Modulation of Mast Cell Activity by a Peptide Agonist of the Thrombin Receptor: Role of Nitric Oxide
S. M. Strukova1*, I. V. Chistov1, B. A. Umarova1, T. N. Dugina1, T. P. Storozhevykh2, V. G. Pinelis2, and E. Glusa3
1Department of Human and Animal Physiology, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; tel.: (095) 939-14162Laboratory of Membranology, Institute of Pediatrics, Russian Academy of Medical Sciences, Lomonosovsky pr. 2/62, Moscow, 117963 Russia; tel.: (095) 134-1445
3Center for Vascular Biology and Medicine, Friedrich Schiller Iena University, Erfurt, Germany
* To whom correspondence should be addressed.
Received June 23, 1998; Revision received November 5, 1998
The effect of a thrombin receptor agonist peptide (TRAP-6) on the release of nitric oxide (NO) and platelet activating factor (PAF) from resting and calcium-ionophore (A23187)-activated rat peritoneal mast cells (RPMC) was studied using a platelet aggregation bioassay. RPMC spontaneously released NO, which inhibited TRAP-6-, ADP-, and PAF-stimulated platelet aggregation. This effect of NO was abolished by the addition of an NO binding agent, oxyhemoglobin (oxyHb), to the platelet suspension. The RPMC-induced suppression of platelet aggregation was completely inhibited by the NO-synthase inhibitor L-NAME. TRAP-6 and its high affinity analog haTRAP stimulated the rapid release of NO from RPMC. The effect of TRAP-6 was inhibited by pretreatment of the RPMC with L-NAME or with the inhibitor of the constitutive NO-synthase isoform (cNOS) calmidazolium. TRAP-6 inhibited PAF release from A23187-activated RPMC via an NO-dependent mechanism. Platelet aggregation induced by PAF release from activated RPMC was also confirmed in experiments using the PAF receptor antagonist ginkgolide B. Thus, TRAP-6 is a rapidly acting modulator of mast cell reactivity; it stimulates NO release and inhibits PAF secretion.
KEY WORDS: thrombin receptor agonist peptide, mast cells, nitric oxide, platelet activating factor
Abbreviations: L-NAME) Nomega-nitro-L-arginine methyl ester; cNOS) constitutive NO-synthase; iNOS) inducible NO-synthase; NO) nitric oxide; oxyHb) oxyhemoglobin; PAF) platelet activating factor; PAR-1) protease-activated receptor; RPMC) rat peritoneal mast cells; PRP) platelet rich plasma; TRAP-6) thrombin receptor agonist peptide; haTRAP-6) high affinity TRAP-6 analog.
Thrombin, a serine protease of the trypsin family, catalyzes limited
proteolysis of blood fibrinogen; it also exhibits regulatory properties
controlling the activation and inhibition of blood clotting, vascular
tone, and the inflammatory and proliferative phases of wound healing
[1-7]. Thrombin stimulates
adhesion of platelets, polymorphonuclear leukocytes, monocytes, and
T-lymphocytes to endothelial cells and cell aggregation and also
increases endothelium permeability and potentiates proliferation of
endothelial cells, smooth muscle cells, fibroblasts, and T-lymphocytes
[6, 7]. The regulatory effects
of thrombin on cells are mediated by its membrane receptors. One of its
cloned receptors, PAR-1 (protease-activated receptor), is a member of a
superfamily of integral membrane proteins, seven-domain receptors
coupled to G-proteins [8, 9].
