Co-expression of Gene 31 and 23 Products of Bacteriophage T4
L. P. Kurochkina and V. V. Mesyanzhinov*
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 336-6022; E-mail: vvm@ibch.siobc.ras.ru* To whom correspondence should be addressed.
Received October 21, 1998; Revision received December 9, 1998
Folding of the major capsid protein of bacteriophage T4 encoded by gene 23 is aided by Escherichia coli GroEL chaperonin and phage co-chaperonin gp31. In the absence of gene product (gp) 31, aggregates of recombinant gp23 accumulate in the cell similar to inclusion bodies. These aggregates can be solubilized with 6 M urea. However, the protein cannot form regular structures in solution. A system of co-expression of gp31 and gp23 under the control of phage T7 promoter in E. coli cells has been constructed. Folding of entire-length gp23 (534 amino acid residues) in this system results in the correctly folded recombinant gp23, which forms long regular structures (polyheads) in the cell.
KEY WORDS: bacteriophage T4, capsid, gene 31 and 23 products, folding, expression vector
Abbreviations: HAP) hydroxylapatite; IPTG) isopropyl-1-thio-beta-D-galactoside.
Folding of many proteins in the cell is aided by chaperons [1-3]. Specifically, in
Escherichia coli cells folding of cytoplasmic proteins is
controlled by a complex of GroEL chaperonin and GroES co-chaperonin.
The bacteriophage T4 genome encodes several chaperons which regulate
folding and assembly of viral proteins. Gene product 31 (gp31) is one
such protein helper; together with E. coli GroEL chaperonin, it
is absolutely necessary for folding of gp23--the major structural
capsid protein of phage T4 [4]. With mutations in
both gene 31 and gene groEL, amorphous aggregates of gp23 accumulate on
the cell membrane [5].
It is known that gp31 (111 amino acid residues, molecular weight ~12 kD) also completely substitutes for E. coli co-chaperonin GroES during bacteriophage lambda and T5 morphogenesis as well as chaperonin-assisted folding of ribulose bisphosphate decarboxylase in vivo and in vitro [6, 7]. Like GroES, gp31 forms a stable complex with GroEL chaperonin in the presence of Mg-ATP and inhibits GroEL ATPase activity in vitro [7]. X-Ray structural data showed that in spite of weak homology of gp31 and GroES amino acid sequences, the spatial structures of these proteins are quite similar [8]. Active gp31 is also a heptamer and has toroidal shape; however, the diameter of the orifice in the gp31 heptamer is larger (26 Å) than in GroES (16 Å). Amino acid residues forming the orifice of the toroid differ in their nature, being preferentially hydrophobic in GroES and hydrophilic in gp31 [9]. The latter property may cause immediate participation of gp31 in gp23 folding. According to NMR data, the mobile loop of gp31 interacting with GroEL is 6 amino acid residues longer than the loop of GroES [8], and this results in increased cage height in the GroEL/gp31 chaperonin complex. This is crucial for the folding of gp23 which, probably, cannot be accommodated within the smaller confines of the cage in the GroEL/GroES complex. By analogy with the functioning of the GroEL/GroES chaperonin complex, a model for GroEL/gp31-assisted folding of gp23 has been recently proposed [4]. According to this model, GroEL interacts with the nascent chain of gp23, preventing its premature aggregation. One of the functions of gp31 seems to be the efficient release of gp23 from the chaperonin with ATP hydrolysis.
Although the spatial structures of GroEL, GroES, and gp31 chaperonins are resolved, major difficulties in interpretation of the gp23 folding mechanism are related to the absence of detailed information on the spatial structure of this protein.
The purpose of this work was to obtain plasmid vectors allowing production of functionally active gp23 in E. coli cells in order to study its folding and the initial stages of polymerization. Formation of the polymeric tubular structures built of gp23, so-called polyheads, may provide evidence for the normal folding and assembly of the active protein in the cells; polyheads can be easily revealed and analyzed by electron microscopy and image processing techniques.
