Interaction of Peroxynitrite and Hydrogen Peroxide with Dinitrosyl Iron Complexes Containing Thiol Ligands in vitro
I. I. Lobysheva*, V. A. Serezhenkov, and A. F. Vanin
Institute of Chemical Physics, Russian Academy of Sciences, ul. Kosygina 4, Moscow, 117977 Russia; fax: (095) 938-2156; E-mail: irlobysh@polymer.chph.ras.ru* To whom correspondence should be addressed.
Received July 21, 1998; Revision received September 21, 1998
The interaction of peroxynitrite with thiolate dinitrosyl iron complexes (DNIC) has been examined and compared with the interaction with H2O2. Peroxynitrite oxidized DNIC containing various thiolate ligands--cysteine, glutathione, and bovine serum albumin. Analysis of the oxidation suggested a two-electron reaction and gave third-order rate constants of (9.3 ± 0.5)·109 M-2·sec-1 for DNIC with BSA, (4.0 ± 0.3)·108 M-2·sec-1 for DNIC with cysteine, and (1.8 ± 0.3)·107 M-2·sec-1 for DNIC with glutathione at 20°C and pH 7.6. Peroxynitrite was more reactive towards DNIC than towards sulfhydryls. Addition of sodium dithionite after the reaction led to significant restoration of the EPR signal of DNIC with cysteine. The reaction of glutathione DNIC with H2O2 was about 600 times slower than with ONOO- and not reversed by sodium dithionite. Thus peroxynitrite, in contrast to hydrogen peroxide, changes the pool of nitrosocompounds which can be responsible for interconversion, storage, and transportation of nitric oxide in vivo.
KEY WORDS: nitric oxide, peroxynitrite, hydrogen peroxide, EPR spectroscopy of iron complexes, thiol oxidation, redox properties of nitrosyl adducts
Dinitrosyl iron complexes (DNIC) with thiol-containing ligands may be involved in various processes providing transportation and stabilization of nitric oxide (NO) in cells and tissue. NO appears to be a major second messenger in different physiological processes such as neurotransmission, vasodilatation, platelet aggregation, etc. [1-3]. The functions of nitric oxide vary greatly--it can be cytostatic or cytotoxic depending on its level provided by various forms of nitric oxide synthase depending on the state of the organism. The effects of NO can are changed significantly due to reactions with oxygen and superoxide (O2-.). The toxic effect of NO results partly from products of very fast reactions between NO and superoxide:peroxynitrite and its conjugate acid, peroxynitrous acid. Peroxynitrite anion (ONOO-) is a relative stable species in alkaline solution and has two distinct pK values, at 7.5-8.0 and 6.8, which could be related to cis and trans conformations [4]. At physiological pH (7.2-7.4) peroxynitrous acid decomposes with a half-life of ~1 sec. The products of decomposition have the reactivity of hydroxyl radical and nitrogen dioxide [5, 6]:
Peroxynitrite and peroxynitrous acid are potent oxidants and can initiate lipid peroxidation [7], DNA fragmentation [8], inactivation of enzymes containing Fe-S clusters or zinc-thiolate centers [9, 10], and oxidation of sulfhydryl groups in proteins and low mass molecules [11]. Formation of peroxynitrite in vivo can be seen by immunohistochemical methods in different tissue [12].
The ability of DNIC and of nitrosothiols to provide a “depot” of nitric oxide in vivo makes interesting the discussion of these adducts in tissue during the diffusion of NO from NO-producing cells such as endothelial cells, hepatocytes, and macrophages to target cells. Thus, investigation of the interaction between these compounds and peroxynitrite or end products of its decay is important. The interaction of DNIC with NO2 was discussed in [13], where decomposition of DNIC initiated by a mixture of gaseous NO and a small amount of NO2 was demonstrated. The stability of resulting nitrosothiols did not change under these conditions.
