Use of Stable Analogs of Myosin ATPase Intermediates for Kinetic Studies of the “Weak” Binding of Myosin Heads to F-Actin
E. V. Rostkova1, L. N. Moiseeva1, M. V. Teplova1, O. P. Nikolaeva2, and D. I. Levitsky1,2*
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, Moscow, 117071 Russia; fax: (095) 954-2732; E-mail: inbio@glas.apc.org2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181
* To whom correspondence should be addressed.
Received October 20, 1998; Revision received December 8, 1998
It is known that ternary complexes of myosin subfragment 1 (S1) with ADP and the Pi analogs beryllium fluoride (BeFx) and aluminum fluoride (AlF4-) are stable analogs of the myosin ATPase intermediates M*·ATP and M**·ADP·Pi, respectively. Using kinetic approaches, we compared the rate of formation of the complexes S1·ADP·BeFx and S1·ADP·AlF4- in the absence and in the presence of F-actin, as well as of the interaction of these complexes with F-actin. We show that in the absence of F-actin the formation of S1·ADP·BeFx occurs much faster (3-4 min) than that of S1·ADP·AlF4- (hours). The formation of these complexes in the presence of F-actin led to dissociation of S1 from F-actin, this process being monitored by a decrease in light scattering. The light scattering decrease of the acto-S1 complex occurred much faster after addition of BeFx (during 1 min) than after addition of AlF4- (more than 20 min). In both cases the light scattering of the acto-S1 complex decreased by 40-50%, but it remained much higher than that of F-actin measured in the absence of S1. The interaction of the S1·ADP·BeFx and S1·ADP·AlF4- complexes with F-actin was studied by the stopped-flow technique with high time resolution (no more than 0.6 sec after mixing of S1 with F-actin). We found that the binding of S1·ADP·BeFx or S1·ADP·AlF4- to F-actin is accompanied by a fast increase in light scattering, but it does not affect the fluorescence of a pyrene label specifically attached to F-actin. We conclude from these data that within this time range a “weak” binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin occurs without the subsequent transition of the “weak” binding state to the “strong” binding state. Comparison of the light scattering kinetic curves shows that S1·ADP·AlF4- binds to F-actin faster than S1·ADP·BeFx does: the second-order rate constants for the “weak” binding to F-actin are (62.8 ± 1.8)·106 M-1·sec-1 in the case of S1·ADP·AlF4- and (22.6 ± 0.4)·106 M-1·sec-1 in the case of S1·ADP·BeFx. We conclude that the stable ternary complexes S1·ADP·BeFx and S1·ADP·AlF4- can be successfully used for kinetic studies of the “weak” binding of the myosin heads to F-actin.
KEY WORDS: myosin subfragment 1, F-actin, “weak” binding of myosin to actin, kinetics, skeletal muscle
Abbreviations: S1) myosin subfragment 1; BeFx, AlF4-) beryllium fluoride and aluminum fluoride anions, respectively.
It is well known that muscle contraction and many other types of
biological motility are based on a cyclic interaction of myosin heads
with actin, this being accompanied by ATP hydrolysis in the heads.
According to recent ideas, the molecular mechanism of actomyosin-driven
biological motility is based on significant conformational changes
occurring in the myosin head during the ATPase reaction [1, 2]. This reaction can be
represented by the following simplified scheme:
M + ATP <--> M*·ATP <--> M**·ADP·Pi <--> M*·ADP <--> M + ADP,
where M is the myosin head and each asterisk reflects a conformational change registered by an increase in tryptophan fluorescence [3-5]. In the absence of ATP the myosin heads are tightly bound to actin, this being the so-called “strong”-state binding of myosin to actin. However, in the presence of ATP, the formation of the intermediate M*·ATP dramatically decreases the affinity of myosin to actin and leads, under physiological conditions, to dissociation of the myosin heads from actin filaments. The complex M**·ADP·Pi formed by the hydrolysis of ATP can bind to actin again, but this low-affinity binding is quite different from that observed in the absence of nucleotides; this is the so-called “weak”-state binding of myosin to actin. Subsequent actin-induced fast dissociation of the M**·ADP·Pi complex returns the actin-bound myosin heads to their initial conformation characteristic for the “strong” actin-binding state with the result that the heads perform mechanical work by moving actin filaments along myosin filaments. Thus, the functioning of the myosin head as a “molecular motor” is explained by significant structural changes which occur in the head due to formation of the intermediates M*·ATP and M**·ADP·Pi during the ATPase reaction, changing the character of the actin--myosin interaction [1, 2].
