Is One Amino Acid Responsible for Substrate Specificity of Monoamine Oxidase A and B?
A. V. Veselovsky, A. S. Ivanov, and A. E. Medvedev*
Institute of Biomedical Chemistry, Russian Academy of Medical Sciences, Pogodinskaya ul. 10, Moscow, 119832 Russia; fax: (095) 245-0857; E-mail: medvedev@ibmh.msk.su* To whom correspondence should be addressed.
Received May 26, 1998; Revision received June 23, 1998
Mutation of Phe-208 for Ile in monoamine oxidase (MAO) A and mutation of Ile-199 for Phe in MAO B inverted substrate specificity and inhibitor selectivity of the mutants towards the opposite form of the enzyme, MAO B and MAO A, respectively (Y. Tsugeno and A. Ito (1997) J. Biol. Chem., 272, 14033-14036). However, according to data presented by Tsugeno and Ito such point mutations did alter kinetic characteristics of oxidative deamination of some but not all substrates. These point mutations did not alter the secondary structure evaluated by four computer methods. Analysis of the efficacy of a few series of inhibitors suggests spatial differences of substrate binding sites of MAO A and MAO B. All these discrepancies indicate that one amino acid can be responsible for binding of some but not all substrates and inhibitors.
KEY WORDS: monoamine oxidases A and B, substrate specificity, point mutation, chimeric molecules, substrate binding site, spatial structure
Monoamine oxidase (MAO; EC 1.4.3.4) plays a central role in the metabolism of important neurotransmitter monoamines. Altered MAO activity observed in numerous neuropsychiatric disorders and the possibility of their correction with MAO inhibitors has made this enzyme a very popular target of basic and clinical studies [1].
In mammals, MAO exists in two forms, MAO A and MAO B, which were initially distinguished by sensitivity to acetylenic inhibitors (clorgyline and deprenil, respectively) and by preferential substrate oxidation (serotonin and phenylethylamine, respectively) [1]. Now it is finally recognized that these enzymes are different proteins encoded by different genes [2]. However, the three-dimensional structure of these enzymes remains unknown due to unsuccessful attempts for their crystallization [3]. MAO A and MAO B are integral proteins of the outer mitochondrial membrane, and they easily aggregate during purification. Thus, the use of procedures of protein crystallization results in formation of unstructured forms [3]. Lack of amino acid sequence homology with proteins with known three-dimensional structure limits the applicability of methods for computer modeling of MAO A and MAO B [4].
So, the structures of MAO A and MAO B are now being analyzed using a few directions. The first one includes a study of soluble (mono)amine oxidases from microorganisms as possible evolutionary precursors of human/mammalian MAO A and MAO B [3]. However, even in spite of successful crystallization of the enzyme from A. niger (so-called monoamine oxidase N) [3], its molecular properties (including amino acid sequence) so significantly differ from those of MAO A and MAO B that it cannot be considered as a direct evolutionary precursor of human/mammalian MAO A and MAO B.
Another direction focuses on essential structural factors, fragments of amino acid sequences, determining differences in substrate specificity and inhibitor selectivity of animal/human MAO A and MAO B. Analysis of deduced amino acid sequence revealed high identity between MAO A and MAO B (~70%) and the existence of a few highly conservative sites [2, 5]. The N-terminus contains a site homologous to the FAD-binding fragment of many flavin-containing enzymes, and the C-terminus accommodates the phosphate group of flavin. The C-terminus is characterized by a high content of hydrophobic and positively charged amino acids and apparently serves for anchoring the enzyme in the membrane [5-8]. In both forms cysteine responsible for covalent anchoring of FAD was recognized. Analysis of the functional importance of cysteines in MAO B revealed that substitution of three of nine cysteine residues (including the FAD-binding one) for serine, alanine, and histidine was accompanied by a loss of enzymatic activity, whereas substitution of FAD-binding cysteine for alanine in MAO A was not accompanied by a total loss of activity [9, 10].