We report here an investigation of the interaction between compounds potentially able to accumulate or donate NO and two oxidative agents: peroxynitrite and hydrogen peroxide.
MATERIALS AND METHODS
Cysteine, reduced glutathione, bovine serum albumin (BSA), Hepes, and sodium dithionite were purchased from Sigma (USA). Ferrous sulfate was from Fluka (UK). NaNO2 and H2O2 were produced in Russia (analytical grade). Dinitrosyl iron complexes with cysteine, glutathione (GS), and phosphate were prepared by treatment of 5.4 mM FeSO4 and 10.8 mM GS or cysteine with gaseous nitric oxide in a Thunberg vessel under pressure 200-300 mm Hg in solutions previously degassed by vacuum and buffered with 15 mM Hepes (pH 7.6) or without thiols in 15 mM sodium phosphate (pH 7.6). Gaseous NO was synthesized in the reaction of FeSO4 with NaNO2 in 0.1 M HCl with subsequent purification in an evacuated system. Complexes with BSA were synthesized by incubation of 0.6 mM BSA solution with 0.5 mM solution of phosphate DNIC.
S-Nitrosoglutathione was synthesized in a Thunberg vessel by treatment of 50 mM glutathione with a mixture of gaseous NO and air for 5 min with subsequent evacuation of excess NO2. The concentration of S-nitrosoglutathione was determined spectrophotometrically at 338 nm (molar extinction coefficient 980 M-1·cm-1).
Peroxynitrite was synthesized by mixing an acidified solution of 0.6 M NaNO2 and 0.6 M H2O2 with subsequent stabilization with 0.9 M NaOH [14]. The concentration of peroxynitrite was determined spectrophotometrically at 302 nm (molar extinction coefficient 1670 M-1·cm-1). This reagent was synthesized every time before an experiment.
Optical spectra were recorded on a Specord spectrophotometer (Germany). X-Band EPR spectra were recorded on a Bruker 106 spectrometer (Germany) under the following conditions: temperature 20°C, microwave power 5 mW, modulation amplitude 0.5 mT. The relative concentration of paramagnetic centers in samples were determined by comparing the intensity of the EPR signal in set of samples prepared under identical conditions in calibrated tubes.
RESULTS
Reactions between peroxynitrite and dinitrosyl iron complexes with glutathione and cysteine. Dinitrosyl iron complexes with glutathione and cysteine are observed in dimeric and monomeric forms depending on the concentration of free thiols in solution [15]. Monomeric forms of DNIC dominate of dimeric forms at high concentrations of low-molecular-weight thiols in solution. The monomeric form is paramagnetic and is characterized by an anisotropic EPR signal in frozen solution which can become isotropic with gav = 2.03 in liquid solution (Fig. 1). Reactions of DNIC with oxidizing agents (peroxynitrite and hydrogen peroxide) were studied by determination of paramagnetic DNIC during reaction in solution. A small amount of peroxynitrite in alkaline solution (25 mM stock solution) was added to 1 mM DNIC in the presence of free thiols at molar ratio DNIC/free thiol < 1:20. The final pH of the reaction solution changed insignificantly (about 0.1-0.2 unit). The initial yellow color of the solution changes. This process correlates with a decrease of the 310-nm band characterizing DNIC optical spectra and the appearance of absorption near 330-340 nm which could be due to nitrite. A solution with peroxynitrite decomposed at pH ~ 3 for 5 min was used as control.