To study the conformational changes occurring in the myosin head upon formation of the intermediates M*·ATP and M**·ADP·Pi during the ATPase reaction, stable analogs of these intermediates are intensively used, i.e., ternary complexes of the isolated myosin head (myosin subfragment 1, S1) with ADP and Pi analogs such as orthovanadate (Vi) [6, 7], beryllium fluoride (BeFx) [7-11], aluminum fluoride (AlF4-) [7, 11], or scandium fluoride (ScFx) [12] anions. In particular, it has been shown in our laboratory using differential scanning calorimetry that the formation of the complexes S1·ADP·Vi, S1·ADP·BeFx, and S1·ADP·AlF4- is accompanied by global conformational changes in the S1 molecule which are reflected in a significant change in the character of S1 thermal denaturation (in particular, in a significant increase in thermal stability of the protein) [13-16]. Until recently it was thought that all these complexes, including S1·ADP·BeFx, correspond to the intermediate M**·ADP·Pi [7-9, 14]. However, recent data from structural studies performed by various methods (EPR spectroscopy [17], low-angle neutron scattering in protein solution [18], differential scanning calorimetry [19], and X-ray analysis of crystals of the isolated motor domain of the head of Dictyostelium discoideum myosin [20]) indicate that the S1·ADP·BeFx complex is different from all other stable ternary complexes of S1 with ADP and Pi analogs; it most likely corresponds to the M*·ATP intermediate state. Thus, comparison of the properties of the S1·ADP·BeFx complex with those of other S1 ternary complexes with ADP and Pi analogs reveals similarities and differences between the myosin ATPase intermediates M*·ATP and M**·ADP·Pi. Among stable analogs of the intermediate state M**·ADP·Pi, the complex S1·ADP·AlF4- is the most suitable for such comparison, since the use of optical methods for studies of the complex S1·ADP·Vi is strongly limited because of the high optical activity of vanadate.
Using kinetic approaches, in the present work we compared the rate of formation of the complexes S1·ADP·BeFx and S1·ADP·AlF4- in the absence and in the presence of F-actin. Moreover, using the methods of transient kinetics, we studied the “weak” binding of these complexes to F-actin and revealed a clear difference between the complexes S1·ADP·BeFx and S1·ADP·AlF4- in the rate of their binding to actin filaments.
MATERIALS AND METHODS
Actin and myosin were prepared from rabbit skeletal muscles. Actin was purified by two cycles of polymerization--depolymerization. One hour before experiments G-actin was polymerized by addition of MgCl2 to final concentration 5 mM and used as F-actin. S1 was obtained by digestion of myosin filaments with alpha-chymotrypsin in the presence of 1 mM EDTA [21]. Concentrations of actin and S1 were determined spectrophotometrically using E1%280 nm = 7.5 cm-1 for S1 and E1%290 nm = 6.3 cm-1 for G-actin. Molar concentrations of S1 and G-actin were calculated using molecular masses of 115 and 42 kD for S1 and actin, respectively. Specific modification of actin at Cys-374 by attachment of the fluorescent pyrene label was performed by incubation of F-actin (1 mg/ml) at room temperature in the dark for 16 h with a 1.5-fold molar excess of N-(1-pyrenyl)iodoacetamide (Molecular Probes, USA) [22]; unbound label was removed by a following cycle of depolymerization--polymerization of actin.
The S1·ADP·BeFx and S1·ADP·AlF4- complexes were obtained by incubation (30-60 min, room temperature) of S1 (1 mg/ml) with 0.2 mM ADP, 5 mM NaF, and 0.2 mM BeCl2 (or AlCl3) in medium containing 30 mM Hepes-KOH (pH 7.3) and 1 mM MgCl2. The formation of the complexes was monitored by the inhibition of the K+-EDTA-ATPase activity of S1. The K+-EDTA-ATPase activity was practically absent (<1%) in the S1·ADP·BeFx complex, and it did not exceed 5-7% of the initial value for the S1·ADP·AlF4- complex. The time course of the formation of the complexes was monitored by measuring an increase in the intrinsic tryptophan fluorescence of S1 using a Hitachi F-3000 spectrofluorimeter (Japan) at 20°C with excitation at 295 nm and emission at 336 nm. In this case the concentration of S1 was 0.1 mg/ml (0.87 µM).