MATERIALS AND METHODS
Bacterial strains. Bacterial strain E. coli Top 10 from Invitrogene (USA) was used for production of plasmid DNA and transformation while cloning. Strain E. coli BL21 (DE3) from Novagen (USA) was used for expression of genes cloned into plasmids under control of the phage T7 promoter.
Polymerase chain reaction. The Dry Block PHC-1 amplifier (Techne, England) was used to perform the polymerase chain reaction (PCR). The reaction mixture (100 µl) for the standard amplification contained ~1 ng of bacteriophage T4 matrix genome DNA, 50 pmoles of each primer, 10 µl of ten-times concentrated buffer from Promega (USA), 200 µM of each dNTP, and 2 units of Taq polymerase from Promega. Mineral oil (25 µl) was overlaid on the mixture. In all cases, the reaction was started by preliminary DNA denaturation at 97°C for 3 min; then, 25 amplification cycles were carried out: 94°C (1 min); Tanneal. (1 min); 72°C (X min). The optimal annealing temperature (Tanneal.) for gene amplification was 54°C for gene 31 and 46°C for gene 23, and the period of time for formation of the primers (X) was 1 and 2 min, respectively. The products of amplification were analyzed in 1% agarose gel by their DNA mobility.
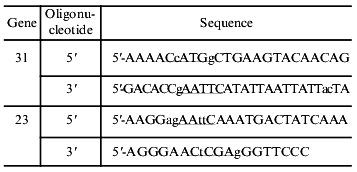
Cloning of genes 31 and 23. Entire-length gene 31 and gene 23 were amplified by the PCR method using synthetic oligonucleotides as primers; point mutations were inserted into their sequences for generation of convenient cloning sites (see table, sites are underlined). The 358-bp fragment containing entire-length gene 31 was cloned into pET-23d(+) vector (Novagen) by NcoI--EcoRI sites (pMPK-31 vector). The 1604-bp fragment containing entire-length gene 23 was cloned into pMPK-31 vector by EcoRI--XhoI sites (pPK-31-23 vector). Clones containing DNA insertions of the proper size were checked by restriction analysis and for protein expression. The insertion containing gene 31 in vector pMPK-31 was checked by DNA sequencing.
Oligonucleotide sequences used for amplification of gene 31 and gene 23
Gene expression in E. coli BL21 (DE3) cells. The genes cloned under phage T7 promoter were expressed according to Studier [10]. Competent BL21 (DE3) cells were transformed with plasmid, plated onto dishes with L-agar containing 1% glucose and ampicillin (100 µg/ml), and then were incubated for 12-18 h at 37°C. The colonies of transformants were inoculated in 4 ml of 2× TY medium containing ampicillin (200 µg/ml) and were grown at 37°C up to optical density A600 ~ 1 (aliquots of transformants were stored with 10% glycerol at -70°C). To induce expression, isopropyl-1-thio-beta-D-galactoside (IPTG) was added to the final concentration of 1 mM, and the mixture was incubated for 3 h at 37°C (or overnight at 30°C). The cells were centrifuged at 4000 rpm for 15 min using a Heraeus Megafuge 2.0 R. For preparative production of recombinant proteins, the volume of culture medium was increased to 100-500 ml.
Electrophoretic analysis of recombinant proteins. After expression, 100 µl of cell culture was mixed with 30 µl of four-times sample buffer (0.25 M Tris-HCl, pH 6.8, 8% SDS, 40% (v/v) glycerol, 20% (v/v) 2-mercaptoethanol, and 0.02% (w/v) Bromphenol Blue), heated for 3 min at 100°C, and in 10-30 µl portions loaded onto the gel. The proteins were electrophoresed under denaturing conditions in 15% polyacrylamide gel in the presence of SDS according to Laemmli [11]. The gels were stained with 0.3% Coomassie R from Sigma (USA) in acetic acid--ethanol--water (1:3:6 v/v) mixture and then destained with solution containing 50% ethanol and 7% acetic acid. A standard set of proteins from Pharmacia and Biotech (Sweden) containing alpha-lactalbumin (14.4 kD), soy bean trypsin inhibitor (20.1 kD), carboanhydrase (30.0 kD), ovalbumin (43.0 kD), BSA (67.0 kD), and glycogen phosphorylase (94.0 kD) was used as the molecular weight marker.