During last 5-7 years a few groups have been constructing chimeric MAO molecules consisting of various fragments of amino acid sequence of MAO A and MAO B. These experiments have revealed that the N-terminus determines FAD-binding, but it does not influence enzyme specificity, whereas the C-terminal fragment of 28 amino acids represents the mitochondrial targeting signal responsible for MAO insertion into the membrane. However, neither the N- nor the C-terminal regions determine substrate specificity and inhibitor selectivity of these two enzymes [2]. Construction of chimeric molecules with substitution of a median fragment of the polypeptide chain resulted in certain changes in catalytic and regulatory properties of enzymes expressed in COS cells [2]. These manipulations exerted more pronounced effects on the inhibitory selectivity. For example, [I]50 (concentration required for 50% inhibition) for inhibition of chimeric MAO A containing amino acid segment 161-375 of MAO B (so-called MAO AB161-375A) by clorgyline, a selective inhibitor of MAO A, was three orders of magnitude higher than that of wild-type MAO A and nearly matched that of MAO B (0.54 ± 0.08 and 0.64 ± 0.15 µM for MAO AB161-375A and MAO B, respectively). Comparison of the inhibition pattern of wild-type and chimeric MAO by a selective MAO B inhibitor also revealed similarity between MAO AB161-375A and MAO B. However, the catalytic properties of the chimeric enzyme with phenylethylamine as substrate (Km, catalytic constant kcat, and ratio kcat/Km) exhibited an intermediary position between the wild-types of MAO A and MAO B. A similar situation was also observed in the case of benzylamine, another selective substrate of MAO B, with the exception that Km and kcat values were significantly higher in the case of chimeric MAO AB161-375A than for wild-types MAO A and MAO B.
In the middle of 1997 a paper by Tsugeno and Ito, "A key amino acid responsible for substrate selectivity of monoamine oxidase A and B”, appeared in The Journal of Biological Chemistry [11]. This was an impressive result by Ito’s group which was sequentially studying chimeric MAO A and MAO B molecules. Two years earlier this group detected fragments of amino acid sequences of both MAO A and MAO B responsible for substrate specificity and inhibitor sensitivity of these enzymes. According to data by Ito’s group, the amino acid segment 120-220 determines substrate specificity whereas amino acid segment 220-400 is responsible for catalytic activity [12]. This is consistent with data by Shih et al. [2]. And now these authors appear to localize a key amino acid determining selective substrate binding at the active sites of MAO A and MAO B. Using the method of site-directed mutagenesis, Tsugeno and Ito substituted Phe-208 in MAO A for the corresponding residue of Ile in MAO B and made "reversed” substitution of Ile-199 in MAO B for Phe. This was accompanied by change in substrate specificity of the chimeric molecules which acquired some properties of substrate specificity typical for the opposite type of MAO (the table).
Km values (in mM) for deamination of some amines
catalyzed by wild and mutant forms of MAO A and MAO B (data are taken
from [12])
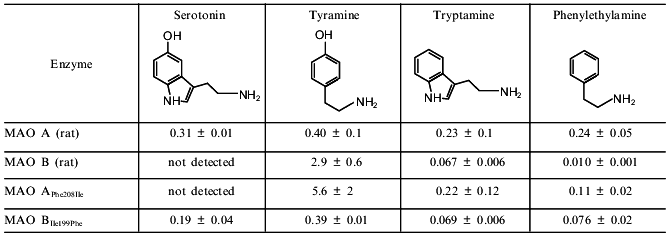
The comparison of Km values for wild-type MAOs and mutants shows that the most dramatic changes were found for deamination of serotonin and tyramine as substrates. Substitution of Phe in MAO A for Ile reduced serotonin deaminating activity so strongly that authors could not even determine Km values. The affinity for tyramine was reduced 14-fold. So, in the case of these two substrates mutant MAO A with the single amino acid substitution behaves similarly to MAO B. Substitution of Ile in MAO B for Phe increased affinity of the mutant enzyme for serotonin and tyramine up to the level observed in MAO A. These mutant forms, MAO APhe208Ile and MAO BIle199Phe demonstrated sensitivity to the "diagnostic” concentration of clorgyline and deprenil which was typical for wild MAO B and MAO A, respectively. So, these data by Ito’s group clearly indicate that in the case of substrates used the mutation of only one amino acid could reverse the type of MAO.
However, in the case of phenylethylamine and tryptamine as substrates the situation is different (the table). Low concentrations of phenylethylamine are selectively deaminated by MAO B, whereas tryptamine is a substrate for both MAOs [13]. Substitution of Phe in MAO A for Ile did not influence Km for tryptamine, whereas the affinity of this mutant (MAO APhe208Ile) to phenylethylamine, the "diagnostic” MAO B substrate, was one order of magnitude less than in wild-type MAO B. Similar changes were found in the mutant MAO BIle199Phe: the enzyme affinity for tryptamine remained unchanged, and it was reduced for phenylethylamine, but nevertheless Km MAO BIle199Phe for phenylethylamine did not reach Km values typical for wild-type MAO A.