Dissociation of the complex of S1 with F-actin was induced by addition of 0.2 mM BeCl2 or 0.2 mM AlCl3 in the presence of 5 mM NaF and 0.2 mM ADP and was monitored by measuring changes in light scattering intensity at 340 nm either using the Hitachi F-3000 spectrofluorimeter or a D-110 stopped-flow spectrophotometer (Durrum, USA) interfaced with a personal computer for automatic data collection. Concentrations of F-actin and S1 were varied within the ranges of 1-2 and 0.5-4 µM, respectively.
Kinetics of the binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin was studied by stopped-flow experiments in the millisecond time range by measuring either an increase in light scattering on the D-110 stopped-flow spectrophotometer or the fluorescence of the pyrene label attached to F-actin on an SF-61MX stopped-flow spectrofluorimeter (Hi-Tech Scientific, UK) with excitation at 365 nm and emission at 390 nm. All stopped-flow experiments were carried out at 20°C. The data were analyzed and plotted using the software package Origin 3.73 (MicroCal Inc., USA).
RESULTS AND DISCUSSION
Kinetics of formation of the S1·ADP·BeFx and S1·ADP·AlF4- complexes. Before beginning experiments on the interaction of the S1·ADP·BeFx and S1·ADP·AlF4- complexes with F-actin, it was important to estimate the rate and completeness of the formation of these complexes. It is known from the literature that the formation of the ternary complexes S1·ADP·BeFx and S1·ADP·AlF4- is accompanied by inhibition of the K+-EDTA-ATPase of S1 and by an increase in the intrinsic tryptophan fluorescence of S1 [7]. Figure 1 shows the time course of the increase in the tryptophan fluorescence intensity of S1 after addition of 0.2 mM BeCl2 or AlCl3 to S1 in the presence of 0.2 mM ADP and 5 mM NaF. It is seen that the formation of the S1·ADP·BeFx complex occurs very rapidly, during 3-4 min (curve 1 in Fig. 1), this being in good agreement with data in the literature [7]. Thirty minutes after addition of BeCl2, no residual K+-EDTA-ATPase activity of the S1 preparation could be measured (the residual activity did not exceed 1% of the initial value); this is indicative of complete formation of the S1·ADP·BeFx complex. In contrast, under the same conditions, the formation of the S1·ADP·AlF4- complex proceeded much more slowly (curve 2 in Fig. 1). This fact was taken into account in subsequent experiments; for complete formation of the S1·ADP·AlF4- complex the incubation at 20°C was extended to 1-2 h with a subsequent 24 h incubation at 4°C prior to experiments. However, even after such treatment the K+-EDTA-ATPase activity of S1 in the presence of ADP, NaF, and AlCl3 was 5-7% of the activity measured in the absence of AlCl3. Thus, the extent of the formation of the S1·ADP·AlF4- complex was about 95% in our experiments.
Another approach for estimating the rate of formation of the S1·ADP·BeFx and S1·ADP·AlF4- complexes is to investigate the dissociation of the actomyosin complex upon addition of BeFx or AlF4- in the presence of ADP. The formation of the S1·ADP·BeFx and S1·ADP·AlF4- complexes in the active sites of S1 molecules attached to F-actin in the presence of ADP (“strong” binding state) leads to dissociation of S1 from F-actin, which can be monitored by a decrease in light scattering. This approach described earlier for studies of the interaction of BeFx with an acto-S1·ADP complex, and it has been shown that the decrease in light scattering proceeds rapidly, within 30 sec, and a transient kinetic method (e.g., the stopped-flow method) is required to record this process [9]. A very similar result was obtained in our stopped-flow experiments on the dissociation of the acto-S1·ADP complex after its mixing with BeFx (Fig. 2a). The experimental kinetic curve is biphasic, and thus it is fitted to two exponentials: a fast decrease (the first 8 sec of the reaction) followed by a slow decrease of light scattering (Fig. 2a). The observed rate constants are 0.63 ± 0.03 and 0.05 ± 0.001 sec-1 for the fast and slow phases of the reaction, respectively. In contrast, upon addition of AlF4- the dissociation of the acto-S1·ADP complex proceeds much more slowly, and thus its recording does not require the stopped-flow method (curve 1, Fig. 2b). In this case the experimental kinetic curve can be fitted to a single exponential; the observed rate constant of the reaction is 0.003 ± 0.0002 sec-1.Fig. 1. Time courses of the increase in intensity of the S1 tryptophan fluorescence observed on formation of the ternary complexes S1·ADP·BeFx (1) and S1·ADP·AlF4- (2). The reaction was initiated by addition of 0.2 mM BeCl2 (1) or 0.2 mM AlCl3 (2) to 0.87 µM S1 in medium containing 30 mM Hepes-KOH (pH 7.3), 1 mM MgCl2, 0.2 mM ADP, and 5 mM NaF. Excitation and emission wavelengths were 295 and 336 nm, respectively. I0, fluorescence intensity of the S1·ADP complex before addition of BeCl2 or AlCl3.
