritoneum) | apoptosis via iNOS; initial report on apoptosis | M. Sarih et al; Biochem. Biophys. Res. Commun., 191, 503-508, 1993 |
| Islet cells | cell lysis by donors; DNA strand breaks | K. Fehsel et al; Diabetes, 42, 496-500, 1993 |
| Islet cells | cell lysis by donors or activated macrophages | K. D. Kröncke et al; Diabetologia, 36, 17-24, 1993 |
| Lymphoblasts (TK6) | strand breaks by NO; mutagenicity of NO | T. Nguyen et al; Proc. Natl. Acad. Sci., 89, 3030-3034, 1992 |
| Islet cells | cell lysis via iNOS; NMMA-inhibition | L. Bergmann et al; FEBS Lett., 299, 103-106, 1992 |
| Fibroblasts (C3H.OL) | cell lysis via iNOS | P. J. Duerksen-Hughes et al; J. Immunol., 149, 2114-2122, 1992 |
| II. Inhibition of NO-mediated apoptosis and/or toxicity that is suppressed by NO | ||
| EC | NO blocked TNF-alpha mediated apoptosis; inhibition of caspases | S. Dimmeler et al; J. Exp. Med., 185, 601-607, 1997 |
| EC | apoptosis of confluent EC is blocked by NO | A. Lopez-Farre et al; Am. J. Physiol., 41, H760-H768, 1997 |
| Hepatocytes | NO upregulated HSP70 blocks TNF-alpha mediated apoptosis | Y. M. Kim et al; J. Biol. Chem., 272, 1402-1411, 1997 |
| B cells (spleen) | apoptosis by purine antimetabolites is blocked by NO donors | S. Hortelano and L. Bosca; Mol. Pharmacol., 51, 414-421, 1997 |
| Carcinoma cells (HeLa G) | apoptosis via iNOS (tr) is blocked by Bcl-2 overexpression | Z. Melkova et al; FEBS Lett., 403, 273-278, 1997 |
| Mesangial cells | modulation of apoptosis by the superoxide--NO interaction | K. Sandau et al; J. Immunol., 158, 4938-4946, 1997 |
| P815 mastocytoma cells | apoptosis by donors, protection by Bcl-2 overexpression | J. E. Albina et al; J. Immunol., 157, 279-283, 1996 |
| L929, NIH3T3 cells | apoptosis via iNOS; protection by Bcl-2 overexpression | K. P. Xie et al; Canc. Immunol. Immunother., 43, 109-115, 1996 |
| Insulinoma RINm5F cells | HSP70 overexpression blocked cell lysis but not DNA damage | K. Bellmann et al; FEBS Lett., 391, 185-188, 1996 |
| RAW 264.7 macrophages | NO-mediated apoptosis is blocked by Bcl-2 overexpression | U. K. Meßmer et al; J. Biol. Chem., 271, 20192-20197, 1996 |
| RAW 264.7 macrophages | cytokine- and NO-mediated protection; p53 downregulation | B. Brüne et al; Biochem. Biophys. Res. Commun., 229, 396-401, 1996 |
| B lymphocytes | apoptosis blocked by NO, cGMP-dependent, Bcl-2 related | A. M. Genaro et al; J. Clin. Invest., 95, 1884-1890, 1995 |
| EC | NO donors block LDL toxicity (cell lysis) | A. T. Struck et al; FEBS Lett., 361, 291-294, 1995 |
| Hepatocytes | NO upregulated Hsp32 blocked cell lysis by NO | Y. M. Kim et al; FEBS Lett., 374, 228-232, 1995 |
| Fibroblasts (NIH3T3) | metallothionein overexpression blocked NO toxicity | M. A. Schwarz et al; Proc. Natl. Acad. Sci., 92, 4452-4456, 1995 |
| Myocytes | NO blocked stretch induced death | W. Cheng et al; J. Clin. Invest., 96, 2247-2259, 1995 |
| Chondrocytes | NO blocked O2--induced apoptosis | F. J. Blanco et al; Am. J. Pathol., 146, 75-85, 1995 |
| Lung fibroblasts (V79) | NO blocked peroxide-mediated toxicity | D. A. Wink et al; Arch. Biochem. Biophys., 319, 402-407, 1995 |
| B lymphocytes | apoptosis blocked by NO, cGMP-independent | J. B. Mannick et al; Cell, 79, 1137-1146, 1994 |
| Lung fibroblasts (V79) | NO blocked peroxide toxicity | D. A. Wink et al; Proc. Natl. Acad. Sci., 90, 9813-9817, 1993 |
Initial reports measured cell lysis as an index of toxicity. NO initiated apoptosis was first realized for peritoneal macrophages and macrophage cell lines such as RAW 264.7 cells [34, 35]. Further studies on apoptotic mechanisms concentrated on insulinoma RINm5F cells [36, 37], thymocytes [38, 39], chondrocytes [40], and rat mesangial cells [41], among others (the table).