Comparison of substrate structures (see the table) shows that mutations influenced only the affinity for substrates possessing a hydroxyl group in the benzene ring (serotonin and tyramine). Substitution of Phe in MAO A for the more hydrophobic amino acid, Ile, reduced the enzyme affinity for hydrophilic serotonin and tyramine but increased the affinity for the more hydrophobic phenylethylamine. Substitution of Ile in MAO B for Phe induced the opposite effect.
Due to lack of changes in tryptamine oxidation, the reasonable question arises whether one amino acid is responsible for substrate specificity of MAO A and B. Monoamine oxidase possess wide substrate specificity [13, 14], and besides the acetylenic inhibitors used as the "diagnostic” probe for determination of A- and B-forms of the enzyme there are many other selective inhibitors of these enzymes [15].
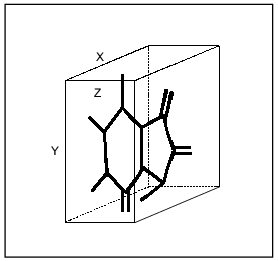
Analysis of the efficacy of MAO A and MAO B inhibition by a few series of reversible inhibitors suggests the existence of different substrate-binding sites of MAO A and MAO B [4, 16, 17]. Using a few of isatin (indole-2,3-dione) analogs characterized by limited rotation of substituents in the molecule (which gives the minimal amount of energetically favorable conformers) we found differences in the sizes of most potent and selective MAO A and MAO B inhibitors. Sizes of selective MAO A inhibitors with planar structure did not exceed 14 Å "in length” and 6 Å "in width” (Fig. 1). Selective MAO B inhibitors were smaller (8.5, and 5.0 Å, respectively) [16, 17]. Since compounds used in that study inhibited both enzymes in a competitive manner with respect to the substrates (serotonin and phenylethylamine, respectively) these differences obviously reflect features of substrate binding sites of MAO A and MAO B. "Rigid” analogs of the short acting antidepressant pirlindole (2,3,3a,4,5,6-hexahydro-8-methyl-1H-pyrazino[3,2,1-j,k]carbazole) selectively inhibiting MAO A had 3-D sizes which did not exceed 13 Å "in length”, 7 Å "in width”, and 4.4 Å "in height” (Medvedev et al. (1998) J. Chem. Inf. Comput. Sci., in press). Flexible pirlindole analogs which could be "packed” into a more compact (and energetically possible) structure effectively inhibited MAO B as well (Fig. 2). The latter agrees with data from Singer’s laboratory on MAO B inhibition by oxodiazolones [18]. Long and flexible oxodiazolones were much more effective MAO B inhibitors than their rigid shorter analogs which possessed sizes (at least along X and Y axes) exceeding limits which we found for selective MAO B inhibitors from other chemical groups [4]. All these data obviously suggest 3-D differences of substrate binding sites of MAO A and MAO B. The Phe-208 residue in MAO A is probably involved into interaction (binding?) with the hydroxyl group of substrates serotonin and tyramine and with the hydrophilic center of clorgyline (and other selective MAO A inhibitors?). The Ile-199 residue in MAO B might be responsible for the interaction with more hydrophobic substrates (phenylethylamine, benzylamine) and with deprenil. This scheme is also applicable to such preferential MAO A substrates as adrenaline and noradrenaline possessing hydroxyl groups in the aromatic ring [13].
Fig. 1. Three-dimensional box for determination of linear sizes of inhibitors (shown using isatin as an example).
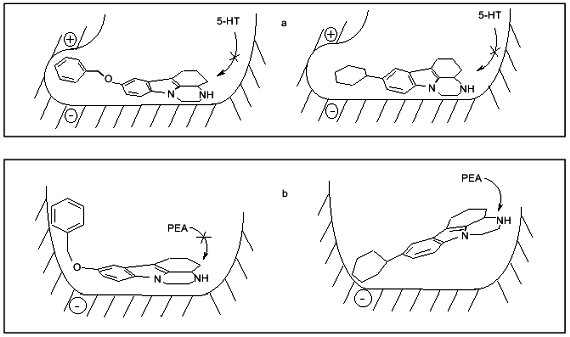
These suggestions do not contradict our 3D-QSAR + CoMFA models of MAO A and MAO B based on pirlindole analogs and showing the existence of favorable regions both for positive and negative charges which can probably explain preferential interaction of serotonin and tyramine with MAO A (Fig. 3).Fig. 2. Scheme of the structure of the active sites of MAO A (a) and MAO B (b) and possible positions of inhibitors, pirlindole analogs, with "flexible” (left) and "rigid” (right) substituents. Arrows show that phenylethylamine (PEA) may displace the inhibitor with a "rigid” but not that with a "flexible” substituent. Serotonin (5-HT) cannot displace either type of molecule.
