Structure of the O-Polysaccharide of the Lipopolysaccharide of Pseudomonas syringae pv. garcae ICMP 8047
E. L. Zdorovenko1, V. V. Ovod2, A. S. Shashkov1, N. A. Kocharova1, Yu. A. Knirel1*, and K. Krohn2
1Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninskii pr. 47, Moscow, 117913 Russia; fax: (095) 135-5328; E-mail: knirel@ioc.ac.ru2Institute of Medical Technology, University of Tampere, P.O. Box 607, Tampere, 33101 Finland; fax: (358-3) 215-7332; E-mail: ltvlov@uta.fi
* To whom correspondence should be addressed.
Received January 11, 1999
The composition and structure of the O-polysaccharide of the lipopolysaccharide of Pseudomonas syringae pathovar garcae ICMP 8047 were studied using methylation analyses, Smith degradation, and 1H- and 13C-NMR spectroscopy, including two-dimensional correlation spectroscopy (COSY), total correlation spectroscopy (TOCSY), nuclear Overhauser effect spectroscopy (NOESY), and H-detected 1H,13C heteronuclear multiple-quantum coherence (HMQC) experiments. The polysaccharide was found to contain L-rhamnose and 3-acetamido-3,6-dideoxy-D-galactose (D-Fuc3NAc) in the ratio 4:1 and to consist of two types of pentasaccharide repeating units. The major (1) and minor (2) repeating units differ from each other only in the position of substitution of one of the rhamnose residues in the main chain. Similar structural heterogeneity has been reported formerly in O-polysaccharides of some other P. syringae strains having a similar monosaccharide composition. A Fuc3NAc residue is attached to the main rhamnan chain as a side chain by a (alpha1-->4) glycosidic linkage; this has not hitherto been described in P. syringae:
KEY WORDS: lipopolysaccharide, O-specific polysaccharide, 3-acetamido-3,6-dideoxy-D-galactose, structural heterogeneity, Pseudomonas syringae
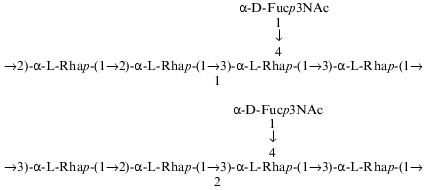
Abbreviations: COSY) correlation spectroscopy; Fuc3NAc) 3-acetamido-3,6-dideoxygalactose; HMQC) heteronuclear multiple-quantum coherence; NOESY) nuclear Overhauser effect spectroscopy; Rha) rhamnose; TOCSY) total correlation spectroscopy.
Phytopathogenic bacteria of the genus Pseudomonas syringae affect
a wide spectrum of wild and cultivated plants. Based on host
specificity, the bacteria are divided into more than 50 pathovars [1]. Strains of P. syringae are also classified
on the basis of immunospecificity [2-5], which is determined by differences in the
structure of the lipopolysaccharide of the outer membrane of the cell
wall [6-9]. Our systematic
chemical and immunochemical studies of the lipopolysaccharides of P.
syringae [5-11] are aimed
at the determination of the molecular basis of the immunospecificity,
clarifying its role in the specific recognition of the host plant, and
estimation of the significance of data on composition and structure of
the lipopolysaccharide for taxonomy and classification of the bacteria.
In the present work, we describe a new structure of the
O-polysaccharide chain of the lipopolysaccharide of P. syringae
pv. garcae ICMP 8047, which causes a disease of coffee
trees (Coffea arabica L.). This pathovar has not been studied
hitherto in respect to the lipopolysaccharide structure.
MATERIALS AND METHODS
Strain P. syringae pv. garcae ICMP 8047 (NCPPB 1399, GSPB 2680) was isolated in Kenya in August 1974. The bacterium was grown on potato dextrose agar (Difco Laboratories, USA) at 22°C for 24 h. Lipopolysaccharide was isolated as described [7] and degraded by hydrolysis with 2% acetic acid for 1.5 h at 100°C. Core oligosaccharide and O-polysaccharide fractions were isolated by gel chromatography of the carbohydrate portion on a column (56 cm × 2.6 cm) of Sephadex G-50 (Pharmacia, Sweden) using 0.05 M pyridinium acetate (pH 4.5) as eluent and monitoring with a differential refractometer (Knauer, Germany).