The extracellular N-terminal PAR-1 segment contains the peptide bond
Arg41-Ser42 which is cleaved by thrombin. The new, thrombin-shortened
N-terminal segment contains (in human receptor) the sequence
S42FLLRNPNDKYEPF, also known as TRAP-14 (thrombin receptor
agonist peptide). It activates the receptor by interacting with sites
on the second extracellular loop [10]. PAR-1 has
been found on platelets, fibroblasts, endothelial cells, smooth muscle
cells, and some other cells. TRAP-14 and its six-amino-acid N-terminal
fragment S42FLLRN (TRAP-6) have been synthesized; they also
exhibit a thrombin-like effect on all of the above-mentioned cell types
[1, 2, 10]. The pro-inflammatory effect of thrombin in
vivo is has been suggested to occur via activation of its receptors
on mast cells; histochemical method have revealed mast cell
degranulation after administration of TRAP-14 to rats [11]. However, these data are insufficient to conclude
that TRAP causes direct degranulation of mast cells and consequent
amine release. New members of the PAR family, PAR-2 and PAR-3, have
recently been recognized. The former is activated by trypsin and mast
cell-secreted tryptase but not by thrombin, whereas the latter is
activated by thrombin. These receptors are expressed by cells of
various tissues [12, 13].
However, these receptors have not been found on mast cells. There are
limited data on the interaction of thrombin with mast cells. Use of
FITC-labeled thrombin revealed the presence of a single class of
binding sites on RPMC with Kd = 1 nM. This binding is
rapid, saturable, and reversible, and it does not depend on the active
site of thrombin [14]. It has also been found that
thrombin can cause degranulation of mast cells and release of heparin
and histamine [15-17].
Thrombin-induced activation of mast cells, platelets, fibroblasts,
endothelial cells, and some other cells clearly involves a few
receptors including PAR-1 [7]. In mast cells,
picomolar concentrations of thrombin increased cGMP and decreased
histamine secretion [17]. However, the mechanisms
of modulation of mast cell reactivity by thrombin require detailed
investigation.
Nitric oxide (NO) might act as an endogenous regulator of mast cell reactivity. It stimulates guanylate cyclase activity, increases cGMP level and inhibits platelet aggregation; it also inhibits histamine and PAF-secretion [18-20]. The release of NO by mast cells has been demonstrated during activation by lipopolysaccharide, gamma-interferon, and interleukin-1beta [18-22].
In the present study, we have investigated the influence of thrombin receptor agonist peptide on NO release by stimulated and non-stimulated rat peritoneal mast cells. Platelet aggregation was used to monitor the release of NO and PAF by mast cells.
MATERIALS AND METHODS
The following chemicals were used in the study: ADP, PAF, A23187, L-NAME, calmidazolium, ginkgolide B, indomethacin, hemoglobin, Ficoll-400 from Serva (Germany); sodium dithionite from Merck (Germany); Sephadex G-25 from Pharmacia (Sweden). TRAP-6 (Ser-Phe-Leu-Leu-Arg-Asn-OH), the high affinity (ha)TRAP (Ala-Phe(pF)-Arg-Cha-Arg-Tyr-NH2 where Cha = Cyclohexyl-Ala), and peptide agonist PAR-2 (Ser-Leu-Ile-Gly-Arg-Leu) were synthesized in the Institute of Molecular Biotechnology (Jena, Germany).
RPMC isolated as described in [23] were purified in a Ficoll gradient (30 and 40%) and washed three times in Tyrodes solution, pH 7.2, containing 145 mM NaCl, 10 mM Na-HEPES, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2·6H2O, 5 mM glucose, and 0.1% albumin. The resulting cell suspension was diluted to the concentration required and kept in ice. Before experiments RPMC were treated with 5 µM indomethacin. To inhibit NO formation, RPMC were incubated with 300 µM L-NAME for 1 h; then 2·104 cells (10 µl) were incubated with 0.1-10 µM TRAP-6, and after selected time intervals aliquots of cell suspension were added to PRP. Samples containing the same number of unstimulated mast cells were used as controls. In a series of experiments, RPMC (3·104) were stimulated with the Ca-ionophore A23187 (1 µM) for 2 min. This ionophore concentration did not induce platelet aggregation. In some experiments, before stimulation by the ionophore, the RPMC were incubated with TRAP-6 (10 µM) for 5-40 min. In other experiments RPMC were treated with the calmodulin antagonist, 4 µM calmidazolium, for 40 min.