During our studies we mainly concentrated on the mouse monocyte/macrophage cell line RAW 264.7, RINm5F cells, and rat mesangial cells. These investigations revealed the following characteristic, NO-mediated alterations: induction of the inducible NOS in RAW 264.7 macrophages by LPS and IFN-gamma produced typical morphological and biochemical apoptotic alterations [35] that were prevented by NMMA, a NOS inhibitor. Further, we observed that NMMA-reduced DNA fragmentation known as the typical apoptotic "DNA-ladder". With the use of several spontaneously decomposing NO donors, known as "NONOates", we showed that the integral of concentration over time accounted for the damage [42]. Comparable to the macrophage system, endogenously generated or exogenously supplied NO initiated apoptotic alterations in RINm5F cells [37]. During NO intoxication in macrophages, cellular NAD+ and ATP levels were preserved. Consistent, membrane integrity assayed by LDH release was assured and inhibitors of poly(ADP-ribose) polymerase, like 3-aminobenzamide, were not effective [35, 42].
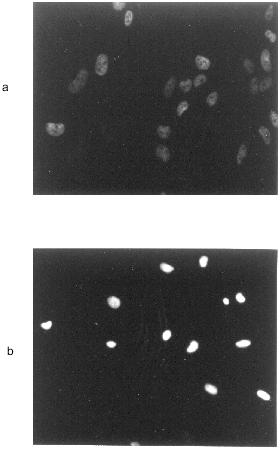
Furthermore, activation of protein kinase C (PKC) or protein kinase A (PKA) suppressed NO-mediated apoptotic alterations [35]. In analogy to several other systems, macrophages were protected by Zn2+, an inhibitor for the Ca2+,Mg2+-dependent endonuclease that participates in DNA cleavage. Figure 1 exemplifies morphological apoptotic alterations in rat mesangial cells upon treatment with the NO donor S-nitrosoglutathione.
NO-mediated accumulation of the tumor suppressor p53 and caspase activation. The tumor suppressor protein p53 maintains genomic integrity by acting as a “guardian of the genome” [43]. Normally, the half-life of p53 is short, thus resulting in undetectable or low amounts of the protein. In response to DNA damage, the protein accumulates, thereby causing a G1 cell cycle arrest. In case of severe DNA damage p53 initiates apoptosis (for references, [44]). For macrophages and insulinoma RINm5F cells we established accumulation of p53 during NO-mediated cell death [45]. Cytokine/LPS induced p53 accumulation was suppressed by NMMA, which indicates an active role of NO. Furthermore, we probed for NO-induced apoptosis in RAW 264.7 macrophages stably transfected with plasmids encoding p53 antisense RNA [46]. These results established a functional role of the tumor suppressor p53 during NO-induced apoptotic cell death. However, p53 antisense experiments and the use of the p53 negative cell line U937 substantiated p53-independent signaling pathways to be operative during NO-mediated apoptosis as well [46].Fig. 1. GSNO-induced DNA fragmentation in rat mesangial cells. Rat mesangial cells (3·103 cells/assay) were stimulated without additions (a) or with 2 mM S-nitrosoglutathione (b) for 16 h. DNA strand breaks were detected by the TUNEL technique. For details see reference [50].
Further, we directed our interest on caspase activation and the degradation of classical substrates such as PARP [30]. PARP cleavage was established in response to endogenously generated or exogenously supplied NO in parallel to DNA fragmentation. This seems logical when we consider the diverse apoptotic signaling mechanisms to be integrated at a point that results in proteolytic events such as caspase activation.
ANTAGONISTIC SIGNALING PATHWAYS DURING NO INTOXICATION
Bcl-2 overexpression. Cell death is controlled, in part, by the classical antiapoptotic protein Bcl-2. During our studies we stably transfected RAW 264.7 macrophages with human Bcl-2 [47]. Transfectants were protected from cell death following NO donor exposure or activation of inducible NOS. Although Bcl-2 blocked apoptosis, p53 expression remained unchanged in transfectants. In contrast, PARP cleavage and thus caspase activation was almost completely blocked [30.. We conclude that Bcl-2 acts downstream of p53 and upstream of caspase activation, presumably nullifying the NO-mediated increase in Bax protein in RAW 264.7 cells. Protection against NO-mediated cytotoxicity by Bcl-2 overexpression has been confirmed by others [48].