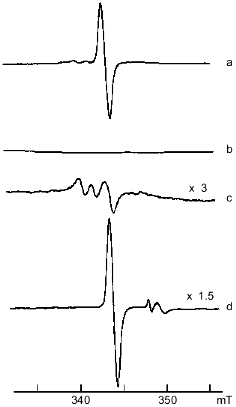
The pH dependence of the interaction between DNIC and peroxynitrite was investigated by mixing 0.5 mM DNIC buffered with 15 mM Hepes with a small amount of peroxynitrite to 0.6 mM final concentration and adjusting the pH. The change of initial pH did not exceed 0.1-0.2 unit. The dependence of relative changes of the EPR signal intensity on initial pH is demonstrated in Fig. 2.Fig. 1. X-Band EPR spectra of 1 mM DNIC after adding 20 mM cysteine in 15 mM Hepes, pH 7.6 (a); 2 mM peroxynitrite was added to the previous sample (b); 20 mM cysteine was added 10 min after starting the reaction with peroxynitrite (c); 10 mM dithionite was added to the end products of the reaction with peroxynitrite (d). Spectra were recorded at 20°C, microwave power 5 mW, modulation frequency 100 kHz, modulation amplitude 0.5 mT, and receiver gain 1·104 (a, b), 3·104 (c), and 1.5·104 (d).
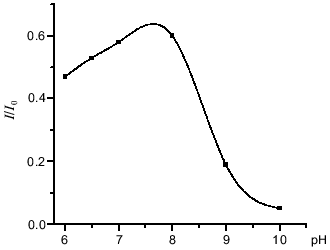
The interaction between peroxynitrite and DNIC with glutathione, cysteine, and albumin was investigated at pH 7.6 and 20°C. The reaction time was very short, so the intensity of the DNIC EPR signal decreased to a level dependent on the concentration of added peroxynitrite during the time of sample preparation (~2 min) and then did not change. Rate constants were estimated as described in [11]. A small amount of peroxynitrite was added to the solution and the EPR signal of final complexes was measured. Under these conditions, the decomposition of peroxynitrite may involve the reaction with DNIC, the reaction with sulfhydryl groups of free thiols, and other decomposition processes including spontaneous decay:Fig. 2. Influence of pH on the peroxynitrite-mediated oxidation of DNIC with glutathione. The reaction was started by addition of 0.56 mM peroxynitrite to 0.5 mM DNIC at 20°C in 15 mM Hepes. I/I0 is the intensity of the DNIC EPR signal 3 min after starting the reaction normalized to initial intensity of the signal.
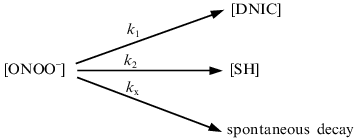
According to this scheme, the decomposition of peroxynitrite may be written as:
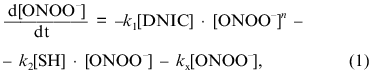
where [DNIC] is the concentration of the complexes in solution, k1 is the rate constant of oxidation of the complexes, n is the order of this rate for peroxynitrite, [SH] is the concentration of free thiols in solution, k2 is the second-order rate constant for the reaction between sulfhydryl and peroxynitrite, and kx is the constant for spontaneous decay. Because under the conditions of the experiments the concentration of free thiols significantly exceeded the concentration of peroxynitrite and DNIC, approximate integration of Eq. (1) gives:
The oxidation of DNIC was determined according to:
Integration of Eq. (3) with time increasing to infinity gives:
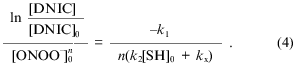
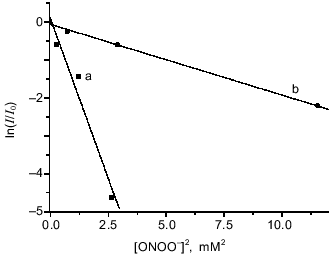
The dependence of relative intensity of the DNIC EPR signal from the left-hand expression on the concentration of added peroxynitrite is demonstrated in Fig. 3 for the complexes with glutathione and cysteine. Analysis of the data gave the best linear fit when n = 2.
