The Carboxy-Terminal Domain Initiates Trimerization of Bacteriophage T4 Fibritin
A. V. Letarov1,2, Yu. Ya. Londer3, S. P. Boudko1, and V. V. Mesyanzhinov1*
1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 336-6022; E-mail: vvm@ibch.siobc.ras.ru2Ivanovsky Institute of Virology, Russian Academy of Medical Sciences, ul. Gamalei 16, Moscow, 123098 Russia
3Bach Institute of Biochemistry, Russian Academy of Sciences, Leninskii pr. 33, Moscow, 117071 Russia
* To whom correspondence should be addressed.
Received November 11, 1998; Revision received March 23, 1999
Bacteriophage T4 fibritin is a triple-stranded, parallel, segmented alpha-helical coiled-coil protein. Earlier we showed that the C-terminal globular domain (foldon) of fibritin is essential for correct trimerization and folding of the protein. We constructed the chimerical fusion protein W31 in which the fibritin foldon sequence is followed by the small globular non-alpha-helical protein gp31 of the T4 phage. We showed that the foldon is capable of trimerization in the absence of the coiled-coil part of fibritin. A deletion mutant of fibritin (NB1) with completely deleted foldon is unable to fold and trimerize correctly. An excess of this mutant protein did not influence the refolding of fibritin in vitro, and the chimerical protein inhibited this process efficiently. Our conclusion is that the trimerization of the foldon is the initial step of fibritin refolding and is followed by the formation of the coiled-coil structure.
KEY WORDS: bacteriophage T4, fibritin, folding, foldon, superhelical protein, coiled coil
Abbreviations: PMSF) phenylmethylsulfonyl fluoride; IPTG) isopropyl beta-D-thiogalactoside.
Fibritin of bacteriophage T4 is a fibrous superhelical protein which
consists of three identical polypeptide chains forming a parallel,
segmented, alpha-helical structure; super helical (coiled-coil)
parts are separated by extended loops [1]. Fibritin
is a convenient model for studying the mechanisms of folding and
oligomerization of the large class of alpha-helical fibrous
proteins, e.g., myosin, tropomyosin, and laminin [2].
Fibritin, encoded by gene wac (whisker antigen control), has a length of about 52 nm and is reported to be a structural protein which forms the collar and the whiskers of a phage particle. The whiskers work as molecular chaperons in the assembly of phage long tail fibers which recognize the surface receptors of the bacterial cell. Interacting with distal and proximal halves of the long tail fibers, the whiskers accelerate their assembly and attachment to the baseplate of the phage tail [3, 4].
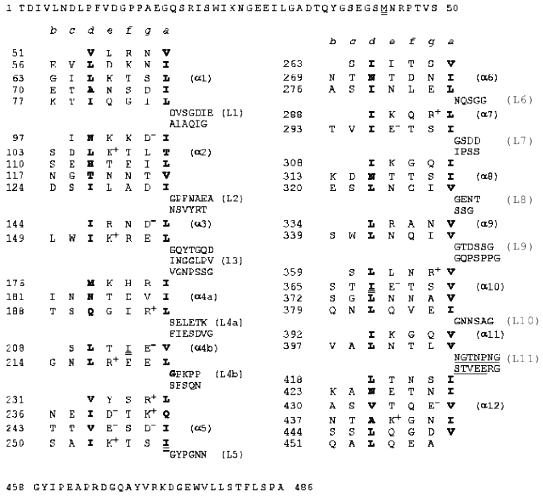
Heptad repeats of amino acid residues in the fibritin sequence are a characteristic feature of coiled-coil proteins (see Fig. 1); in the heptad pattern (a-b-c-d-e-f-g)n positions (a) and (d) are occupied by hydrophobic amino acid residues [5]. This suggests that fibritin is a superhelical protein [6, 7]. However, the sequence analysis showed that the superhelical parts are divided by sequences which resemble globular protein loops. Also, two small globular domains with no heptad repeats in their sequence flank the molecule. The N-terminal domain of fibritin is essential for the attachment of the protein to the phage particle, while the C-terminal domain is necessary for correct folding and assembly of the homotrimer [1]. Some mutants with point mutations in the C-terminal domain of fibritin (called the foldon) are not able to form trimeric proteins; a number of such mutants produce insoluble aggregates (inclusion bodies), which often indicate misfolding [7].