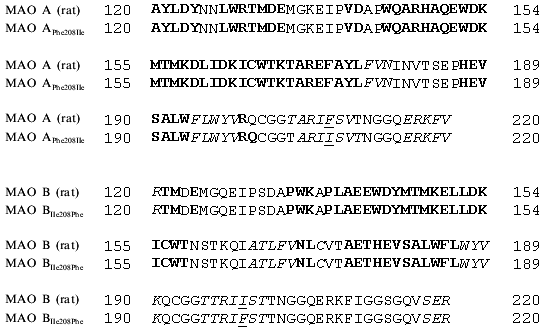
Our analysis of series of MAO inhibitors suggests significant differences in the active site structures of these enzymes. It is possible that point mutations caused local changes in the active site structure of these enzymes. However, using four methods of computer prediction of the secondary structure based on amino acid sequence (methods by Levin, Gilbrat, Garnier, and the homology method) did not reveal changes in the secondary structures in the regions of substituted amino acids (Fig. 4). All these methods predict positions of mutated amino acids of either MAO A or MAO B in beta-sheets and point substitution influenced neither the type of secondary structure nor number of amino acids in it. This suggests that the point mutations were not accompanied by significant structural rearrangements, and alterations in substrate specificity are probably due to local changes in the substrate binding site of the active center.Fig. 3. Contour maps of distribution of steric (a, c) and electrostatic (b, d) fields of 3D-QSAR + CoMFA models of MAO A (a, b) and MAO B (c, d) for pirlindole derivatives. For steric fields solid lines show unfavorable regions for the position of substituents and broken lines show favorable regions for the position of substituents, which can increase the inhibitory activity of molecules. For electrostatic fields solid lines show regions where positively charged substituents increase inhibitory effect, broken lines designate favorable regions for negatively charged groups. The pirlindole molecule is used as the example.
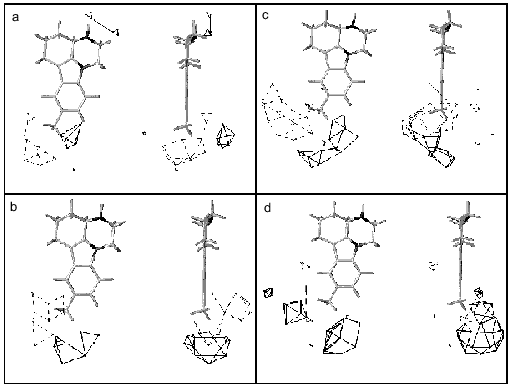
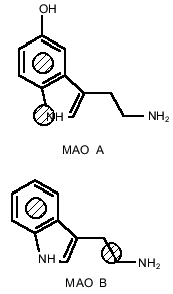
Using analysis of the main selective substrates and inhibitors of MAO A and MAO B, it is possible to develop a pharmacophore model for their substrates (Fig. 5). A pharmacophore model represents a number of points in space characterized by certain physicochemical properties and distances between them. This model determines the minimal number of functional groups and their properties and mutual interposition which must be present in the ligand molecule to be bound to the target molecule. The benzene ring and pyrrole nitrogen atom are pharmacophore points for MAO A, whereas in the case of MAO B the benzene ring and a hydrophobic region of the side chain are the pharmacophore points. These pharmacophore models suggest that at least two amino acids are involved into substrate binding at the active site. If the interaction with the benzene ring may be determined by the same amino acid the second amino acids must be different and may be probably located at different positions of the polypeptide chain of the MAO. In this connection, it should be mentioned that two amino acids (Thr-158 and His-382) essential for catalytic activity of MAO B have been identified [20].Fig. 4. Prediction of secondary structures by method of Gilbrat et al. [19] in MAO A and MAO B (wild and mutant strains) in the region determining substrate binding. Amino acids of alpha-helix and beta-sheet are marked in bold and italic, respectively. Mutated amino acids are underlined.