For sugar analysis, the O-specific polysaccharide washydrolyzed with 2 M CF3COOH (120°C, 2 h), and the monosaccharides were identified by GLC as their alditol acetates [12] on an Ultra 2 (Hewlett-Packard) capillary column using a Hewlett-Packard 5880 instrument and by GLC/MS on a Hewlett Packard 5890 chromatograph equipped with a NERMAG R10-10L mass spectrometer. Temperature gradients used were 160°C (1 min) to 290°C at 10°C/min and 160°C (3 min) to 250°C at 3°C/min, respectively. Sugar ratios are reported as detector response ratios. The absolute configurations of the monosaccharides were determined by GLC of acetylated glycosides with (+)-2-octanol by a modified method [13, 14].
Methylation was carried out with methyl iodide in dimethyl sulfoxide in the presence of solid sodium hydroxide [15]. Hydrolysis of the methylated polysaccharide was performed as in sugar analysis, and the partially methylated monosaccharides were converted into alditol acetates and analyzed by GLC and GLC/MS using the conditions described above.
For Smith degradation, the polysaccharide (12 mg) was oxidized with 0.1 M sodium metaperiodate in the dark for 48 h at 20°C; after adding an excess of ethylene glycol, the product was reduced by an excess of sodium borohydride and desalted on a column (80 cm × 1.6 cm) of a TSK HW-40 (S) gel (Merck, Germany) in water. The resulting modified polysaccharide was hydrolysed with 2% acetic acid for 2 h at 20°C, reduced with an excess of sodium borohydride, and fractionated on a TSK HW-40 (S) gel column in water. After additional purification on the same column, the yields of oligosaccharide-glycerols 3 and 4 were 1.6 and 1 mg, respectively.
Samples were deuterium-exchanged by freeze-drying from 2H2O. 1H- and 13C-NMR spectra were recorded with a Bruker DRX-500 spectrometer (Germany) in solutions in 2H2O at 326 and 303 K for the polysaccharide and oligosaccharide-glycerols 3 and 4, respectively, using acetone (deltaH 2.225, deltaC 31.45) as internal standard. Two-dimensional spectra were run using standard Bruker software. Mixing times of 200 and 100 msec were used in TOCSY and NOESY experiments, respectively.
RESULTS AND DISCUSSION
Mild acid degradation of the lipopolysaccharide from P. syringae pv. garcae ICMP 8047 produced an O-specific polysaccharide which was isolated by gel chromatography. The elution profile of the carbohydrate portion of the degraded lipopolysaccharide showed that S-type lipopolysaccharide was predominant in the studied strain, whereas the content of the R-type lipopolysaccharide with a carbohydrate moiety restricted to the core oligosaccharide was low.
Sugar analysis of the polysaccharide after full acid hydrolysis revealed rhamnose (Rha), 3-amino-3,6-dideoxygalactose (Fuc3N), glucose, and 2-amino-2-deoxyglucose in the ratios 4.0:1.0:0.16:0.06. Fuc3N was identified by GLC/MS using the authentic compound from the O-specific polysaccharide of P. syringae pv. coriandricola GSPB 2028 (W-43) [11]. Taking into account published data on the lipopolysaccharide composition of some other P. syringae pathovars [6, 16], it could be suggested that the O-specific polysaccharide of the studied strain contains Rha and Fuc3N, whereas Glc and GlcN are, most likely, components of the core oligosaccharide. Determination of the absolute configurations of the main monosaccharides by GLC of acetylated 2-octyl glycosides [13, 14] showed that Rha is present in the L form and Fuc3N in the D form.
Methylation analysis of the polysaccharide revealed 3,4-di-O-methylrhamnose, 2,4-di-O-methylrhamnose, and 2-O-methylrhamnose in the ratios 1.7:1.1:1.0, as well as 3,6-dideoxy-2,4-di-O-methyl-3-(N-methyl)acetamidogalactose. The methylation data showed that the polysaccharide is branched and includes 2-substituted, 3-substituted, and 3,4-disubstituted Rha residues and a terminal Fuc3N residue in the side chain.