Human blood PRP was used for the investigation of platelet aggregation, which was determined in a BIOLA dual-channel laser aggregometer (BIOLA Ltd., Russia). A suspension of RPMC (10 µl) or Tyrode's solution was added to a cuvette containing 300 µl of PRP. The mixture was incubated at 37°C for 1 min with constant mixing, and then 10 µl of an aggregation inducing compound (5 µM ADP, 10 nM PAF, or 60 µM TRAP-6) was added. Platelet aggregation assayed by the standard turbidimetric method or the size of aggregates determined by the fluctuations in optical density of the platelet suspension were monitored for 6 min [24]. In some experiments PRP was preincubated for 2 min with 10 µM oxyHb (that binds NO) or with 30 µM ginkgolide B. The latter blocks platelet aggregation induced by PAF but does not influence ADP- or TRAP-6-stimulated aggregation.
Oxyhemoglobin (oxyHb) was prepared by oxidation of hemoglobin with sodium dithionite (Na2S2O4) [25] and purified by gel-filtration on a Sephadex G-25 column. OxyHb concentration was determined spectrophotometrically using epsilon576 = 15.99 mM-1·cm-1 [25].
The data was statistically treated using Student's method. The results are means (±m) of three to nine independent experiments. The differences were considered as statistically significant at p < 0.05.
RESULTS
Influence of non-stimulated RPMC on TRAP-6-induced platelet aggregation. RPMC (from 2.0·104 to 3.5·105) dose-dependently inhibited platelet aggregation induced by 60 µM TRAP-6. Maximal inhibition (by 55%) was observed at 3.5·105 cells. Mast cells treated with an inhibitor of NO formation (L-NAME) did not suppress TRAP-6-induced platelet aggregation. Preincubation of platelets with an NO-binding agent (oxyHb) significantly attenuated the inhibitory effect of mast cells on TRAP-6-induced platelet aggregation (Fig. 1).
These results support reports in the literature [18-20] on the release of NO by mast cells, which inhibits thrombin-induced platelet aggregation.Fig. 1. Inhibition of platelet aggregation by non-stimulated RPMC. TRAP-6 (60 µM) causing 80-90% of maximal platelet aggregation was used as the inducer. A suspension (10 µl) containing 2.0·104 (1), 4.0·104 (2), 2.0·105 (3), or 3.5·105 (4) RPMC was added to 300 µl of PRP, incubated in the cuvette of the aggregometer for 1 min at 37°C, and then platelet aggregation was stimulated by 60 µM TRAP-6; 5) preincubation of 2.0·104 RPMC with L-NAME (300 µM) for 1 h completely inhibited the effect of RPMC; 6) antiaggregation effect of RPMC attenuated after preincubation of PRP during 2 min with oxyHb (10 µM) before adding RPMC (2.0·104). The data are means (±m) of 5-9 independent experiments; *p < 0.01 (versus 1); #p < 0.05 (versus control without RPMC).

Influence of TRAP-6-activated RPMC on platelet aggregation. Incubation of RPMC with TRAP-6 inhibited aggregation of stimulated platelets. The effect depended on peptide concentration (1-10 µM) and the incubation time (Fig. 2). In these experiments the effect of RPMC (2.0·104) causing 32% inhibition of platelet aggregation (see Fig. 1) was defined as 100%. With 10 µM TRAP-6, inhibition by 65% was observed after 5 min incubation. TRAP-6-activated RPMC also inhibited platelet aggregation stimulated by 10 nM PAF or by 5 µM ADP. Mast cells activated by 1 µM haTRAP (high affinity analog of TRAP-6) also inhibited platelet aggregation (by 35, 64, and 66% after 5, 15, and 30 min incubation of haTRAP with RPMC). Preincubation of PRP with oxyHb sharply attenuated the antiaggregatory effect of TRAP-6-treated RPMC. In contrast to TRAP-6, the peptide agonist of PAR-2, which did not influence platelet aggregation, was also ineffective in NO release from RPMC (data not shown). These data suggest that the interaction of TRAP-6 with PAR-1 stimulates NO production by mast cells.