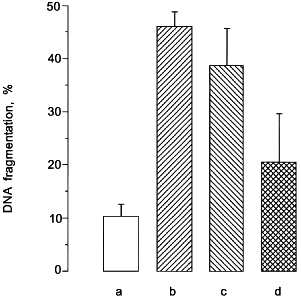
The superoxide--nitric oxide radical interaction. Both NO· and superoxide (O2-) are important inflammatory mediators. Notably, mesangial cells (MC) [49] or macrophages not only generate NO· but also O2- after cytokine stimulation. Therefore, we investigated the interrelation between NO· and O2- in MC or macrophage cell death by generating radicals with the use of NO donors (GSNO, spermine--NO) and O2- generating systems (2,3-dimethoxy-1,4-naphthoquinone or hypoxanthine--xanthine oxidase). Both radicals induced apoptosis and/or necrosis in a concentration dependent manner. Noteworthy is that the coincubation of NO· and O2- was cross-protective, whereby maximum protection required the existence of a timely coincident and balanced NO·/O2- ratio [50, 51]. Exemplified in Fig. 2, the formation of O2- or NO· produced DNA fragmentation, while the response was significantly lowered by radical cogeneration.
Suggestively, a balanced ratio of reactive nitrogen and oxygen species participates in regulating apoptosis after radical (O2- and/or NO·) formation. Although NO can be cytotoxic, there are situations where NO functions as a protective signal. Examples are ischemia-reperfusion, peroxide-induced toxicity, lipid peroxidation, or myocardial injury (see the table for details). Despite the likely formation of ONOO- in some of these studies is assumed, a direct damaging effect of this NO·/O2- combination product appeared negligible under those experimental conditions [52].Fig. 2. DNA fragmentation in response to NO and/or superoxide. Mesangial cells (2.5·105 cells/assay) were incubated without any addition (a), with systems hypoxanthine (50 µM)--xanthine oxidase (0.5 U/ml) (b) and spermine--NO (500 µM) (c), or the combination of both (d). DNA-fragmentation was quantitated after 24 h by the diphenylamine assay as outlined in reference [50]. Results are mean values ± SEM of at least three separate experiments; *p < 0.02 versus incubations with NO· and O2- alone.
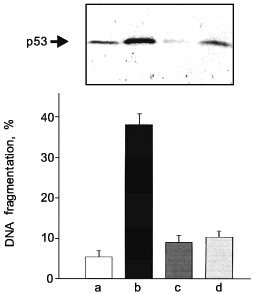
Protection by low dose NO and/or cytokine prestimulation. Pretreatment of macrophages with LPS/IFN-gamma in the presence of NMMA, imparts resistance to apoptotic cell death, normally elicited by high dose exogenously supplied GSNO. Similarly, pretreatment with low, and thus non-toxic doses of GSNO (25-200 µM) conferred resistance from a second exposure to a higher dose of GSNO (1 mM) [53]. Protection is seen at the level of DNA fragmentation and accumulation of the tumor suppressor protein p53. Thus, inducible protective mechanisms that suppress NO-initiated apoptosis are upregulated in response to NO delivering compounds and by LPS, cytokine preactivation.
Protection against exogenously added GSNO by cytokine- or low-dose NO-prestimulation is shown in Fig. 3 by measuring DNA fragmentation.
In some analogy, NO-induced autoprotection in association with suppression of NO- or TNF-alpha-mediated hepatocyte or endothelial cell death has been reported with the assumption that heme oxygenase and/or heat shock protein induction or caspase inhibition are involved (see the table for details). Moreover, initial observations that NO blocked B lymphocyte apoptosis has been reported, with the diverse argumentation that protection is either cGMP-dependent [54] or -independent [55].Fig. 3. Cellular protection by pretreatment with LPS/IFN-gamma. RAW 264.7 macrophages were stimulated with 1 mM GSNO (b, d), the combination of LPS (10 µg/ml), IFN-gamma (100 U/ml), NMMA (1 mM) (c, d), or all agonists (d). DNA fragmentation was quantitated 8 h after the addition of GSNO by the diphenylamine assay as described in reference [53]. Western blot analysis of p53 was measured 4 h after the addition of GSNO. Cells were preincubated with LPS/IFN-gamma/NMMA for 15 h before the apoptotic agonist (1 mM GSNO) was supplied; a) control (no additions).