Deletion mutants of fibritin with the intact C-terminal domain and consequently shortened superhelical part of the molecule keep the ability for correct assembly and formation of trimeric proteins [1, 7]. For two such N-truncated fibritins E and M (119 and 78 C-terminal amino acid residues, respectively), the three-dimensional structure was solved recently to 2.3 and 1.8 Å resolution [8, 9]. The X-ray analysis confirmed fibritin to actually be a superhelical segmented protein with the C-terminal domain having a stable beta-propeller structure. Analysis of loop insertion in fibritin E allowed us to predict 12 coiled-coil segments in the full length protein (Fig. 1).
Fibritin and deletion mutants lacking the N-terminal part are able to renature rapidly to the native structure after heat denaturation in vitro [1, 4]. If we subject a mixture of different-length fibritin mutants to the renaturation procedure we get heterotrimers. Fibritin is resistant to 2% SDS at room temperature, so unheated protein samples give rise to a band on SDS-PAGE corresponding to the higher molecular weight of the trimeric structure.
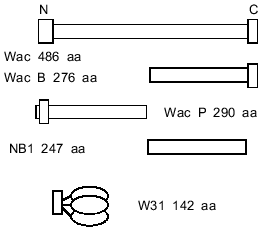
It has remained unclear whether the trimerization of the C-terminal part of fibritin is the first step of the folding of the protein, or whether the alpha-helical parts are capable of self-trimerization and formation of superhelical parts, the role of the foldon being to stabilize the final structure. In the first case C-terminal domain trimerization should not depend on the alpha-helical segments. It is difficult to test this idea because of the small molecular weight of the foldon (~3 kD). Thus, we engineered a chimera in which the foldon of fibritin was attached to gene product 31 of phage T4 (see Fig. 2). SDS-PAGE analysis showed that the protein formed SDS-resistant oligomers due to the foldon sequence.
Furthermore, a shortened fibritin derivative (NB1, 247 amino acid residues) which does not contain the C-terminal domain (Fig. 2) is soluble when expressed in E. coli, and unheated samples on SDS-PAGE show only the band related to the monomeric form. The conformation of the NB1 polypeptide chain apparently conforms to a monomeric folding intermediate of fibritin. In renaturation experiments the NB1 mutant did not form trimers and did not inhibit the trimerization of fibritin containing the C-terminal domain. These data indicate that the coiled coil-forming fragments of the polypeptide chain do not take part in initiation of the trimerization process before the interaction of the C-terminal domains.
MATERIALS AND METHODS
E. coli strains and plasmids. The strain Top 10 (Invitrogen, USA) was used for the selection of recombinant clones and plasmid DNA production. Protein expression was done in the strain JM 109 (DE3) (Promega, USA) containing a chromosomal copy of the T7 RNA polymerase gene under lac UV5 control, and expression was induced by the addition of isopropyl beta-D-thiogalactoside (IPTG). All plasmids were created on the basis of pET19b(+) (Novagen, USA) allowing transcription of cloned DNA sequences from T7 RNA polymerase promoter and containing the ribosome-binding site for effective translation in the cell.
Polymerase chain reaction (PCR) was done in the Tertsik-MS amplifier (DNA-Technology, Russia) using Taq-polymerase as described in [10]. In both cases the cycle parameters were: 94°C 0.5 min, 48°C 1 min, 72°C 1 min; number of cycles, 30.
Gene cloning was done using the common techniques described in [11].
SDS-PAGE analysis was carried out as described by Laemmli [12].
Engineering of a deletion mutant of fibritin B lacking the C-terminal domain (NB1). A fragment of the coiled-coil region was amplified between primers I and II (table) on the template of phage T4 DNA and cloned into sites Nco I and BamH I of the plasmid pET19b(+). The end of the coiled-coil region determined from the X-ray structure is the last residue of the alpha-helix, Gly-456 [9].
Primers used for polymerase chain reaction
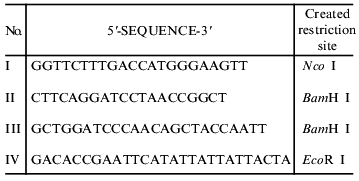
Engineering of chimerical protein W31. A DNA fragment containing the sequence of gene 31 was obtained by the PCR technique between primers III and IV (table) on the template of T4 genomic DNA. For cloning of this fragment, we used the vector pWAC P previously produced in our laboratory [13]. The plasmid pWAC P expresses fibritin P with the foldon transposed to the N-terminus. Fibritin P in turn was made on the basis of a vector with fibritin sequence lacking the N-terminal globular domain and part of the coiled-coil segments (Fig. 2). The fragment BamH I--EcoR I coding the coiled-coil part of fibritin P was cut out from the plasmid pWAC P. The linear vector was ligated with a DNA fragment coding gp31 previously restricted with the same enzymes. As a result, the reading frame was preserved and the plasmid coded the fusion protein (named W31): the foldon (a sequence of 36 amino acids, f-MQALQEAGYIPEAPRDGQAYVRKDGEWVLLSTFLSPA) and gp31 (109 amino acid residues).