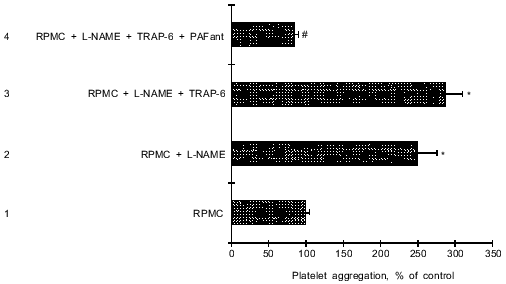
Influence of NO synthase inhibitor on the inhibition of platelet aggregation by TRAP-6-activated RPMC. To inhibit NO formation in mast cells, the cells were incubated with L-NAME for 1 h, activated with 10 µM TRAP-6 for 5 min, and then added to the platelet suspension. Figure 3 shows that such RPMC actually significantly stimulated (by 187%) rather than inhibited platelet aggregation. Since blockade of NO formation by L-NAME causes RPMC activation and secretion of inflammatory mediators, we investigated PAF secretion after stimulation of mast cells with TRAP-6. Platelet PAF-receptors were blocked with the specific inhibitor ginkgolide B (30 µM). This completely blocked stimulation of platelet aggregation by RPMC which were pretreated with L-NAME and incubated with TRAP-6 (Fig. 3).Fig. 2. Effect of TRAP-6 activated RPMC on platelet aggregation. 1) Control, platelet aggregation stimulated by 60 µM TRAP-6 or 10 nM PAF after the addition of 2.0·104 non-stimulated RPMC (in 10 µl); 2, 3, 4) RPMC preincubated with 10 µM TRAP-6 for 1 (2), 5 (3), or 15 (4) min; 5, 6) RPMC preincubated with 1 µM (5) or 10 µM (6) TRAP-6 for 5 min. The data are means (±m) of 5-6 independent determinations. *p < 0.05 versus 1.

Thus, blockade of NO synthesis and activation of L-NAME-treated mast cells by TRAP-6 led to secretion of PAF, and this caused increased platelet aggregation.Fig. 3. Influence of TRAP-6 on PAF release by L-NAME-treated mast cells. Platelet aggregation was investigated in the presence of 2.0·104 RPMC (10 µl). 1) Non-stimulated RPMC (control); 2) RPMC pretreated with 300 µM L-NAME for 1 h before addition to PRP; 3) RPMC pretreated with L-NAME (300 µM) as in (2) but activated with 10 µM TRAP-6 for 5 min before addition to PRP; 4) RPMC preincubated with L-NAME and activated with TRAP-6 as in (3) but added to PRP pretreated with 30 µM ginkgolide B (PAF antagonist). In all cases aggregation was stimulated by 60 µM TRAP-6 or 5 µM ADP. The data are means (±m) of 5 independent determinations. *p < 0.01 versus 1; #p < 0.01 versus 3.
Influence of TRAP-6 on RPMC activated by the calcium ionophore A23187. These experiments were carried out to investigate the role of TRAP-6 in the modulation of RPMC activity and their secretion of PAF initiated by the calcium ionophore A23187.
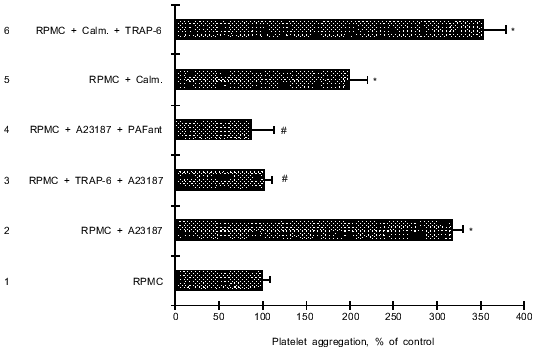
Mast cells preincubated with 1 µM A23187 for 2 min were added to a platelet suspension. After equilibration for 1 min, an aggregation inducer was added. A23187-pretreated RPMC caused a significant increase in platelet aggregation (by 218%) compared with the aggregation in the presence of non-activated RPMC (Fig. 4). This effect of ionophore-activated mast cells on platelet aggregation was completely blocked by pretreatment of platelets with the PAF-receptor antagonist ginkgolide B. These results together with data from the literature [20] suggest that the Ca-ionophore activates PAF secretion.