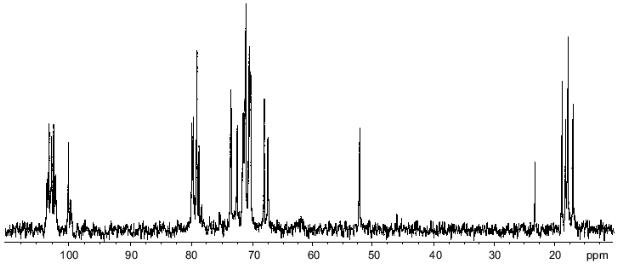
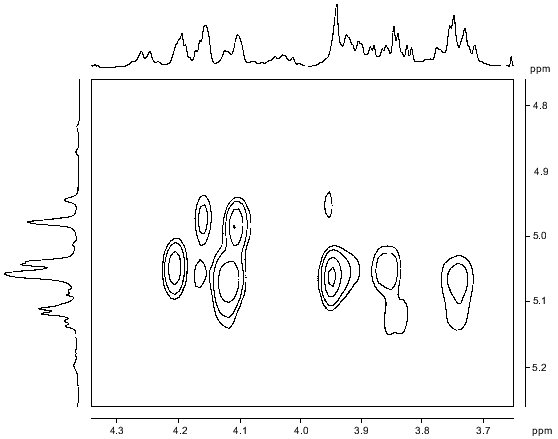
In the resonance region of anomeric carbons in the 13C-NMR spectrum of the polysaccharide (Fig. 1), there were two series of signals with integral intensity ratio ~3:1. The major series included five signals at delta 100.0, 102.0, 102.3, 102.6, and 103.0. The minor series contained four well-resolved signals at delta 99.7, 102.8, 103.2, and 103.3, and the fifth signal could be suggested to be superimposed with the major signal at delta 102.0, which had higher intensity and width as compared to other signals of the main series. Only a few minor signals were observed in other regions of the spectrum, evidently due to multiple coincidences with signals of the major series. These data showed the presence in the polysaccharide of two types of repeating units with different structures, and, therefore, the polysaccharide lacks strict regularity.
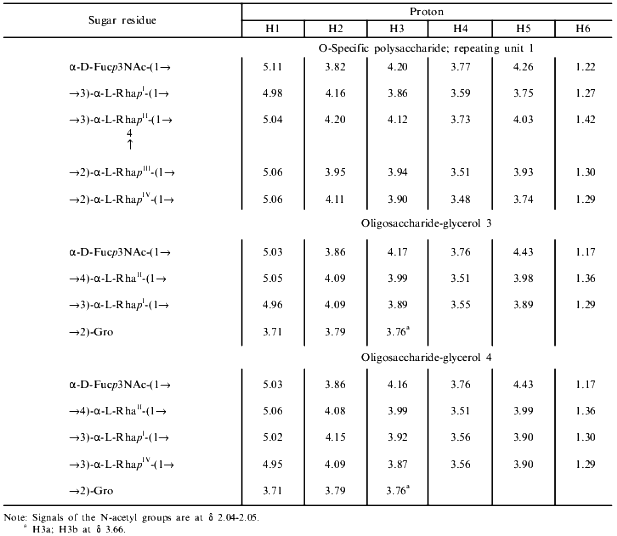
In the 1H-NMR spectrum of the polysaccharide there were signals of different intensities as well, including the anomeric proton signals for four rhamnose residues at delta 4.98, 5.04, and 5.06 (2H) (broadened singlets) and one Fuc3NAc residue at delta 5.11 (doublet). Only two anomeric proton signals of the minor series were clearly observed, at delta 4.94 (broadened singlet, Rha H1) and 5.13 (doublet, Fuc3NAc H1). Judging from the ratio of the signal intensities in the major and minor series, the ratio of the repeating units of the two types is ~3.5:1.Fig. 1. 13C-NMR spectrum of the O-polysaccharide.
In addition to the five anomeric signals, the major series of the 13C-NMR spectrum contained signals for five CH3--C groups (C6) of 6-deoxy sugars at delta 17.0 (Fuc3N), 17.8, 17.9, 18.3, and 18.9 (4 Rha), one carbon bearing nitrogen (Fuc3N C3) at delta 52.2, 19 sugar ring carbons linked to oxygen at delta 67.6-79.9, and one N-acetyl group (Me at delta 23.3, CO at delta 175.4).