Estimation of the third-order rate constant from the curves in Fig. 3 gave k1cys = (4.0 ± 0.3)·108 M-2·sec-1 for DNIC with cysteine and k1GS = (1.8 ± 0.3)·107 M-2·sec-1 for DNIC with glutathione when it was assumed that k2 = 5900 ± 480 M-1·sec-1 and kx = 0.39 ± 0.02 sec-1 as reported in the literature [5, 11].Fig. 3. Oxidation of DNIC with glutathione and cysteine by peroxynitrite. Reactions were started by addition of the indicated amounts of peroxynitrite to 0.5 mM DNIC in the presence of 20 mM cysteine (a) or 20 mM glutathione (b) at 20°C in 15 mM Hepes, pH 7.6. I/I0 is the intensity of DNIC EPR signal 3 min after starting the reaction normalized to the intensity of the initial EPR signal.
The interaction of the DNIC dimer with peroxynitrite was similar to that of the monomer.
Reduction by addition of 10 mM dithionite to the solution of oxidized DNIC with cysteine restored 90% of the initial EPR signal at pH ~ 7.6 (Fig. 1); the reduction of DNIC with glutathione did not achieve this level. No restoration of the EPR signal of paramagnetic complexes decreased by peroxynitrite was observed after the addition of cysteine or reduced glutathione.
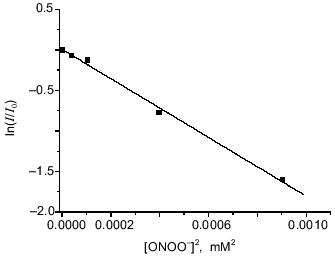
Reaction between peroxynitrite and dinitrosyl iron complexes with BSA. Iron is coordinated by one sulfhydryl group of the protein in dinitrosyl iron complexes with BSA, Fe(NO)2(BSA), and due to the structure of BSA the complex does not form diamagnetic dimers; it shows the anisotropic EPR signal characterized by g1 = 2.051, g2 = 2.043, and g3 = 2.01 in frozen and liquid solution [16]. Addition of peroxynitrite immediately decomposes the complex with BSA. The dependence of the EPR signal intensity on concentration of added peroxynitrite is demonstrated in Fig. 4. Small amounts of peroxynitrite were added in accordance with the conditions described above. Analogous analysis of the interaction between DNIC with BSA and peroxynitrite gives for determination of the complex oxidation constant:
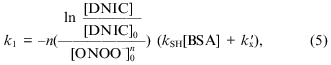
where [BSA] is the concentration of albumin, because the protein contains only one sulfhydryl group, kSH is the second-order rate constant of oxidation of albumin sulfhydryl groups, and kx´ is the constant for the sum of processes of peroxynitrite decomposition including other interactions with albumin.
The best linearization of the curve from Fig. 4 is achieved for n = 2; this allows calculation of the third-order rate constant between peroxynitrite and BSA-containing DNIC--k1BSA = (9.3 ± 0.5)·109 M-2·sec-1 at 20°C and pH 7.6. For this calculation we used data from the literature [11]: kSH = 2800 ± 180 M-1·sec-1 and kx´ = 0.911 ± 0.059 sec-1.Fig. 4. Oxidation of DNIC with bovine serum albumin by peroxynitrite. Reactions were started by addition of the indicated amounts of peroxynitrite to 0.2 µM DNIC with albumin in presence of 0.6 mM BSA in 15 mM Hepes, pH 7.6, and 20°C. I/I0 is the intensity of the DNIC EPR signal 3 min after starting the reaction normalized to the intensity of the initial EPR signal.
The oxidation was irreversible; addition of dithionite did not regenerate the EPR signal of the complex.