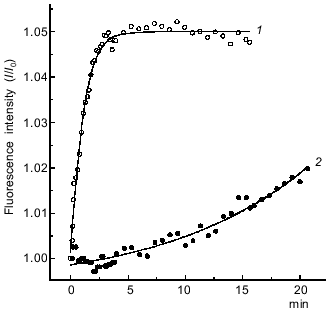
Thus, both in the presence and in the absence of F-actin the formation of the S1·ADP·BeFx complex proceeds much faster than that of the S1·ADP·AlF4- complex.Fig. 2. Kinetics of light scattering changes observed on addition of BeFx or AlF4- to the complex of S1 with F-actin in the presence of ADP. a) Stopped-flow measurement. One syringe contained BeFx (BeCl2 + NaF), and the other contained F-actin--S1·ADP complex. Final concentrations of components: 2 µM S1, 1 µM F-actin, 0.2 mM ADP, 0.2 mM BeCl2, 5 mM NaF, 1 mM MgCl2, 30 mM Hepes-KOH (pH 7.3). b) Steady-state measurements after addition of 0.2 mM AlCl3 (1) or 0.2 mM BeCl2 (2) to the complex of S1 (2 µM) with F-actin (1 µM) in the presence of 0.2 mM ADP and 5 mM NaF. Other conditions were the same as in Fig. 1. Curve 3 was observed under the same conditions after addition of AlF4- (or BeFx) to F-actin in the absence of S1. Light scattering was monitored at 340 nm. All kinetic curves were normalized to the initial light scattering of the acto-S1·ADP complex (I0). The smooth solid lines are computer fits consisting of two exponentials.
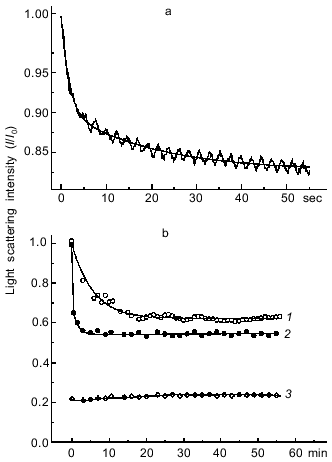
It is important to note that during steady-state measurements the level to which the light scattering intensity of the complex of S1 with F-actin decreased after addition of AlF4- in the presence of ADP was almost half of the initial value, but nevertheless it was much higher than the light scattering intensity of F-actin measured under the same conditions but in the absence of S1 (curve 3, Fig. 2b). Under these conditions the light scattering of S1 solution is negligible. We also observed a similar effect on dissociation of the complex of S1 with F-actin after addition of BeFx, although in this case the time course of the light scattering decrease could not be recorded under steady-state conditions because of high rate of the process (curve 2, Fig. 2b). These data suggest that after the light scattering decrease induced by addition of BeFx or AlF4-, the system comes to some equilibrium state at which some fraction of the S1 molecules remains bound to F-actin. We suggest that such an equilibrium is determined by several simultaneous processes: 1) dissociation of the actomyosin complex, this being accompanied by the decrease in light scattering; 2) “weak” binding to F-actin of newly formed ternary complexes S1·ADP·BeFx and S1·ADP·AlF4-, this probably being reflected in an increase in light scattering; 3) dissociation of the S1·ADP·BeFx and S1·ADP·AlF4- complexes induced by F-actin, with the result that nucleotide-free S1 binds again to F-actin with an increase in light scattering. The goal of our following studies was to comparatively analyze the kinetics of the binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin. To record this binding, it was necessary to distinguish it from other simultaneously proceeding kinetic processes described above. It is known that dissociation of the S1·ADP·BeFx complex upon its mixing with F-actin occurs rather rapidly, this process being measured in seconds, whereas under the same conditions the dissociation of the S1·ADP·AlF4- complex proceeds much more slowly, being measured in minutes or even hours [7, 9, 11]. Very similar time ranges are characteristic for the formation of the S1·ADP·BeFx and S1·ADP·AlF4- complexes (Fig. 1) as well as for dissociation of the actomyosin complex (Fig. 2). Therefore, to study the binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin, we used the stopped-flow method over a very short time range not exceeding 1 sec after mixing of the solutions, i.e., under conditions when F-actin-induced dissociation of these complexes either does not yet begin or is so negligible that it can be ignored.