Preparation of cell lysates. After expression, the cells were centrifuged and frozen in liquid nitrogen; then they were suspended in 4-10 ml of TE buffer and lysed in the presence of 0.5 mg/ml lysozyme for 20 min at room temperature. The cell suspension was sonicated for 2-3 min (15 sec with 15 sec intervals) using a UD-20 ultrasonic disintegrator (Techpan, Poland). Cellular debris was removed by centrifugation at 12,000g for 10 min. Streptomycin sulfate (30%, w/v) was added to the supernatant at the ratio 1:10, and the mixture was incubated with stirring for 15 min at 4°C. The precipitate was isolated by centrifugation under the same conditions. To precipitate the protein, a saturated ammonium sulfate solution was slowly added to the supernatant to a final concentration of 30%, and the mixture was kept in the cold for 1 h. After centrifugation, the protein-containing precipitate was dissolved in the appropriate buffer.
Purification of gp31. Protein solution in 10 mM Tris-HCl buffer, pH 7.6, was applied onto a 1 × 5-cm column with hydroxylapatite (HAP) from Bio-Rad (USA) equilibrated with 10 mM sodium phosphate buffer, pH 7.6. The protein was eluted by step gradient of sodium phosphate buffer concentrations (from 10 to 50 mM), pH 7.5; the fractions were analyzed by electrophoresis.
Fractionation of gp23 and gp31 protein mixture. Protein solution in 50 mM Tris-HCl buffer, pH 8.5, was applied onto a 1 × 7-cm column with DEAE-cellulose from Pharmacia equilibrated with 50 mM Tris-HCl buffer, pH 8.6. The proteins were eluted by step gradient of NaCl concentrations (from 50 to 500 mM) in the same buffer; the fractions were analyzed by electrophoresis.
Electron microscopy. Ultrathin sections of E. coli cells expressing gp31 and gp23 were prepared for electron microscopy. Cells were sequentially fixed with 2.5% glutaraldehyde (30 min) and 0.5% osmium tetroxide (30 min) in 0.1 M sodium phosphate buffer, pH 7.0, containing 0.02 M sucrose. After dehydration, the preparations were polymerized in a mixture of resins containing Epon-812, DDSA, MNA, and DMP. The cell sections were contrasted with 2% uranyl acetate for 1 h and 0.0002% lead citrate for 2 min. Microphotographs were obtained using a Hitachi 11 electron microscope.
RESULTS AND DISCUSSION
We have constructed a set of vectors allowing production of recombinant proteins gp23 and gp31.
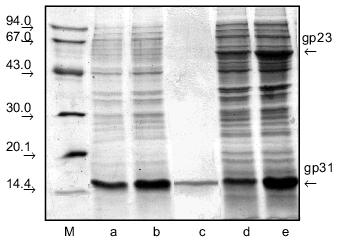
The 358-bp DNA fragment containing entire-length gene 31 was cloned into pET-23d(+) vector by NcoI/EcoRI sites. Expression vector pMPK-31 containing entire-length gene 31 is a superproducer allowing production of large amounts of a soluble recombinant protein with apparent molecular mass ~16 kD in E. coli BL21 (DE3) strain; this correlates with the anomalous electrophoretic mobility of gp31 in the SDS-containing gel (Fig. 1, a and b). For preparative isolation of recombinant gp31, cells from 100 ml of culture were lysed and ultrasonicated with subsequent centrifugation. As a result of such treatment, the protein remains in the supernatant. After salting-out, the protein was dissolved in 10 mM Tris-HCl buffer, pH 7.6, and fractionated on a HAP column. Electrophoretically pure protein fraction was eluted with 10 mM sodium phosphate, pH 7.6 (Fig. 1c). The yield of protein was about 20 mg per 100 ml of cell suspension.