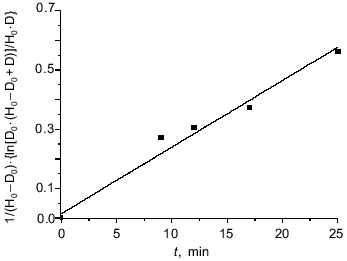
Influence of H2O2 on DNIC with glutathione and cysteine. The reaction of hydrogen peroxide with DNIC at pH 7.6 was much slower than the reaction with peroxynitrite. The time-dependent decrease of the DNIC EPR signal after addition of H2O2 is demonstrated in Fig. 5. Hydrogen peroxide (0.5 mM) was added to 0.7 mM DNIC in presence of 14 mM glutathione. This dependence is quite well fitted by the second-order rate law. The apparent second-order rate constant kn´ was determined from the curve linearized in accordance with:
where H0 and D0 are the initial concentration of H2O2 and DNIC respectively, and D is the concentration of DNIC after time t. The apparent rate constant was 0.37 ± 0.05 M-1·sec-1 for DNIC with glutathione at 20°C and pH 7.6 as calculated from the data of Fig. 5.
The addition of 10 mM sodium dithionite did not change the intensity of the EPR signal after the reaction. The addition of 20 mM of reduced glutathione to the reaction mixture 18 min after starting the oxidation increased the EPR signal intensity from 14 to 51% of the initial signal. This fact pointed to the conversation of about one half of the total DNIC to diamagnetic forms through dimerization due to oxidation of free thiols by hydrogen peroxide; another part of the DNIC underwent irreversible oxidation.Fig. 5. Interaction between DNIC with glutathione and hydrogen peroxide. Reactions were started by addition of 5 mM H2O2 to 0.7 mM DNIC with glutathione in the presence 20 mM glutathione in 15 mM Hepes, pH 7.6, and 20°C. Aliquots were taken from the reaction mixture and EPR signals were measured at different times. H0 and D0 are the initial H2O2 and DNIC concentrations and D is the DNIC concentration in solution at time t from the beginning of the reaction.
The reaction between DNIC dimers (when the ratio of DNIC/glutathione was about 1:2) and hydrogen peroxide supported this suggestion (data not shown).
The DNIC with cysteine was oxidized irreversibly by hydrogen peroxide too, and mononitrosyl complexes Fe(NO)(cys)4 were oxidized simultaneously.
Reaction of nitrosothiols with peroxynitrite and hydrogen peroxide. The oxidative reaction of nitrosoglutathione was studied spectrophotometrically by following the absorption band characterizing nitrosothiols near 338 nm (epsilon = 980 M-1·cm-1) in solution buffered by 15 mM Hepes, pH 7.6, and 20°C. Small amounts of concentrated peroxynitrite solution were added to the control and experimental cuvettes. Nitrosoglutathione (GSNO), which is stable at pH 7.6, was oxidized quickly and completely by peroxynitrite when the ratio GSNO/peroxynitrite was less than 1:1.5 (t < 0.5 min). The absorption characteristic of nitrite anion was found instead the band of nitrosothiols in the UV range.
Hydrogen peroxide also reduced the absorption band near 338 nm. But the concentration of GSNO decreased by only 50% when the GSNO/H2O2 ratio was about 1:60. The process was not accompanied by the appearance of new bands in the UV range.
DISCUSSION
Peroxynitrite oxidizes DNIC containing thiolates as well as nitrosothiols effectively. Our study demonstrates the oxidative ability of both protonated (ONOOH) and deprotonated (ONOO-) peroxynitrous species with respect to DNIC (Fig. 2). The oxidative properties of both forms have been studied in numerous previous papers [8, 11]. It was shown that peroxynitrite oxidizes thiol groups through one- and two-electron pathways, but the two-electron pathway which was mediated by peroxynitrite anion (ONOO-) prevailed quantitatively over the one-electron pathway [17], though a number of substrates underwent one-electron oxidation by activated species derived from peroxynitrous acid (ONOOH*) [8]. If peroxynitrite was added to DNIC in approximately equal concentrations, the oxidative ability of peroxynitrite increased twofold at pH values near the pK of one peroxynitrite conformation [17]. Data are shown in Fig. 2. Kinetic analysis of the oxidative reactions between DNIC and peroxynitrite at pH 7.6 suggests the following mechanism for the oxidation of the complex:

Because the oxidizing ability of peroxynitrite increased twofold as mentioned above, we suggest that DNIC undergoes two-electron oxidation in the reaction with peroxynitrite: two molecules of ONOOH oxidize the complex by a one-electron mechanism under acid conditions and one molecule of peroxynitrite anion oxidizes the complex by a two-electron process at alkaline pH.