Kinetics of the “weak” binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin. First of all, we had to be convinced that only the “weak” binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin occurs within the recorded time range (less than 1 sec after mixing), while dissociation of the complexes and the subsequent “strong” binding of S1 to F-actin do not begin. For this purpose we used special tests.
According to a well-known kinetic model proposed by Geeves [23, 24], the myosin head binds to actin via a three-step process:
A + M·N <--> A~M·N <--> A--M·N <--> A**·M--N,
where A is actin, M is myosin head, and N is nucleotide. Collision of S1 with F-actin is followed by the fast formation of the A-state (the state of “weak” binding of S1 to F-actin, in which the nucleotide is strongly trapped in the active site of the S1 ATPase), which then isomerizes to the R-state (the state of “strong” binding of S1 to F-actin, in which the affinity of the nucleotide to S1 decreases dramatically). The stopped-flow method allows recording of both “weak” binding of S1 to F-actin (formation of the A-state) by the increase in light scattering and its transition to the “strong” binding state (A --> R isomerization). This isomerization is accompanied by a significant quenching of the fluorescence of both pyrene label covalently attached to Cys-374 of actin [22, 23] and methylanthranilyl group covalently attached to the 2´ or 3´ hydroxyl of the ribose of ADP bound in the active site of the S1 ATPase [25]. Thus, “weak” binding of S1 to F-actin can be monitored by the increase in light scattering, while its transition to the “strong” binding state can be monitored by the quenching of fluorescence of the labels specifically attached to actin or to the ribose of ADP.
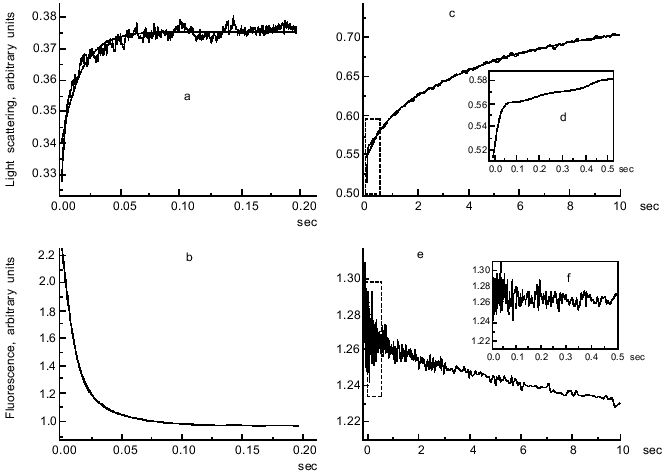
Figure 3 presents stopped-flow records of the fluorescence of pyrene label attached to F-actin and the light scattering obtained after mixing of S1·ADP or S1·ADP·BeFx with F-actin. It is seen that within a short time (0.2-0.5 sec after mixing) the fluorescence of labeled F-actin decreases significantly (by 55%) upon addition of S1·ADP (Fig. 3b), but it does not change upon addition of the S1·ADP·BeFx complex (Fig. 3f). We also observed no changes in the fluorescence within this time range on mixing of unlabeled F-actin with the S1·ADP·BeFx complex containing 2´(3´)-O-(N-methylanthraniloyl)-ADP (mantADP) instead of ADP. However, only 0.2 sec after mixing of the S1·ADP·BeFx with F-actin a significant increase in light scattering (by 10%) was observed (Fig. 3, c and d), this being similar to the increase (by 15%) observed within this time range on mixing of S1·ADP with F-actin (Fig. 3a). According to the interpretation proposed by Geeves [23], the increase in light scattering with no changes in the fluorescence is indicative of the “weak” binding of the myosin heads to F-actin. Thus, we conclude from these data that within the very short time range used (0.5 sec) only the “weak” binding of the S1·ADP·BeFx complex to F-actin occurs, while the processes of dissociation of this complex and the subsequent “strong” binding of S1 to F-actin monitored by fluorescence changes do not yet begin. These processes begin later and proceed much more slowly: we observed a noticeable quenching of the fluorescence only in a second time range (2-10 sec after mixing of the S1·ADP·BeFx complex with F-actin) (Fig. 3e).