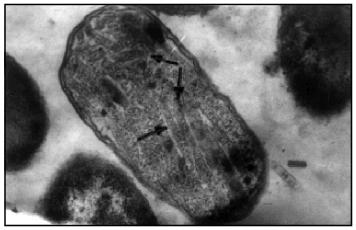
In the next stage we constructed the pPK-31-23 expression vector containing both genes 31 and 23. For this we cloned the 1604-bp fragment containing entire-length gene 23 into the pMPK-31 expression vector by sites EcoRI and XhoI. pPK-31-23 expression vector is characterized by a high level of expression in cells of both proteins, gp31 and gp23 (Fig. 1, d and e). Studies of ultrathin sections of bacteria by electron microscopy showed that gp23 forms regular structures (polyheads) in E. coli cells (Fig. 2). Polyheads, tubular structures, were first discovered during infection with phage T4 mutants defective in several genes of the head, namely, genes 20, 21, 22, or 24 [12]. Assembly of polyheads provides evidence for normal folding of functionally active protein in E. coli cells.Fig. 1. Extracts of E. coli BL21 (DE3) cells expressing gp31 of bacteriophage T4 (pMPK-31 vector) before (a) and after (b) induction with IPTG and purification on HAP (c). Extracts of E. coli BL21 (DE3) cells co-expressing gp31 and gp23 (pPK-31-23 vector) before (d) and after (e) induction with IPTG. Cell extracts were analyzed in 15% polyacrylamide gel containing SDS; M, protein markers.
After ultrasonication of the cell suspension and subsequent centrifugation, gp31 and gp23 remained in the supernatant. For their fractionation, after salting-out with 30% ammonium sulfate the cell lysate was fractionated on a DEAE-cellulose column with a stepwise change of NaCl concentrations (0-500 mM) in 50 mM Tris-HCl buffer, pH 8.5. Most of the gp23 is eluted at 200 mM NaCl, and most of the gp31 is eluted at 300 mM NaCl. The electron-microscopic study of gp23 after chromatography showed that in solution most of the recombinant protein also exists as polyheads (Fig. 3).Fig. 2. Electron microphotograph of a lengthwise section of E. coli BL21 (DE3) cells producing gp23. The assembled polyheads are indicated by arrows.
During dialysis of a solution containing 1-2 mg/ml polyheads against the low ionic strength buffer (1 mM phosphate buffer, pH 7.0) at 4°C, the polyheads dissociate to gp23 monomers. On long standing at low temperature and low ionic strength, monomeric gp23 denatures giving an amorphous precipitate.Fig. 3. Electron microphotographs of polyheads in solution (1.8 cm = 1000 Å).

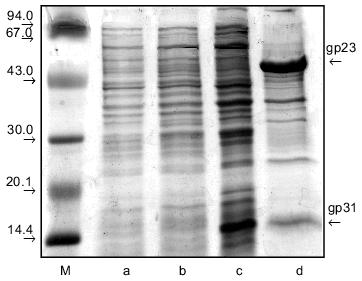
To ascertain the role of gp31 in gp23 folding, we constructed a new vector, pPK-31*-23, expressing the entire-length gp23 with defective gp31. For this, we removed a 225-bp fragment within gene 31 from the pMPK-31 plasmid vector by the sites NcoI--Bsp119I, and then the 1604-bp fragment containing entire-length gene 23 was cloned into the resulting intermediate plasmid by the sites EcoRI--XhoI. Thus, in E. coli cells the fragment of gene 31 was positioned under the T7 promoter encoding only the 36 C-terminal amino acid residues, the distal 75-residue fragment containing the highly flexible mobile loop responsible for binding of gp31 with GroEL [8]. In contrast to the expression system of pPK-31-23, a lower level of gene 23 expression is typical of this construction (Fig. 4b). Analysis of the solubility of the cell lysate demonstrated that pPK-31*-23 vector produces a protein contained in the precipitate, not in the supernatant (Fig. 4, c and d). For this reason, we conclude that gp23 is insoluble and is likely to form an amorphous "lumps" on the cell membrane. It is clear that in the absence of entire-length gp31, gp23 folds improperly, and the protein loses the capability for assembly in the regular supramolecular structure--polyheads.