Suggestively, cellular susceptibility towards apoptotic signaling pathways can effectively be regulated by cytokine or low level NO pretreatment. The activation or deactivation of these pathways will prevent that NO acts cytotoxic or, if not operative, gives NO the ability to kill cells by apoptotic mechanisms.
CONCLUSIONS
The toxicity of NO is modulated by the cellular biological redox-milieu, especially by superoxide cogeneration. Relative rates of NO· formation, redox-reactions, the combination with oxygen, superoxide, and other biomolecules will determine the signaling pathway of nitric oxide. For macrophages, insulinoma RINm5F cells, and many other systems (see the table), activation of the inducible NOS generated sufficient amounts of NO to promote cell lysis or apoptotic cell death. NO-mediated apoptosis is characterized by p53 accumulation and caspase activation in association with the occurrence of typical morphological and biochemical apoptotic parameters. However, not all cellular systems which exhibit inducible NOS upregulation after cytokine treatment enter the apoptotic or necrotic pathway. Inhibition of NO-initiated apoptotic cell death is accomplished by Bcl-2 overexpression, the balanced cogeneration of superoxide, or induction of protective proteins such as heme oxygenase, metallothionein, or heat shock proteins (see the table). Inhibition of NO-mediated apoptosis or an interceptive role of NO in blocking toxicity by other agents such as TNF-alpha, is transmitted by either cGMP-dependent or -independent pathways. The understanding of the interplay between these signaling mechanisms will help to reveal how NO affects life and death of cellular systems.
We thank the Deutsche Forschungsgemeinschaft and in part the European Community for their support.
REFERENCES
1.Nathan, C. (1992) FASEB J., 6,
3051-3064.
2.Fleming, I., Bara, A. T., and Busse, R. (1996)
J. Biol. Chem., 33, 225-234.
3.Moncada, S., Palmer, R. M. J., and Higgs, E. A.
(1991) Pharmacol. Rev., 43, 109-142.
4.Noack, E., and Murphy, M. (1991) in Oxidative
Stress: Oxidants and Antioxidants (Sies, H., ed.) Academic Press,
San Diego, pp. 445-489.
5.Vanin, A. F. (1995) Biochemistry (Moscow),
60, 225-230.
6.Boese, M., Mordvintcev, P. I., Vanin, A. F., Busse,
R., and Mülsch, A. (1995) J. Biol. Chem., 270,
29244-29249.
7.Schmidt, H. H. H. W., and Walter, U. (1994)
Cell, 78, 919-925.
8.Schmidt, H. H. H. W. (1992) FEBS Lett.,
307, 102-107.
9.Stamler, J. S. (1994) Cell, 78,
931-936.
10.Brüne, B., Meßmer, U. K., and Sandau,
K. (1995) Toxicol. Lett., 82/83, 233-237.
11.Krönke, K.-D., Fehsel, K., and
Kolb-Bachofen, V. (1995) Biol. Chem. Hoppe-Seyler, 376,
327-343.
12.Wyllie, A. H., Kerr, J. F. R., and Currie, A. R.
(1980) Int. Rev. Cytol., 68, 251-306.
13.Thompson, C. B. (1995) Science,
267, 1456-1462.
14.Kerr, J. F. R., Wyllie, A. H., and Currie, A. R.
(1972) Br. J. Cancer, 26, 239-257.
15.Reed, J. (1994) J. Cell Biol., 124,
1-6.
16.Tsujimoto, Y., Cossman, J., Jaffe, E., and Croce,
C. M. (1985) Science, 228, 1440-1443.
17.Gajewski, T. F., and Thompson, C. B. (1996)
Cell, 87, 589-592.
18.Steller, H. (1995) Science, 267,
1445-1449.
19.Nagata, S. (1997) Cell, 88,
335-365.
20.Patel, T., Gores, G. J., and Kaufmann, S. H.
(1996) FASEB J., 10, 587-597.
21.Zhivotovsky, B., Burgness, D. H., Vanags, D. M.,
and Orrenius, S. (1997) Biochem. Biophys. Res. Commun.,
230, 481-488.
22.Yuan, J., and Horvitz, H. R. (1990) Dev.
Biol., 138, 33-41.
23.Yuan, J., Shaham, S., Ledoux, S., Ellis, H. M.,
and Horvitz, H. R. (1993) Cell, 75, 641-652.
24.Alnemi, E. S., Livingston, D. L., Nicholson, D.
W., Salvesen, G., Thornberry, N. A., Wong, W. W., and Yuan, J. (1996)
Cell, 87, 171.
25.Takahasi, A., and Earnshaw, W. C. (1996) Curr.