In the next series of experiments before treatment with A23187 mast cells were preincubated with 10 µM TRAP-6 for 5-40 min. After incubation for 5 min with TRAP-6, activation of RPMC with the ionophore did not result in enhanced platelet aggregation (Fig. 4). Moreover, RPMC preincubation with TRAP-6 before activation by A23187 caused sharp (threefold) decrease in platelet aggregation compared with the aggregation in the presence A23187-activated mast cells (Fig. 4). Prolongation of the incubation between RPMC and TRAP-6 before activation of the cells with the ionophore did not cause further reduction in platelet aggregation.Fig. 4. Influence of A23187-activated RPMC on platelet aggregation: the effect of TRAP-6. 1) Platelet aggregation in the presence of 3.0·104 RPMC (in 10 µl) (control); 2) RPMC were preincubated with 1 µM A23187 for 2 min before addition to a platelet suspension; 3) RPMC were preincubated with 10 µM TRAP-6 for 5 min before treatment with A23187; 4) platelets were treated with 30 µM ginkgolide B for 2 min before addition of A23187-activated mast cells; 5) RPMC were treated with 4 µM calmidazolium for 40 min before addition to platelet suspension; 6) RPMC treated with 4 µM calmidazolium as in (5) were incubated with 10 µM TRAP-6 for 10 min before addition to a platelet suspension. In all experiments aggregation was induced by 5 µM ADP. The data are means (±m) of 3-5 independent determinations. *p < 0.05 versus 1; #p < 0.01 versus 2.
These results suggest that TRAP-6 can rapidly modulate A23187-activated RPMC reactivity by blocking ionophore-induced PAF release from these cells and by stimulating NO production by them.
Influence of TRAP-6 on RPMC with inhibited constitutive NO-synthase (cNOS). The calmodulin antagonist calmidazolium inhibits cNOS in rat aorta smooth muscle cells [26]. IL-1beta-induced increase in NO production by mast cells may be due to activation of cNOS [20]. Taking into consideration these data, we investigated mast cell activation by TRAP-6 under conditions of cNOS inhibition by calmidazolium. Treatment of non-stimulated RPMC with 4 µM calmidazolium for 40 min blocked reduction of ADP-stimulated platelet aggregation induced by these mast cells (Fig. 4). However, pretreatment of RPMC with calmidazolium followed by incubation of the cells with TRAP-6 for 10 min augmented ADP-induced platelet aggregation rather than attenuating it (Fig. 4).
Thus, stimulation of NO release by RPMC pretreated with TRAP-6 is apparently due to the activation of cNOS. iNOS may also be involved in the effect of the peptide on mast cells.
DISCUSSION
In spite of great interest in inflammatory processes, little is known about the mechanisms of non-immune activation and regulation of the activity of mast cells which release inflammation mediators--histamine, PAF, and chemoattractants [27]--and also nitric oxide which can inhibit platelet aggregation and neutrophil activation and cause vessel relaxation; these effects of NO are due to activation of soluble guanylate cyclase resulting in accumulation of cGMP and activation of cGMP-dependent protein kinase (G kinase) [28]. The mechanism of inhibition of platelet aggregation by NO may involve phosphorylation of the cytoplasmic C-terminal domain of the TXA2 receptor by a G kinase [29].
We have found that the interaction of the thrombin receptor agonist TRAP-6 with thrombin receptor (obviously PAR-1) on the membrane of peritoneal mast cells results in the inhibition of PAF release from these cells via an NO-dependent mechanism.