We attempted to dissolve gp23 obtained by expression of the pPK-31*-23 vector. For this, we dissolved the precipitate of the cell extract in 6 M urea and then performed stepwise refolding of protein, gradually lowering urea concentration in the dialysis buffer. About 90% of the protein can be dissolved by this procedure. Electron microscopy demonstrated that the renatured protein is incapable of forming polyheads in vitro. It seems that although in this case in vitro refolding produces a soluble protein, it proceeds improperly and gives a form incapable of assembly into polyheads.Fig. 4. Extract of E. coli BL21 (DE3) cells co-expressing gp31* and gp23 (pPK-31*-23 vector) before (a) and after (b) induction with IPTG. The supernatant (c) and precipitate (d) of the lysate of the same cells; M, protein markers.
It is possible that GroEL/gp31 complex which forms in E. coli expression strains gives gp23 dimers detected on dissociation of polyheads [12], and then these dimers form a lattice of procapsid on the core surface or assemble in polyheads. Also, earlier several mutants in gene 23 of phage T4 were detected; in them the normal folding of gp23 proceeds in the absence of gp31 co-chaperonin [13]. It is possible that gp31 does not participate in the binding of gp23 with GroEL, but forms a local supersecondary structure of gp23 which interacts with GroEL. We plan to continue the studies of gp23 folding mechanisms in the cell using the system of co-expression presented in this work. Point and also deletion mutants in gene 23 may also help to solve the problem of gp23 folding.
We thank B. F. Poglazov for his kind assistance in electron-microscopic studies.
This study was supported by the Russian Foundation for Basic Research (grant No. 97-04-49708), the Howard Hughes Medical Institute (grant No. 75197-544204), and also by the grant “Priority Directions of Genetics”.
REFERENCES
1.Hartl, F. U. (1996) Nature, 381,
571-580.
2.Martin, J. (1998) Biochemistry (Moscow),
63, 444-452 (Russ.).
3.Kurochkina, L. P., and Mesyanzhinov, V. V. (1996)
Uspekhi Biol. Khim., 36, 49-86.
4.Van der Vies, S. M., Gatenby, A. A., and
Georgopoulos, C. P. (1994) Nature, 368, 654-656.
5.Laemmli, U. K., Beguin, F., and Kellenberger-Gujer,
K. G. (1970) J. Mol. Biol., 47, 69-85.
6.Georgopoulos, C. P., Hendrix, R. W., Kaiser, A. D.,
and Wood, W. B. (1972) Nature (London) New Biol.,
239, 38-41.
7.Landry, S. J., Zeilstra-Ryalls, J., Fayet, O.,
Georgopoulos, C. P., and Gierasch, L. M. (1993) Nature,
364, 255-258.
8.Hunt, J. F., van der Vies, S. M., Henry, L., and
Deisenhofer, J. (1997) Cell, 90, 361-371.
9.Marusich, E. I., Kurochkina, L. P., and
Mesyanzhinov, V. V. (1998) Biochemistry (Moscow), 63,
473-482 (Russ.).
10.Studier, F. W., and Moffatt, B. A. (1986) J.
Mol. Biol., 189, 113-130.
11.Laemmli, U. K. (1970) Nature, 227,
680-685.
12.Muller, M., Mesyanzhinov, V. V., and Aebi, U.
(1994) J. Struct. Biol., 112, 199-215.
13.Simon, L. D., and Randolf, B. (1984) J.
Virol., 51, 321-328.