Opin. Genet. Dev., 6, 50-55.
26.Orth, K., O'Rourke, K., Salvesen, G. S., and
Dixit, V. M. (1996) J. Biol. Chem., 271, 20977-20980.
27.Whyte, M. (1996) Trends Cell Biol.,
6, 245-248.
28.Kaufmann, S. H., Desnoyers, S., Ottaviano, Y.,
Davidson, N. E., and Poirier, G. G. (1993) Cancer Res.,
53, 3976-3985.
29.Kaufmann, S. (1989) Cancer Res.,
49, 5870-5878.
30.Meßmer, U. K., Reimer, D. M., Reed, J., and
Brüne, B. (1996) FEBS Lett., 384, 162-166.
31.Zhang, J., Dawson, V. L., Dawson, T. M., and
Snyder, S. H. (1994) Science, 263, 687-689.
32.Heller, B., Wang, Z.-Q., Wagner, E. F., Radons,
J., Bürkle, A., Fehsel, K., Burkart, V., and Kolb, H. (1995) J.
Biol. Chem., 270, 11176-11180.
33.Hibbs, J. B., Taintor, R. R., and Vavrin, Z.
(1987) Science, 235, 473-476.
34.Albina, J. E., Cui, S., Mateo, R. B., and
Reichner, J. S. (1993) J. Immunol., 150, 5080-5085.
35.Meßmer, U. K., Lapetina, E. G., and
Brüne, B. (1995) Mol. Pharmacol., 47, 757-765.
36.Kaneto, H., Fujii, J., Seo, H. G., Suzuki, K.,
Matsuoka, T., Nakamura, M., Tatsumi, H., Yamasaki, Y., Kamada, T., and
Taniguchi, N. (1995) Diabetes, 44, 733-738.
37.Ankarcrona, M., Dypbukt, J. M., Brüne, B.,
and Nicotera, P. (1994) Exp. Cell Res., 213, 172-177.
38.Fehsel, K., Kröncke, K.-D., Meyer, K. L.,
Huber, H., Wahn, V., and Kolb-Bachofen, V. (1995) J. Immunol.,
155, 2858-2865.
39.Sandau, K., and Brüne, B. (1996) Cell.
Signal., 8, 173-177.
40.Blanco, F. J., Ochs, R. L., Schwarz, H., and
Lotz, M. (1995) Am. J. Pathol., 146, 75-85.
41.Mühl, H., Nitsch, D., Sandau, K.,
Brüne, B., Varga, Z., and Pfeilschifter, J. (1996) FEBS
Lett., 382, 271-275.
42.Meßmer, U. K., and Brüne, B. (1996)
Arch. Biochem. Biophys., 327, 1-10.
43.White, E. (1994) Nature, 371,
21-22.
44.Smith, M. L., and Fornace, A. J., Jr. (1996)
Am. J. Pathol., 148, 1019-1022.
45.Meßmer, U. K., Ankarcrona, M., Nicotera,
P., and Brüne, B. (1994) FEBS Lett., 355, 23-26.
46.Meßmer, U. K., and Brüne, B. (1996)
Biochem. J., 319, 299-305.
47.Meßmer, U. K., Reed, J. C., and Brüne,
B. (1996) J. Biol. Chem., 271, 20192-20197.
48.Albina, J. E., Martin, B.-A., Henry, W. L., Jr.,
Louis, C. A., and Reichner, J. S. (1996) J. Immunol.,
157, 279-283.
49.Pfeilschifter, J. (1995) Kidney Int.,
48, S50-S60.
50.Brüne, B., Götz, C., Meßmer, U.
K., Sandau, K., Hirvonen, M.-R., and Lapetina, E. G. (1997) J. Biol.
Chem., 272, 7253-7258.
51.Sandau, K., Pfeilschifter, J., and Brüne, B.
(1997) J. Immunol., 158, 4938-4946.
52.Wink, D. A., Hanbauer, I., Murali, C. K.,
DeGraff, W., Gamson, J., and Mitchell, J. B. (1993) Proc. Natl.
Acad. Sci. USA, 90, 9813-9817.
53.Brüne, B., Gölkel, C., and von Knethen,
A. (1997) Biochem. Biophys. Res. Commun., 229,
396-401.
54.Genaro, A. M., Hortelano, S., Alvarez, A.,
Martinez-A, C., and Bosca, L. (1995) J. Clin. Invest.,
95, 1884-1890.
55.Mannick, J. B., Asano, K., Izumi, K., Kieff, E.,
and Stamler, J. S. (1994) Cell, 79, 1137-1146.