Accordingly, the main series in the 1H-NMR spectrum of the polysaccharide contained intense signals for five CH3--C groups (H6) of 6-deoxy sugars at delta 1.22-1.42 and one N-acetyl group at delta 2.05. These data further confirmed that the major repeating unit of the polysaccharide is a pentasaccharide containing four Rha residues and one residue of Fuc3NAc.
The signals of the major series in the 1H-NMR spectrum of the polysaccharide (Table 1) were assigned using two-dimensional COSY (Fig. 2) and TOCSY experiments. The Rha residues were enumerated according to their sequence in the repeating unit (see below). The J1,2 coupling constant value of 3.8 Hz indicated that the Fuc3NAc residue is linked by the alpha-glycosidic linkage. The four Rha residues are alpha-linked as well, as followed from the chemical shifts delta 3.74-4.03 for H5 and delta 70.4-70.7 for C5 (compare the H5 chemical shift delta 3.86 and 3.39 [17] and the C5 chemical shift delta 70.0 and 72.3 [18] in alpha-Rha and beta-Rha, respectively).
Table 1. 1H-NMR data (chemical
shifts in ppm)
The major series of signals in the 13C-NMR spectrum of the polysaccharide (Table 2) was assigned using a two-dimensional H-detected 1H,13C-HMQC experiment (Fig. 3). Correlation between the proton at the carbon carrying nitrogen (H3) and the corresponding carbon (C3) at delta 4.20/52.2 confirmed the position of the acetamido group in the Fuc3NAc residue. Low-field positions at delta 78.7-79.9 of the signals for C2 of RhaIII and RhaIV, C3 of RhaI, and C3 and C4 of RhaII, as compared to their positions in the spectra of nonsubstituted alpha-Rha at delta 71.3-73.5 [18], allowed determination of the modes of substitution of the monosaccharide residues in the major repeating unit of the polysaccharide.Fig. 2. Part of a COSY spectrum of the O-polysaccharide. The corresponding parts of the 1H-NMR spectrum are displayed along the axes.
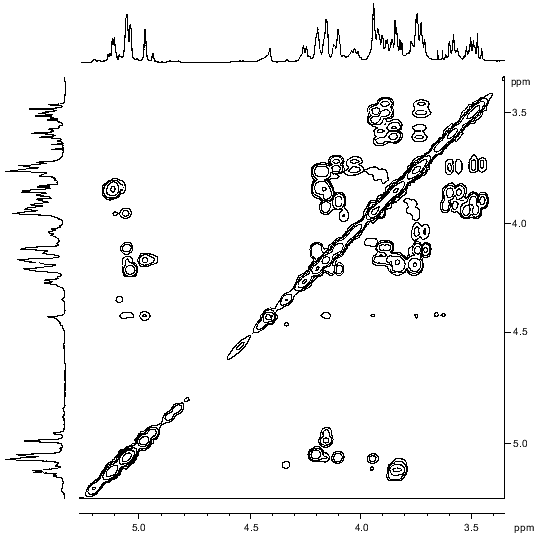
Table 2. 13C-NMR data (chemical
shifts in ppm)
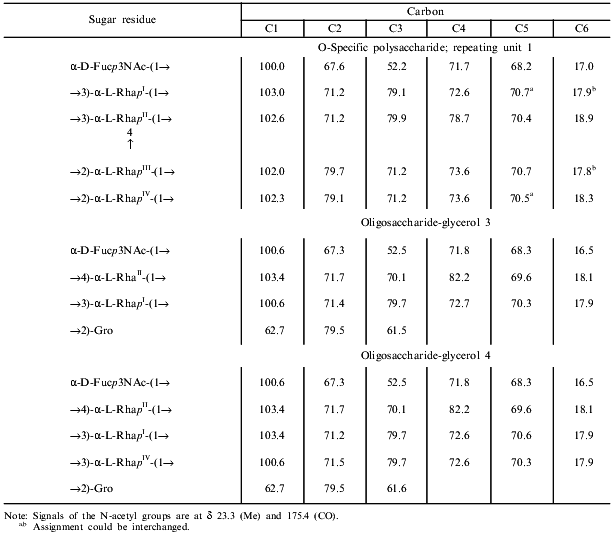
A two-dimensional NOESY experiment (Fig. 4) revealed the following interresidue correlations between transglycosidic protons in the major series: RhaI H1/RhaIV H2, RhaII H1/RhaI H3, RhaIII H1/RhaII H3, RhaIV H1/RhaIII H2, and Fuc3NAc H1/RhaII H3 and H4 at delta 4.98/4.11, 5.04/3.86, 5.06/4.12, 5.06/3.95, 5.11/3.73, and 5.11/4.12, respectively. The presence of an additional interresidue Fuc3NAc H1/RhaII H6 cross peak at delta 5.11/1.42 confirmed the Fuc3NAc-(1-->4)-RhaII fragment [19]. An intense cross peak at delta 5.06/3.74 was interpreted as a superposition of two interresidue cross peaks RhaIII H1/RhaIV H5 and RhaIV H1/RhaI H5, which are typical of alpha1-->2-linked rhamnose residues [11]. In accordance with the alpha configuration of the glycosidic linkages, intraresidue H1/H2 cross peaks were observed for all five monosaccharide residues. Thus, the NOESY data were in agreement with the methylation analysis and 13C chemical shift data and showed that the major repeating unit of the polysaccharide has the structure 1:Fig. 3. Part of a 1H,13C-HMQC spectrum of the O-polysaccharide. The corresponding parts of the 1H- and 13C-NMR spectra are displayed along the horizontal and vertical axes, respectively.