Thus, we believe that one amino acid may determine the specificity of binding of some selective substrates and inhibitors of MAO A and MAO B, but it cannot account for the number of other basic differences between these enzymes such as different affinity for oxygen and distinct effect of substrates on kinetic parameters of half-reactions [21].Fig. 5. Pharmacophore models for MAO A and MAO B. Hatched points indicate sites of preferential binding in the active site.
This work was supported by the Ministry of Science of the Russian Federation (grant No. 04.01.06.96), sub-program "Design of New Drugs by Methods of Chemical and Biological Synthesis" within the Federal program "Studies and Developments of Priority Directions in the Development of Civil Science and Technology”.
REFERENCES
1.Gorkin, V. Z., and Medvedev, A. E. (1995) in
Proteins and Peptides [in Russian], Vol. 1, Nauka, Moscow, pp.
83-88.
2.Shih, J. C., Chen, K., and Geha, R. M. (1998) J.
Neural Transm., 52 (Supl.), 1-8.
3.Singer, T. P., Yankovskaya, T. P., Bernard, S.,
Cronin, C., and Sablin, S. O. (1997) Vopr. Med. Khim.,
43, 440-456.
4.Veselovsky, A. V., Ivanov, A. V., and Medvedev, A.
E. (1997) Vopr. Med. Khim., 43, 527-536.
5.Bach, A. W. J., Lan, N. C., Johnson, D. L., Abell,
C. W., Bembeneck, M. E., Kwan, S. W., Seeburg, P. H., and Shih, J. H.
(1988) Proc. Natl. Acad. Sci. USA, 85, 4934-4938.
6.Wu, H.-E., Chen, K., and Shih, J. C. (1993) Mol.
Pharmacol., 43, 888-893.
7.Kwan, S. W., Lewis, D. A., Zhou, B. P., and Abell,
C. W. (1995) Arch. Biochem. Biophys., 316, 385-391.
8.Zhou, B. P., Lewis, D. A., Kwan, S. W., Kirksey, T.
J., and Abell, C. W. (1995) Biochemistry, 34,
9526-9531.
9.Hiro, I., Tsugeno, Y., Ogata, F., and Ito, A.
(1996) J. Biochem., 120, 759-765.
10.Gottowik, J., Malherbe, P., Lang, G., Daprada,
M., and Cesura, A. M. (1995) Eur. J. Biochem.,230,
934-942.
11.Tsugeno, Y., and Ito, A. (1997) J. Biol.
Chem., 272, 14033-14036.
12.Tsugeno, Y., Hirashiki, I., Ogata, F., and Ito,
A. (1995) J. Biochem. (Tokyo), 118, 974-980.
13.Suzuki, O., Katsumata, Y., and Oya, M. (1982) in
Monoamine Oxidase: Basic and Clinical Frontiers (Kamijo, K.,
Usdin, E., and Nagatsu, T., eds.) Excrepta Medica, Amsterdam, pp.
74-86.
14.Gorkin, V. Z. (1983) Amine Oxidases in
Clinical Research, Pergamon Press, Oxford.
15.Cesura, A. N., and Pletscher, A. (1992) Prog.
Drug Res., 38, 171-297.
16.Medvedev, A. E., Ivanov, A. S., Kamyshanskaya, N.
S., Kirkel, A. Z., Moskvitina, T. A., Gorkin, V. Z., Li, N. Y., and
Marshakov, V. Yu. (1995) Biochem. Mol. Biol. Int., 36,
113-122.
17.Medvedev, A. E., Ivanov, A. S., Veselovsky, A.
V., Skvortsov, V. S., and Archakov, A. I. (1996) J. Chem. Inf.
Comput. Sci., 36, 664-671.
18.Kruger, M. J., Mazouz, F., Ramsay, R. R.,
Milcent, R., and Singer, T. P. (1995) Biochem. Biophys. Res.
Commun., 206, 556-562.
19.Gilbrat, J. F., Garnier, J., and Robson, B.
(1987) J. Mol. Biol., 198, 425-443.
20.Cesura, A. M., Gottowik, J., Lahm, H. W., Lang,
G., Imhof, R., Malherbe, P., Rothlisberger, U., and DaPrada, M. (1996)
Eur. J. Biochem., 236, 996-1002.
21.Ramsay, R. (1997) Vopr. Med. Khim.,
43, 457-470.