The situation with the S1·ADP·AlF4- complex was more complicated. In this case a noticeable quenching of the fluorescence of pyrene label attached to F-actin and of mantADP bound to S1 was sometimes observed within a very short time range (up to 0.5 sec) after mixing the S1·ADP·AlF4- complex with F-actin, although the amplitude of these changes did not exceed 30% of the control value measured in the absence of AlF4- [26]. It was proposed from these data that on fast binding of the S1·ADP·AlF4- complex to F-actin, this complex is able to form a small fraction of the R-state (i.e., the state of “strong” binding to F-actin) [26]. However, it was shown in subsequent experiments that this is not the case. The point is that the S1·ADP·AlF4- complex is formed much less completely than the S1·ADP·BeFx complex (Fig. 1), and as a result it almost always contains of about 5% of S1 molecules free of AlF4-. At the same time, the fluorescence was measured with a significant molar excess of S1 (i.e., at S1/F-actin molar ratio of 5-30) to suppress the reversibility of the reaction. Under these conditions, the presence of even a small admixture of S1 molecules free of AlF4- and having a high affinity to F-actin in the S1·ADP·AlF4- preparation was enough to mimic, at least partially, the “strong” binding of the S1·ADP·AlF4- complex to F-actin. Our recent experiments showed that under changed conditions (e.g., with the complete formation of the S1·ADP·AlF4- complex or with equimolar concentrations of S1 and F-actin) the S1·ADP·AlF4- complex behaves like the S1·ADP·BeFx complex, i.e., its mixing to F-actin leads to a fast increase in light scattering but it is not accompanied by any change in the fluorescence of the pyrene label attached to F-actin during 0.5 sec after mixing (Fig. 3f). We conclude from these data that within a very short time range, not exceeding 1 sec after mixing, both ternary complexes (S1·ADP·BeFx and S1·ADP·AlF4-) bind to F-actin only in the “weak” binding state. Thus, this approach (i.e., measurements in a very short time range not exceeding 1 sec after mixing of the solutions) allows probing the “weak” binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin separately from other processes proceeding much more slowly (actin-induced dissociation of the complexes, dissociation of S1 from F-actin, etc.).Fig. 3. Stopped-flow records of light scattering (a, c, d) and fluorescence of pyrene label attached to F-actin (b, e, f) obtained after mixing of F-actin with S1·ADP (a, b) or S1·ADP·BeFx (c-f). One syringe contained F-actin, and the other contained S1 in the complex with ADP or ADP·BeFx. Conditions: 0.5 µM F-actin, 5 µM S1, 0.2 mM ADP, 2 mM MgCl2, 20 mM MOPS (pH 7.0), and also (in the case of the complex S1·ADP·BeFx) 5 mM NaF and 0.2 mM BeCl2 (final concentrations of the components are listed). The smooth lines are computer fits to single exponentials. Insets (d) and (f) correspond to initial parts of records (c) and (e) shown with an expanded time scale from 0 to 0.5 sec; these parts are shown by dashed lines on panels (c) and (e).
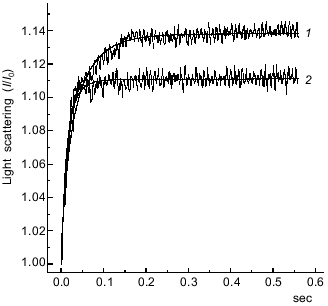
We compared the rates of the “weak” binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin by recording the change in the light scattering within a very short time range (0.6 sec after mixing of the solutions). To prevent possible artifacts caused by the presence of a small admixture of S1 molecules free of AlF4- in the preparation of S1·ADP·AlF4-, we performed the measurements at similar molar concentrations of S1 and F-actin. The binding of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin were studied in the same experiment, i.e., one syringe contained F-actin, and the other syringe was in turn filled either with S1·ADP·BeFx or S1·ADP·AlF4-. As a result, we obtained pairs of curves and subjected them to computer analysis; a typical pair of such kinetic curves for the binding of the S1·ADP·BeFx and S1·ADP·AlF4- to F-actin is presented in Fig. 4. The relative change in light scattering (I/I0) measured 0.6 sec after mixing with F-actin was always higher in the case of S1·ADP·BeFx (1.137 ± 0.007) than in the case of S1·ADP·AlF4- (1.111 ± 0.005).