As in the one-dimensional 1H-NMR spectrum, most cross peaks of the minor series in the two-dimensional COSY, TOCSY, and NOESY spectra of the polysaccharide coincided with cross peaks of the major series. In the NOESY spectrum, H1 of only one rhamnose residue of the minor series gave a clear cross peak, at delta 4.94/3.95 with H2 of another Rha residue. This cross peak corresponded to the RhaIV H1/RhaIII H2 correlation peak from the major repeating unit.Fig. 4. Part of a NOESY spectrum of the O-polysaccharide. The corresponding parts of the 1H-NMR spectrum are displayed along the axes.
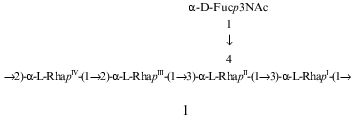
In the two-dimensional 1H,13C-HMQC spectrum (Fig. 3), H1 at delta 4.94 correlated with C1 at delta 103.3; hence, the corresponding Rha residue is 3-substituted. Indeed, glycosylation of this residue at position 2 would cause an upfield displacement of the C1 signal by 1-1.5 ppm [18] (e.g., compare the C1 chemical shifts delta 102.0 and 102.3 in the 2-substituted RhaIII and RhaIV residues in the major repeating unit of the polysaccharide (Table 2)). Correspondingly, two other C1 signals in the minor series at delta 102.8 and 103.2 also belonged to 3-substituted Rha residues, and that at delta 102.0 to a 2-substituted Rha residue.
These data allowed suggestion that the minor repeating unit of the polysaccharide is a pentasaccharide having structure 2, which differs from structure 1 of the major repeating unit in the position of substitution of RhaIV (1 and 2 contain 2-substituted and 3-substituted RhaIV, respectively). This suggestion is in agreement with previous findings which showed that all P. syringae O-specific polysaccharides with four L-Rha residues in the repeating unit are characterized by the same type of structural heterogeneity in the main chain ([8, 10, 11, 20, 21] and our unpublished data):
In order to confirm independently the structures 1 and 2, Smith degradation of the polysaccharide was performed. As a result, two oligosaccharide-alditols 3 and 4 were obtained and separated by gel chromatography.
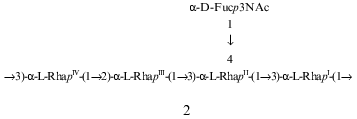
The 1H- and 13C-NMR spectra of the products 3 and 4 were assigned using two-dimensional COSY and 1H,13C-HMQC experiments, respectively (Tables 1 and 2). The data showed that 3 consists of one Fuc3NAc residue, two Rha residues, and glycerol, whereas 4 includes one Rha residue more. In both oligosaccharide-glycerols, Fuc3NAc is the terminal residue, one of the rhamnose residues (RhaII) is 4-substituted, and the others (RhaI in 3, RhaI and RhaIV in 4) are 3-substituted. This followed from down-field displacements to delta 79.5-82.2 of the signals for C4 of RhaI and C3 of RhaI and RhaIV, compared to their positions at delta 71.3-73.5 in nonsubstituted alpha-Rha [18]. Therefore, both oligosaccharide-glycerols are linear.