Preparation of extracts from cells expressing recombinant proteins. The cell culture (strain JM 109 (DE3) carrying the proper plasmid) was grown at 37°C in 200 ml of 2x TY medium [11] until the density reached 0.6 OD (600 nm). The expression of the protein was induced by addition of IPTG to 1 mM final concentration. Incubation was continued for 12 h at 25°C with vigorous aeration. The cells were collected by centrifugation at 4,000 rpm at 4°C for 15 min and then resuspended in 10 ml 50 mM Tris-HCl (pH 7.4). The cells were lysed by ultrasonic disintegration for 40 sec on ice using a UD-20 unit (Techpan, Poland). Cell debris was pelleted by centrifugation at 15,000 rpm for 10 min at 4°C. Saturated ammonium sulfate was continuously added (dropwise with vigorous mixing) to the supernatant until the beginning of precipitation. Then the solution was incubated for 30 min at 4°C. The precipitate was centrifuged in the same way. The pellet was dissolved in 3 ml of 10 mM sodium phosphate buffer (pH 7.4) and centrifuged at 10,000g to remove insoluble particles. The precipitation by ammonium sulfate was repeated. In the final stage PMSF was added to 0.5 mM concentration. The resulting extracts contained more than 70-80% of the target protein and were used in experiments without further purification.
Purification of fibritin B. The protein was purified as described above. In addition, after salt precipitation the pellet was dissolved in 3 ml of 50 mM Tris-HCl (pH 7.6), and the solution was applied to a hydroxyapatite column (BioRad, USA) equilibrated with 10 mM sodium phosphate buffer (pH 7.6). The protein was eluted using the same buffer.
Trypsinolysis procedure. The protein was treated with trypsin in 50 mM Tris-HCl buffer (pH 7.4) containing 1 mM CaCl2. All protein samples (not containing PMSF) were at 1 mg/ml concentration. Final concentrations of trypsin were 0.05, 0.025, 0.0125, and 0.006 mg/ml. The samples were incubated for 30 min at 37°C. Trypsinolysis was stopped by addition of PMSF to 1 mM concentration. Then Laemmli buffer for SDS-PAGE was added.
RESULTS
Properties of fibritin NB1, a deletion mutant lacking the C-terminal domain. The protein NB1 is a variant of fibritin B with the C-terminal domain deleted (Figs. 1 and 2). It is produced in E. coli up to ~30% of total cell protein. After ultrasonic disintegration of the expressing culture and subsequent centrifugation this protein is found in the supernatant. It can be precipitated by (NH4)2SO4 at 20% saturation. After the precipitation the protein easily dissolves in 50 mM Tris-HCl buffer (pH 7.6), but it partially aggregates when stored at 4°C for a long time.
Fig. 1. Heptad repeats in the amino acid sequence of fibritin. Positions (a) and (d) are occupied by hydrophobic residues (shown in bold). Heptad repeats are separated by fragments corresponding to loops.
The electrophoretic mobility of the protein in SDS-PAGE corresponds to expected molecular weight of 27 kD. In the gel pattern of unheated samples no bands corresponding to the trimeric form were observed (Fig. 3).Fig. 2. Schematic structures of proteins used in this work.
Fibritin NB1 was found to be highly sensitive to tryptic digestion. At trypsin concentrations above 0.0125 mg/ml (protein/enzyme ratio ~100:1) the protein was fully hydrolyzed in 1 h at 37°C. Fibritin B is resistant to digestion under the same conditions (Fig. 4, lanes 1-5).Fig. 3. SDS-PAGE analysis of partially purified extracts containing chimerical protein W31 and fibritin NB1: 1) W31 unheated sample; 2) W31 heated sample; 3) NB1 unheated sample; 4) NB1 heated sample. M, protein markers. Molecular masses of protein markers (in kD) are indicated on the left.