In the present report NO release was registered by inhibition of platelet aggregation. Using three experimental approaches which independently blocked the NO effect at different levels (inhibition of NO formation by L-NAME, NO binding by oxyHb, and use of calmidazolium, an inhibitor of calmodulin and Ca-dependent cNOS), we have demonstrated that the inhibition of platelet aggregation by TRAP-6-activated RPMC is really due to NO release.
Using high affinity TRAP (haTRAP) for activation of RPMC, we have confirmed that TRAP can activate NO release by mast cells. Addition of TRAP-6 to RPMC with blockaded NO synthesis resulted in increased secretion of the inflammation mediator PAF. The use of the antagonist of PAF-receptors, ginkgolide B, in our experiments convincingly supports this conclusion.
Although a role of thrombin in the regulation of the inflammatory process is not completely elucidated, it is known that it promotes inflammatory reactions. Thrombin activates leukocyte chemotaxis and adhesion of platelets and leukocytes to endothelium; it increases endothelial permeability, expression of P-, E-, and L-selectins, and histamine release from bone marrow mast cells [2, 3, 6, 15, 30, 31]. However, experiments with vascular smooth muscle cell culture revealed that thrombin regulates (at the transcriptional level) the expression of iNOS via proteolytic activation of PAR-1 [32].
Mechanical activation of mast cells (by vigorous mixing) or their activation by lipopolysaccharide, immunoglobulin E, interleukin 1beta, or gamma-interferon results in release of NO. The latter inhibits secretion of inflammatory mediators (PAF) and blocks platelet aggregation [18-22]. However, we did not find in the literature any indication of the thrombin-induced release of NO by mast cells. Only indirect evidence exists suggesting that thrombin can activate this process: very low thrombin concentrations increased cGMP content in mast cells, and this was accompanied by reduction of histamine secretion [17]. Our results provide further evidence that thrombin (via its interaction with PAR-1) can act as a non-immune modulator of mast cell activity by stimulating NO formation and inhibiting the secretion of inflammation mediators. In other words, thrombin can act as a modulator of the inflammatory process regulator NO.
Contact of thrombin, the main enzyme of the blood clotting system, with mast cells localized along blood vessels can probably occur at sites of tissue damage. Although liver is the major site of prothrombin synthesis, thrombin mRNA is found in macrophages, some cells of the nervous system, and developing mouse skeletal muscles [33-35]. These data suggest the possibility of generation of tissue thrombin. The latter may regulate the activity of connective tissue cells apart from blood vessels and participate in inflammation processes and wound healing. Like thrombin, factor Xa (but not factor IXa) stimulates receptor-mediated NO release by endothelial cells, thus causing vasodilation and hypotension in rats [36].
Inhibition of NO formation in mast cells by L-NAME was shown to activate release of inflammatory mediators: PAF (responsible for rapid increase of endothelial permeability) and histamine (which causes delayed increase of endothelial permeability) [20, 30, 31]. Inhibition of NO synthesis increases mast cell-dependent induction of leukocyte adhesion to endothelium and the development of the inflammatory process [31].
The calcium ionophore A23187, causing increase of [Ca2+]in, induced secretion of PAF by mast cells which was blocked by TRAP-6. The latter effect was accompanied by activation and degranulation of these cells.
Stimulation of NO production in mast cells by TRAP-6 suggests that modulation of activated mast cells is due to NO release in response to TRAP-6 and thrombin.
It remains unclear which NOS isoform [28] is responsible for thrombin-induced increase in NO formation in mast cells. Lack of NO production in TRAP-6-stimulated mast cells which were pretreated (for more than 40 min) with the cNOS inhibitor calmidazolium suggests the involvement of cNOS. Rapid NO formation (within minutes) during the TRAP-6 effect on mast cells seems to support this hypothesis. Our results are consistent with data from the literature showing the presence of cNOS in mast cells [31], although the involvement of iNOS in NO production by mast cells activated by thrombin or other proteases cannot be ruled out.