Computer-assisted analysis of the 13C-NMR spectra was used to establish the structures of 3 and 4. This approach is based on calculation of chemical shifts for all theoretically possible structures of an oligosaccharide with the given sugar composition and selection of a structure (or structures) for which the calculated spectrum is closest to the experimental spectrum [18, 22]. As a result, for each oligosaccharide-glycerol only one linear structure was revealed; they matched the experimental 13C chemical shift data (Table 2) and the positions of substitution of the monosaccharide residues determined from the 1H,13C-HMQC spectrum. These structures were characterized by the least sum of squared deviations of chemical shifts in the calculated and experimental spectra (S = 0.3 after normalization to one sugar residue for each oligosaccharide-glycerol), whereas other theoretically possible structures with the given sugar composition showed significantly higher deviations (S > 1.2).
Therefore, the Smith degradation products 3 and 4 were derived from the major and minor repeating units 1 and 2, respectively. Indeed, 3 and 4 have the same sequence of the 3- and 4-substituted Rha residues as would be expected from structures 1 and 2. It should be mentioned that the structure of the minor repeating unit 2 could be determined unambiguously from structure 4, taking into account the way by which 4 was obtained and the methylation analysis data of the polysaccharide.
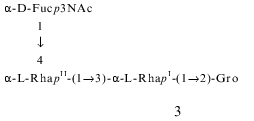
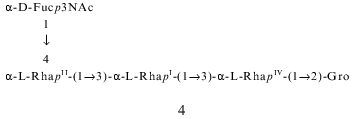
These data confirmed finally the structures of the major and minor repeating units of the O-specific polysaccharide of P. syringae pv. garcae ICMP 8047, which differ from each other in the position of substitution of one of the rhamnose residues, namely, of RhaIV. As mentioned above, this unique type of structural heterogeneity in the main chain has been previously found in O-specific polysaccharides of a number of other P. syringae strains with an L-rhamnan main chain and a residue of either Fuc3NAc [8, 10, 11, 20, 21] or GlcNAc ([8] and our unpublished data) as the lateral substituent. Of them, the O-specific polysaccharide of P. syringae pv. coriandricola GSPB 2028 (W-43) [11] is most similar to the polysaccharide studied in this work. Both polysaccharides have the same composition and the same sequence of the Rha residues in both major and minor repeating units, and they differ only in the site of attachment of the Fuc3NAc residue to RhaII (at position 2 or 4 in P. syringae pathovars coriandricola and garcae, respectively).
Previously, in studies of the O-specific polysaccharides of P. syringae pv. syringae IMV 8300 and NCPPB 281, which also belong to the same structural group, the major and minor repeating units have been demonstrated to enter into the same polysaccharide chain [20, 21]. This could be fulfilled making use of a different behavior of the major and minor repeating units towards Smith degradation: only the former was oxidized, whereas the latter was stable. Unfortunately, in P. syringae pv. garcae ICMP 8047, as in P. syringae pv. coriandricola GSPB 2028 (W-43), both major and minor repeating units were oxidized by periodate, and this approach could not be used to solve the problem. One can only speculate that biosynthesis of all O-specific polysaccharides of this group proceeds by the same, yet unknown, mechanism and that both repeating units occur in the same polysaccharide chain.
This work was supported by a grant from the Tampere University Hospital Medical Research Fund and grant INTAS-Ukraine 95-0142. Strain P. syringae pv. garcae ICMP 8047 was a kind gift of Professor K. Rudolph (Institute of Plant Pathology and Plant Defense, University of Göttingen, Germany).
REFERENCES
1.Young, J. M., Saddler, G. S., Takikawa, Y., DeBoer,
S. H., Vauterin, L., Gardan, L., Gvozdyak, R. I., and Stead, D. E.
(1996) Rev. Plant Pathol., 75, 721-763.
2.Otta, J. D., and English, H. (1971)
Phytopathology, 61, 443-452.
3.Pastushenko, L. T., and Simonovich, I. (1979)
Mikrobiol. Zh., 41, 330-338.