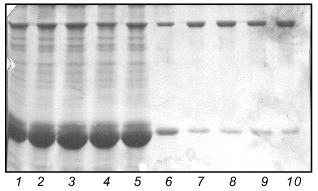
We carried out a renaturation of fibritin B with NB1 protein in excess after heat denaturation. The proteins were mixed in 30 µl volume in mass ratio ~1:5 and denatured by heating at 100°C for 3 min. In a control experiment NB1-containing extract was replaced by the same volume of buffer (fibritin NB1 does not form SDS resistant oligomers after annealing, see Fig. 4, lanes 6 and 7). All tubes were incubated at room temperature for 30 min, then Laemmli buffer was added and the samples were applied to an SDS gel (Fig. 5). It is highly probable that fibritin NB1 has an intermediate conformation of fibritin. To preserve this structure, we did an additional experiment in which a hot solution of denatured fibritin B was added to an ice-cold fibritin NB1 solution (data not shown).Fig. 4. Properties of fibritin B, fibritin NB1, and gp31: 1) fibritin B before trypsin treatment; 2) fibritin B after trypsin treatment (protein/enzyme ratio 20:1); 4 and 5) sample of NB1 protein before and after trypsin treatment, respectively (conditions same as for fibritin B); 6) unheated sample of NB1; 7) the same sample subjected to an annealing procedure (100°C for 3 min, 20°C for 10 min) before Laemmli buffer was added; 8 and 9) heated and unheated samples of gp31 protein.
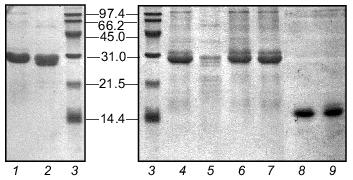
Under various conditions no significant inhibition of fibritin trimerization was observed. This result indicates that no interaction of the coiled coil-forming segments of the fibritin B molecule occurs during the initial step of the in vitro renaturation process. This means that the first step of trimerization and subsequent folding of fibritin should be an interaction of C-terminal sequences with the formation of a stable structure.Fig. 5. Renaturation of fibritin B in the presence of an excess of NB1. Lanes: 1-5) 1, 2, 3, 5, and 10 min after heating; 6-10) analogous samples containing fibritin B only (control).
Properties of the chimerical protein W31. To demonstrate that the C-terminal domain of fibritin is able to form trimers without the coiled-coil part of the molecule, we engineered the chimerical protein W31 (Fig. 2). This protein is expressed with the same efficiency as fibritin NB1 and is soluble. If the protein is analyzed by SDS-PAGE immediately after cell disintegration, there is no oligomeric band in the gel pattern of unheated samples (data not shown). However, after 24-h storage at 4°C (in the presence of 1% (v/v) mercaptoethanol and 1 mM PMSF) up to 80% of the total protein was found in oligomeric form (Fig. 3). It should be noted that protein W31 was constructed on the basis of gp31 of bacteriophage T4. The gp31 protein is a heptameric co-chaperonin [14]. However, the heptamers of gp31 are sensitive to SDS, and unheated samples of this protein do not form a slowly migrating band during SDS-PAGE (Fig. 4, lanes 8 and 9).
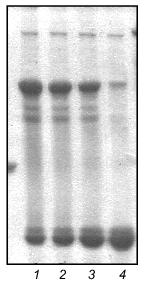
The molecular weight of W31 oligomers estimated from the SDS-PAGE mobility is 50 kD. This is close to the expected electrophoretic mobility (the mobility of the monomer corresponds to 17 kD molecular weight). In sample buffer containing 2% SDS oligomers dissociate completely within 2 h (Fig. 6). Fibritin B trimers are stable under such conditions for a long time at room temperature.
The slow rate of oligomerization indicates that protein W31 is not able to reassemble rapidly after heat denaturation. Nevertheless, after annealing (heating at 100°C for 3 min with subsequent incubation at 4°C for 15 min) about half of the total amount of the protein was found in oligomeric form. The presence of 1% mercaptoethanol (v/v) does not influence the trimerization efficiency, but 1% SDS completely inhibits this process (Fig. 7).Fig. 6. Dissociation of the chimerical protein W31 visualized by SDS-PAGE. Lanes: 1-4) 1, 30, 60, and 120 min incubation with 2% SDS at room temperature.