Good evidence exists that thrombin (TRAP-6 modulates its action) is the non-immune regulator of mast cell reactivity. The thrombin effect is mediated by PAR-1, and this is accompanied by reduction of PAF secretion by activated mast cells.
Since TRAP-6 can activate both mast cells and platelets, we suggest that the N-terminal peptide comprised of 41 amino acids (MGPRRLLLVAACFSLCGPLLSARTRARRPESKATNATLDPR) which is cleaved by thrombin from PAR-1 and acts as potent agonist of platelet aggregation [37] may also activate mast cells. The absence of this peptide probably explains the apparent discrepancy between the efficacy of TRAP-6 and thrombin in the activation of platelets and mast cells: the same effect required three orders of magnitude less thrombin concentration than that of TRAP-6 [1, 2, 7, 10, 17].
Thus, thrombin exhibits dual regulatory effects on inflammatory processes. It increases the release of inflammatory mediators, leading to acute inflammatory reactions (increase of permeability, edema, etc.), but stimulating NO release, thrombin inhibits release of inflammatory mediators and the development of inflammatory process.
We are grateful to A. F. Vanin for his interest in this work and his helpful advice.
This work was supported by the Russian Foundation for Basic Research (98-04-48217) and the State Program for Science and Technology (Engineering Enzymology, 97-04-202/0).
REFERENCES
1.Strukova, S. M., and Kireeva, E. G. (1995)
Vestnik Mosk. Univ. Ser. 16, Biol., No. 1, 3-13.
2.Strukova, S. M., Kireeva, E. G., and Dugina, T. N.
(1997) Vestnik Mosk. Univ. Ser. 16, Biol., No. 1,
8-16.
3.Fenton, J. W., II (1995) Thromb. Haemost.,
74, 493-498.
4.Van Obberghen-Schilling, E., Vouret-Craviari, V.,
Chen, Y-H., Grall, D., Chambard, L-C., and Pouyssegur, J. (1995)
Ann. NY Acad. Sci., 166, 431-441.
5.Glusa, E., Paintz, M., and Bretschneider, E. (1996)
Semin. Thromb. Hemost., 22, 261-266.
6.Carney, D. H., Redin, W., and McCroskey, L. (1992)
Semin. Thromb. Hemost., 18, 91-103.
7.Strukova, S. M., Dugina, T. N., Chistov, I. V.,
Markvicheva, E. A., Kuptsova, S. V., Kolokolchikova, E. G., Rumsh, L.
D., Zubov, L. D., and Glusa, E. (1998) Bioorg. Khim., 24,
288-292.
8.Vu, T-K., Hung, D., Wheaton, V., and Coughlin, S.
(1991) Cell, 64, 1057-1068.
9.Rasmussen, U., Vouret-Craviari, V., Jallot, S.,
Schlesinger, Y., Pages, G., Parirani, A., Lecocq, J-P., Pouyssegur, J.,
and Van Obberghen-Schilling, E. (1991) FEBS Lett., 288,
123-128.
10.Brass, L. (1997) Coronary Artery Disease,
8, 49-58.
11.Cirino, G., Cicala, C., Bucci, M. R., Sorrentino,
L., Maraganore, J. M., and Stone, S. R. (1996) J. Exp. Med.,
183, 821-827.
12.Molino, M., Barnathan, E., Numerof, M., Clark,
J., Dreyer, M., Cumashi, A., Hoxi, J., Schechter, N., Woolkalis, M.,
and Brass, L. (1997) J. Biol. Chem., 272, 4043-4049.
13.Ishihara, H., Connolly, A., Zeng, D., Kahan, M.,
Zheg, Y., Timmons, C., Tram, T., and Coughlin, S. (1997) Nature,
386, 502-506.
14.Strukova, S. M., and Khlgatian, S. V. (1991)
Thromb. Res., 64, 795-796.