Comparison of the oxidizing ability of peroxynitrite with respect to thiols and DNIC containing these thiols as a source of sulfhydryl groups shows the greater oxidizing ability of peroxynitrite toward DNIC. The half-life of DNIC with BSA reacting with 10 µM peroxynitrite is ln2/k1[ONOO]2 ~ 0.8 sec. This result is comparable with one of the fastest reactions of peroxynitrite--its reaction with yeast alcohol dehydrogenase. The half-time for inactivation of the enzyme was about ln2/k1[ONOO] ~ 0.3 sec under similar conditions [10]. The half-time for oxidation of the sulfhydryl of BSA was ln2/k1[ONOO] ~ 25 sec calculated for the same conditions according to data from the literature [11].
The DNIC with glutathione and BSA are more stable than the DNIC with cysteine because the stability of DNIC in solution is generally dependent on the resistance of the thiolate ligand to air oxidation [18]. Thus, complexes with free thiols that are paramagnetic in solution undergo gradually dimerization upon thiol oxidation in air. Dimers are diamagnetic, more stable to oxidation in air, and return to the monomeric paramagnetic form after addition of reduced thiols to the solution. This process does not play a significant role in the oxidizing reaction with peroxynitrite because oxidation of thiols by peroxynitrite is less effective than oxidation of DNIC. Dimeric DNIC also actively reacts with peroxynitrite. Thus, the resistance of the complexes to the action of peroxynitrite may depend on the accessibility of the nitrosyl groups. DNIC containing two large glutathione molecules is more stable than the complex with cysteine when interacting with peroxynitrite--kGS = (1.8 ± 0.3)·107 M-2·sec-1 and kcys = (4.0 ± 0.3)·108 M-2·sec-1. The largest rate constant of oxidation is exhibited by DNIC with BSA, kBSA = (9.3 ± 0.5)·109 M-2·sec-1, where the dinitrosyl iron complex is connected with only one sulfhydryl group of the protein which is located close to the surface [16].
The reaction between NO and ONOO- has been investigated [5] and was shown to be quite significant--k = 9.1·104 M-1·sec-1, so t1/2 ~ 0.8 sec for 10 µM ONOO-. This result is in good agreement with the rate of oxidation of DNIC with BSA (t1/2 ~ 0.8 sec for 10 µM ONOO-). Because of this similarity, we propose that the oxidation occurs through the interaction with nitrosyl group as follows:

Complete decomposition of the complexes with release of nitrosyl groups from DNIC as nitrite anions would be the result of reaction (8b); partially stabilization of the complexes in the oxidized state can occur in reaction (8a). The latter reaction may prevail in oxidation of DNIC with cysteine, this being consistent with the restoration of the DNIC EPR signal after addition of sodium dithionite to the reaction mixture. The formation of RSNOx as possible oxidation products in the reaction of peroxynitrite with thiols was proposed previously [17].
The interaction of DNIC with hydrogen peroxide is less effective. The reaction of DNIC containing glutathione with hydrogen peroxide at 20°C and pH 7.6 would be 600 times slower than the reaction with peroxynitrite if equal concentrations of the oxidant agents were used. Because the oxidation of thiols by hydrogen peroxide is more effective than the oxidation of DNIC (k = 4.7 M-1·sec-1 for oxidation of cysteine by H2O2 [11]), DNIC dimerization follows the oxidation due to the decrease of free thiol in solution. Thus, the addition of cysteine increases the EPR signal of DNIC. Sodium dithionite does not restore the initial signal. These facts mean that oxidation by hydrogen peroxide is significantly different than the reaction with peroxynitrite in the character of the reaction as well as of the end products.