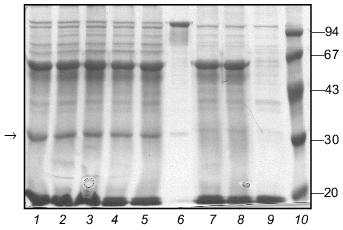
The ability of the W31 protein to reassemble after being heated allowed us to use it in renaturation experiments with fibritin B as described above for fibritin NB1. In contrast to fibritin NB1, an excess (3-5-fold) of protein W31 dramatically decreased the yield of the trimeric form of fibritin B (Fig. 8). Moreover, a significant amount of fibritin B was found in the monomeric form after co-annealing. This result can be explained by the formation of some SDS-sensitive heterooligomers. In the SDS-PAGE patterns of co-annealed samples there are several extra bands between the bands of fibritin B and W31 trimers. These bands obviously correspond to heterooligomeric forms that differ in composition and conformation. The existence of such a large number of heterooligomers with differing electrophoretic mobilities can be explained by steric reasons. Steric problems are likely to arise from the globular conformation of W31 subunits and the effect of the opposite foldon location (at the C-terminus of fibritin B and at the N-terminus of protein W31) (Fig. 2).Fig. 7. Renaturation of the chimerical protein W31: 1) heated sample; 2) unheated sample; 3) renaturation in the presence of 1% mercaptoethanol; 4) renaturation in the presence of 1% SDS; 5) renaturation in 50 mM Tris-HCl buffer (pH 7.6) with no additives.
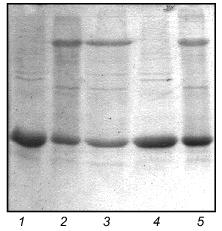
Fig. 8. Renaturation of fibritin B in the presence of excess protein W31. Lanes: 1-5) renaturation times 1, 2, 3, 5, and 10 min, respectively; 6) renaturation of fibritin B for 10 min; 7) renaturation of chimeric protein W31 without fibritin B for 10 min; 8 and 9) unheated and heated W31 samples, respectively. The arrow marks the band of the monomer of fibritin B. The band in lanes 1-5 and 7-9 located slightly lower than the band of the fibritin B trimer is apparently due to an impurity in the W31 preparation. The molecular masses of protein markers are indicated on the right (kD).
DISCUSSION
Significant differences in features of fibritin B and fibritin NB1 suggest that the protein with deleted C-terminal domain is not capable of trimerization and correct folding. Inability to form a compact structure leads to the high sensitivity of fibritin NB1 to tryptic digestion. In contrast, fibritin B is resistant to trypsinolysis despite the numerous potential sites of digestion in its sequence.
The ability of protein NB1 to aggregate indicates that the hydrophobic residues normally forming the internal core of fibritin are partially exposed to the outside. Thus, physical state of the fibritin NB1 polypeptide chain could be the same as in a putative monomeric intermediate of folding. This intermediate must occur in vivo if trimerization is initiated at the C-terminus. The correct assembly of various N-truncated fibritin mutants [7] practically excludes the possibility of co-translational trimerization and folding during the synthesis of the fibritin polypeptide chains.
In the samples of fibritin B and fibritin NB1 denatured by heating the state of the coiled coil-forming fragments of polypeptide chains should be identical (random coil). Therefore, if some interactions of this fragments occur during the initial step of trimerization (immediately after the temperature is changed), NB1 molecules should take part in these interactions with the same efficiency as fibritin B molecules.
According to this model, an excess of NB1 protein should significantly inhibit the refolding of fibritin B. However, no inhibition was observed in our experiments. This result indicates that the coiled coil-forming sequences do not initiate the trimerization. This means that the initial step of fibritin folding must be the trimerization of C-terminal domains. In this case a polypeptide consisting of the C-terminal domain only should be able to form trimers in the absence of the coiled coil-forming region. The low molecular weight (~3 kD) of such a peptide complicates work with it. Therefore, we constructed the chimerical protein W31 consisting of the fibritin foldon sequence and the “load” sequence of a small globular protein which contains no coiled-coil structure. For this construction we chose protein gp31 of phage T4 containing 109 amino acid residues. According to X-ray data [14], the N-terminal domain of gp31 is exposed on the surface of the globule. This circumstance determined our choice of the N-terminal position of the foldon (as opposed to the C-terminal position in fibritin). The ability of this protein to form SDS-resistant oligomers is direct evidence for our model suggesting that fibritin folding initiation is begun from the C-terminal domain.