These kinetic curves (Fig. 4) reflect the attachment of the S1·ADP·BeFx and S1·ADP·AlF4- complexes to F-actin which we supposed to proceed under the conditions used (i.e., at similar molar concentrations of S1 and F-actin) in accordance with the mechanism of a reversible bimolecular reaction:Fig. 4. Time course of the increase in relative intensity of light scattering after mixing of F-actin with the S1·ADP·BeFx (curve 1)or S1·ADP·AlF4- (curve 2) complexes. One syringe contained F-actin and the other contained preformed S1·ADP·BeFx or S1·ADP·AlF4- complex. Final concentrations of the proteins were: 1 µM actin, 1.5 µM S1. Final concentrations of other components of the reaction were the same as in Fig. 2. I0 and I are light scattering intensities at the initial and current moment of the recording, respectively. The smooth lines are computer fits to the equation for a reversible bimolecular reaction (see text for more details).
F-actin + S1·ADP·BeFx <--> F-actin--S1·ADP·BeFx;
F-actin + S1·ADP·AlF4- <--> F-actin--S1·ADP·AlF4-,
which is described by the following equation:
where [A] and [B] are the initial molar concentrations of F-actin and S1·ADP·BeFx or S1·ADP·AlF4-, respectively; [AB] is the equilibrium molar concentration of the complexes F-actin*--S1·ADP·BeFx or F-actin--S1·ADP·AlF4-; I0 and I are the light scattering intensities at the initial and current moment, respectively; DeltaI is a maximal change in light scattering (DeltaI = It-->. - I0); k is the association rate constant; t is time.
The second-order rate constants of S1 binding to F-actin calculated using this equation shows that S1 in the S1·ADP·AlF4- complex binds to F-actin faster (k = (62.8 ± 1.8)·106 M-1·sec-1) than it does in the S1·ADP·BeFx complex (k = (22.6 ± 0.4)·106 M-1·sec-1). Moreover, this equation can be used to calculate the equilibrium molar concentration of the complexes of F-actin with S1·ADP·BeFx or S1·ADP·AlF4-; in both cases this concentration was 0.9 ± 0.001 µM. Taking into account that the initial molar concentration of actin was 1 µM, we conclude that within the studied time range (0.6 sec) almost full saturation of F-actin filaments with S1 molecules in the S1·ADP·BeFx or S1·ADP·AlF4- complexes occurs, and the difference between these complexes is mainly revealed in the rate of their binding to actin filaments.
Thus, we found a not large but quite clear difference between the S1·ADP·BeFx and S1·ADP·AlF4- complexes in the rate of their binding to actin filaments: the S1·ADP·AlF4- complex binds to F-actin somewhat faster than the S1·ADP·BeFx complex does. Also, it is possible that the difference between the S1·ADP·BeFx and S1·ADP·AlF4- complexes in the size of their light scattering changes on binding to F-actin (Fig. 4) reflects a difference in the character of their attachment to F-actin in the “weak” binding state. Similar (although much more pronounced) differences in the size of the maximal light scattering changes were observed earlier when comparing the F-actin-binding properties of S1 isoforms with different alkali light chains; for these S1 isoforms significant differences in sedimentation coefficients of actin filaments decorated with these isoforms were also revealed [27, 28].
The data indicate that the stable ternary complexes S1·ADP·BeFx and S1·ADP·AlF4- can be successfully used for modeling different stages of the myosin ATPase cycle. Taking into account that the S1·ADP·BeFx complex is a stable analog of the intermediate M*·ATP of the myosin ATPase reaction, while the S1·ADP·AlF4- complex mimics the intermediate M**·ADP·Pi, we propose that these intermediates “weakly” bind to F-actin with different rates: M**·ADP·Pi binds to F-actin somewhat faster than M*·ATP does. Taking into consideration recent data on structural differences between the S1·ADP·BeFx and S1·ADP·AlF4- complexes [17-20], we also propose that the M*·ATP and M**·ADP·Pi intermediates differ from each other in the character of their binding to F-actin in the “weak” binding state. These differences between the M*·ATP and M**·ADP·Pi intermediates can explain some features of the mechanism of the interaction of myosin heads with actin coupled with ATP hydrolysis, namely the fact that the heads detached from actin filaments on account of ATP binding bind again to actin in the “weak” binding state after ATP hydrolysis with formation of the M**·ADP·Pi intermediate. Apparently, M**·ADP·Pi has some advantage over M*·ATP upon interaction with actin filaments in the “weak” binding state.