The production of peroxynitrite in biological systems is difficult to assess. Nitric oxide reacts with superoxide anion in a near diffusion-limited reaction with rate constant k = (4.5-6.4)·109 M-1·sec-1. The production of nitric oxide could reach values from 0.05 to 8 µM/min depending on the conditions in vivo. The intracellular steady-state production of O2-. could reach from 1 to 10 nM/sec in normalcy and pathology [19]. Thus, the production of peroxynitrite in vivo especially in prolonged pathophysiological processes such as sepsis, acute inflammation, or ischemia--reperfusion could be significant. Our data indicate that this can change the pool of nitrosoadducts in cells and in contrast to hydrogen peroxide can disturb the processes of storage and transportation of nitric oxide.
This work was supported by the Russian Foundation for Basic Research (grant number 96-04-48066).
REFERENCES
1.Moncada, S., Palmer, R. M. J., and Higgs, E. A.
(1991) Pharmacol. Rev., 43, 109-142.
2.Vanin, A. F. (1991) FEBS Lett., 284,
1-3.
3.Stamler, J. S. (1994) Cells, 78,
931-936.
4.Beckman, J. S., Beckman, T. W., Chen, J., Marshall,
P. A., and Freeman, B. A. (1990) Proc. Natl. Acad. Sci. USA,
87, 1620-1624.
5.Pfeiffer, S., Gorren, A. C. F., Schmidt, K.,
Werner, E. R., Hansert, B., Bohle, D. S., and Mayer, B. (1997) J.
Biol. Chem., 272, 3465-3470.
6.Gatty, R. M., Alvarez, B., Vasquez-Vivar, J., Radi,
R., and Augusto, O. (1998) Arch. Biochem. Biophys., 349,
36-46.
7.Radi, R., Beckman, J. S., Bush, K. M., and Freeman,
B. A. (1991) Arch. Biochem. Biophys., 288, 481-489.
8.Salgo, M. G., Bermudez, E., Squadrito, G. L., and
Pryor, W. A. (1995) Arch. Biochem. Biophys., 322,
500-505.
9.Bouton, C., Hirling, H., and Drapier, J.-C.
(1997) J. Biol. Chem., 272, 19969-19975.
10.Crow, J. P., Beckman, J. C., and McCord, J. M.
(1995) Biochemistry, 34, 3544-3552.
11.Radi, R., Beckman, J. S., Bush, K. M., and
Freeman, B. A. (1991) J. Biol. Chem., 266, 4244-4250.
12.Myatt, L., Rosenfield, R. B., Eis, A. L.,
Brockman, D. E., Gree, I., and Lyall, F. (1996) Hypertension,
28, 488-493.
13.Vanin, A. F., Malenkova, I. V., and Serejenkov,
V. A. (1997) Nitric Oxide: Biol. Chem., 1, 191-203.
14.Koppenol, W. H., Kissner, R., and Beckman, J. S.
(1996) Meth. Enzymol., 269, 296-302.
15.Vanin, A. F., Stukan, R. A., and Manukhina, E. B.
(1996) Biochim. Biophys. Acta, 1295, 5-12.
16.Boese, M., Mordvintcev, P. I., Vanin, A. F.,
Busse, R., and Mulsch, A. (1995) J. Biol. Chem., 270,
29244-29249.
17.Scorza, G., and Minetti, M. (1998) Biochem.
J., 329, 405-413.
18.Lobysheva, I. I., Serezhenkov, V. A., Stukan, R.
A., Bowman, M. K., and Vanin, A. F. (1997) Biochemistry
(Moscow), 62, 934-942 (Russ.).
19.Czapski, G., and Goldstein, S. (1995) Free
Rad. Biol. Med., 19, 785-794.