As shown earlier [1], the renaturation curve of fibritin follows second-order kinetics. This means that trimerization follows the formation of a dimeric intermediate. Since a dimeric state of fibritin has never been directly observed, we supposed that dimers are very unstable and rapidly dissociate to monomers or associate with monomers to form trimers. In contrast, the structure of fibritin trimer is very stable. Thus, the equilibrium F2 + F . F3 should be shifted strongly to the right. However, in the case of the W31 protein the reverse reaction evidently takes place more rapidly. Because SDS inhibits the forward reaction, it leads to gradual dissociation of trimers in SDS-PAGE sample buffer. Therefore, the gel patterns of unheated samples of W31 always contain the monomeric band.
On the base of all the data presented, we conclude that the folding of fibritin B (at least in vitro) is initiated by C-terminal domain trimerization and is followed by the formation of coiled coil (probably in zipper fashion). Thus, the C-terminal domain is a true foldon, i.e., an independently folding unit nucleating the folding of the whole molecule. Using genetic techniques or peptide synthesis it can be isolated as a separate molecule with preserved ability to form a trimeric beta-propeller structure.
There are some data [15-17] suggesting that C-terminal fragments of other fibrous superhelical proteins (myosin, paramyosin, collagen, and others) are also important for assembly of their molecules. The C-terminal position of a fragment which initiates folding is probably a common feature for all (or many) fibrous proteins.
Our work was supported by the grants from the program “Universities of Russia” (2608-415/a8), the Howard Hughes Medical Institute (No. 75197-544204), and the Russian Foundation for Basic Research (No. 99-04-48430).
REFERENCES
1.Efimov, V. P., Nepluev, I. V., Sobolev, B. N.,
Zurabishvili, T. G., Shulthess, T., Lustig, A., Engel, J., Haener, M.,
Aebi, U., Venyaminov, S. Y., Potekhin, S. A., and Mesyanzhinov, V. V.
(1994) J. Mol. Biol., 242, 470-486.
2.Mesyanzhinov, V. V. (1997) Mol. Biol.
(Moscow), 31, 389-397.
3.Wood, W. B., and Eiserling, F. A. (1994) in
Molecular Biology of Bacteriophage T4 (Karam, J. D., ed.)
American Society for Microbiology, Washington D.C., pp. 282-291.
4.Gorbatova, L. P., Selivanova, G. N., Serysheva, I.
I., Turkina, E. V., Belous, E. A., Mesyanzhinov, V. V., Poglazov, B.
F., and Selivanov, N. A. (1979) Dokl. Akad. Nauk SSSR,
247, 738.
5.McLachlan, A. D., and Stewart, M. (1975) J. Mol.
Biol., 98, 293-304.
6.Sobolev, B. N. (1993) Modelling of Structure of
Bacteriophage T4 Fibrillar Proteins: Candidate's dissertation [in
Russian], Ivanovsky Institute of Virology, Moscow.
7.Sobolev, B. N., Shneider, M. M., Marusich, E. I.,
and Mesyanzhinov, V. V. (1995) in Evolutionary Biochemistry and
Related Areas of Physiocochemical Biology (Poglasov, B. F., et al.,
eds.) Bach Institute of Biochemistry and ANKO, Moscow, pp. 351-377.
8.Strelkov, S. V., Tao, Y., Shneider, M. M.,
Mesyanzhinov, V. V., and Rossmann, M. G. (1998) Acta Crystallogr. D.
Biol. Crystallogr., 54, 805-816.
9.Tao, Y., Strelkov, S. V., Mesyanzhinov, V. V., and
Rossmann, M. G. (1997) Structure, 5, 789-798.
10.PCR Protocols (1990) (Innis, M. A., Gelfand, D.
H., and Sninsky, J. J., eds.) Academic Press, London.
11.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning, Cold Spring Harbor Press.
12.Laemmli, U. K. (1970) Nature, 227,
680-685.
13.Londer, Yu. Ya., and Mesyanzhinov, V. V. (1998)
in Int. Symp. Protein Structure, Stability and Folding. Book of
Program and Abstracts, ONTI PNC RAN, Pushchino, p. 174.
14.Hunt, J. F., van der Vies, S. M., Henry, L., and
Deisenhofer, J. (1997) Cell, 90, 361-371.
15.Cohen, C. (1998) J. Struct. Biol.,
122, 3-9.
16.Parry, D. A., and North, A. C. (1998) J.
Struct. Biol., 122, 67-75.
17.Sturmann, N., Heins, S., and Aebi, U. (1998)
J. Struct. Biol., 122, 42-66.