4.Saunier, M., Malandrin, L., and Samson, R. (1996)
Appl. Environ. Microbiol., 62, 2360-2374.
5.Ovod, V., Rudolph, K., and Krohn, K. (1997) in
Proc. 5th Int. Working Group on Pseudomonas syringae Pathovars and
Related Pathogens, Berlin, 1995 (Rudolph, K., Burr, T. J.,
Mansfield, J. W., Stead, D., Vivian, A., and von Kietzell, J., eds.)
Kluwer Academic Publishers, Dordrecht-Boston-London, pp. 526-531.
6.Zdorovenko, G. M., Gubanova, N. Y., Solyanik, L.
P., Knirel, Y. A., Yakovleva, L. M., and Zakharova, I. Y. (1991) in
Proc. 4th Int. Working Group on Pseudomonas syringae Pathovars,
Florence, Italy, 1991, pp. 391-401.
7.Ovod, V., Rudolph, K., Knirel, Y. A., and Krohn, K.
(1996) J. Bacteriol., 178, 6459-6465.
8.Knirel, Y. A., and Zdorovenko, G. M. (1997) in
Proc. 5th Int. Working Group on Pseudomonas syringae Pathovars and
Related Pathogens, Berlin, 1995 (Rudolph, K., Burr, T. J.,
Mansfield, J. W., Stead, D., Vivian, A., and von Kietzell, J., eds.)
Kluwer Academic Publishers, Dordrecht-Boston-London, pp. 475-480.
9.Ovod, V., Knirel, Y. A., and Krohn, K. (1997) in
Proc. 5th Int. Working Group on Pseudomonas syringae Pathovars and
Related Pathogens, Berlin, 1995 (Rudolph, K., Burr, T. J.,
Mansfield, J. W., Stead, D., Vivian, A., and von Kietzell, J., eds.)
Kluwer Academic Publishers, Dordrecht-Boston-London, pp. 532-537.
10.Knirel, Y. A., Ovod, V., Zdorovenko, G. M.,
Gvozdyak, R. I., and Krohn, K. (1998) Eur. J. Biochem.,
258, 657-661.
11.Knirel, Y. A., Ovod, V., Paramonov, N. A., and
Krohn, K. (1998) Eur. J. Biochem., 258, 716-721.
12.Sawardeker, J. S., Sloneker, J. H., and Jeanes,
A. (1965) Anal. Chem., 37, 1602-1603.
13.Leontein, K., Lindberg, B., and Lnngren, J.
(1978) Carbohydr. Res., 62, 359-362.
14.Shashkov, A. S., Senchenkova, S. N., Nazarenko,
E. L., Zubkov, V. A., Gorshkova, N. M., Knirel, Y. A., and Gorshkova,
R. P. (1997) Carbohydr. Res., 303, 333-338.
15.Ciucanu, I., and Kerek, F. (1984) Carbohydr.
Res., 131, 209-217.
16.Gross, M., Mayer, H., Widemann, C., and Rudolph,
K. (1988) Arch. Microbiol., 149, 372-376.
17.Jansson, P.-E., Kenne, L., and Widmalm, G. (1989)
Carbohydr. Res., 8, 169-191.
18.Lipkind, G. M., Shashkov, A. S., Knirel, Y. A.,
Vinogradov, E. V., and Kochetkov, N. K. (1988) Carbohydr. Res.,
175, 59-75.
19.Lipkind, G. M., Shashkov, A. S., Mamyan, S. S.,
and Kochetkov, N. K. (1988) Carbohydr. Res., 181,
1-12.
20.Knirel, Y. A., Zdorovenko, G. M., Shashkov, A.
S., Yakovleva, L. M., Gubanova, N. Y., and Gvozdyak, R. I. (1988)
Bioorg. Khim., 14, 172-179.
21.Knirel, Y. A., Zdorovenko, G. M., Shashkov, A.
S., Mamyan, S. S., Gubanova, N. Y., and Solyanik, L. P. (1988)
Bioorg. Khim., 14, 180-186.
22.Kochetkov, N. K., Vinogradov, E. V., Knirel, Y.
A., Shashkov, A. S., and Lipkind, G. M. (1992) Bioorg. Khim.,
18, 116-125.