15.Rarin, E., and Marx, G. (1984) J.
Immunol., 133, 3282-3285.
16.Umarova, B. A., Khlgatian, S. V., and Strukova,
S. M. (1989) Byul. Eksp. Biol. Med., 107, 131-133.
17.Strukova, S. M., Dugina, T. N., Khlgatian, S. V.,
Redkozubov, A. E., Redkozubova, G. P., and Pinelis, V. G. (1996)
Semin. Thromb. Hemost., 22, 145-150.
18.Salvemini, D., Masini, E., Anggard, E.,
Mannaioni, P. F., and Vane, J. (1990) Biochem. Biophys. Res.
Commun., 169, 596-601.
19.Masini, E., Mannioni, P. F., Pistelli, A.,
Salvemini, D., and Vane, J. (1991) Biochem. Biophys. Res.
Commun., 177, 1178-1182.
20.Hogaboam, C., Befus, A., and Wallace, J. (1993)
J. Immunol., 151, 3767-3774.
21.Eastmond, N. C., Banks, E. M., and Coleman, J. W.
(1997) J. Immunol., 159, 1444-1450.
22.Mannaioni, P. F., Bello, M. G., DiBello, M. G.,
Mirabella, C., Gai, P., Schunack, W., and Masini, E. (1997) Int.
Arch. Allergya. Immunology, 113, 297-299.
23.Thon, I. L., and Uvnas, B. (1967) Acta
Physiol. Scand., 71, 303-315.
24.Gabbasov, Z. A., Popov, E. G., Gavrilov, I. Y.,
and Pozin, E. Y. (1989) Thromb. Res., 54, 215-223.
25.Salvemini, D., deNucci, G., Gryglevski, R., and
Vane, G. (1989) Proc. Natl. Acad. Sci. USA, 86,
6328-6332.
26.Schini, V. B., and Vanhoutte, P. M. (1992) J.
Pharmacol. Exp. Ther., 261, 553-557.
27.Landry, Y., Bronner, C., Mousli, M., Fisher, T.,
and Valle, A. (1992) Bull. Inst. Pasteur, 90, 83-98.
28.Scsmidt, H. H. H. W., Hofmann, H., and Ogilvie,
P. (1995) The Role of Nitric Oxide in Physiology and
Pathophysiology, Springer, Heidelberg.
29.Wang, G-R., Zhu, Y., Halushka, P. V., Lincoln, T.
M., and Mendelsohn, M. E. (1998) Proc. Natl. Acad. Sci. USA,
95, 4888-4893.
30.Gaboury, J., Yiao-rei, N., and Kubes, P. (1996)
Circulation, 93, 318-320.
31.Kubes, P., and Neil Granger, D. (1996)
Cardiovasc. Res., 32, 699-708.
32.Schini-Kerth, V. B., Fisslthaler, B., Andersen,
T. T., Fenton, J. W., II, Vanhoutte, P. M., and Busse, R. (1995)
Thromb. Haemost., 74, 980-986.
33.Lindahl, U., Peiler, G., Bozgwald, J., and
Seljelid, R. (1989) Arch. Biochem. Biophys., 273,
180-188.
34.Zoubine, M. N., Ma, J. Y., Smirnova, I. V.,
Citron, B. A., and Festoff, B. W. (1996) Develop. Biol.,
179, 447-457.
35.Citron, B. A., Smirnova, I. V., Zoubine, M. N.,
and Festoff, B. W. (1997) Thromb. Res., 87, 303-313.
36.Papapetropoulos, A., Piccardoni, P., Cirino, G.,
Bucci, M., Sorrentino, R., Cicala, C., Johnson, K., Zachariou, V.,
Sessa, W., and Altieri, D. (1998) Proc. Natl. Acad. Sci. USA,
95, 4738-4742.
37.Furman, M., Liu, L., Benoit, S., Becker, R.,
Barnard, M., and Michelson, A. (1998) Proc. Natl. Acad. Sci.
USA, 95, 3082-3087.