We are very grateful to Prof. B. I. Kurganov and Prof. M. A. Geeves for their help in analysis of the results and for helpful discussion. This work was supported in part by the Russian Foundation for Basic Research (grant 97-04-48043) and the Program of Support for Leading Scientific Schools of the Russian Federation (grant 96-15-98019).
REFERENCES
1.Rayment, I., and Holden, H. M. (1994) Curr.
Opin. Struct. Biol., 3, 944-952.
2.Spudich, J. A. (1994) Nature, 372,
515-518.
3.Bagshaw, C. R., and Trentham, D. R. (1974)
Biochem. J., 141, 331-349.
4.Johnson, K. A., and Taylor, E. W. (1978)
Biochemistry, 17, 3432-3442.
5.Werber, M. M., Szent-Gyorgyi, A. G., and Fasman, G.
D. (1972) Biochemistry, 11, 2872-2883.
6.Goodno, C. C. (1982) Meth. Enzymol.,
85, 116-123.
7.Werber, M. M., Peyser, Y. M., and Muhlrad, A.
(1992) Biochemistry, 31, 7190-7197.
8.Phan, B. C., and Reisler, E. (1992)
Biochemistry, 31, 4787-4793.
9.Phan, B. C., Faller, L. D., and Reisler, E. (1993)
Biochemistry, 32, 7712-7719.
10.Henry, G. D., Maruta, S., Ikebe, M., and Sykes,
B. D. (1993) Biochemistry, 32, 10451-10456.
11.Maruta, S., Henry, G. D., Sykes, B. D., and
Ikebe, M. (1993) J. Biol. Chem., 268, 7093-7100.
12.Gopal, D., and Burke, M. (1995) J. Biol.
Chem., 270, 19282-19286.
13.Levitsky, D. I., Shnyrov, V. L., Khvorov, N. V.,
Bukatina, A. E., Vedenkina, N. S., Permyakov, E. A., Nikolaeva, O. P.,
and Poglazov, B. F. (1992) Eur. J. Biochem., 209,
829-835.
14.Bobkov, A. A., Khvorov, N. V., Golitsina, N. L.,
and Levitsky, D. I. (1993) FEBS Lett., 332, 64-66.
15.Bobkov, A. A., and Levitsky, D. I. (1995)
Biochemistry, 34, 9708-9713.
16.Levitsky, D. I., Bobkov, A. A., Golitsina, N. L.,
Nikolaeva, O. P., Pavlov, D. A., and Poglazov, B. F. (1996)
Biofizika, 41, 64-72.
17.Ponomarev, M. A., Timofeev, V. P., and Levitsky,
D. I. (1995) FEBS Lett., 371, 261-263.
18.Mendelson, R. A., Schneider, D. K., and Stone, D.
B. (1996) J. Mol. Biol., 256, 1-7.
19.Golitsina, N. L., Bobkov, A. A., Dedova, I. V.,
Pavlov, D. A., Nikolaeva, O. P., Orlov, V. N., and Levitsky, D. I.
(1996) J. Muscle Res. Cell Motil., 17, 475-485.
20.Fisher, A. J., Smith, C. A., Thoden, J., Smith,
R., Sutoh, K., Holden, H. M., and Rayment, I. (1995)
Biochemistry, 34, 8960-8972.
21.Weeds, A. G., and Taylor, R. S. (1975)
Nature, 257, 54-57.
22.Criddle, A. H., Geeves, M. A., and Jeffries, T.
(1985) Biochem. J., 232, 343-349.
23.Geeves, M. A. (1991) Biochem. J.,
274, 1-14.
24.Geeves, M. A., and Conibear, P. B. (1995)
Biophys. J., 68, 194s-201s.
25.Woodward, S. K. A., Eccleston, J. F., and Geeves,
M. A. (1991) Biochemistry, 30, 422-430.
26.Nikolaeva, O. P., and Geeves, M. A. (1996) J.
Muscle Res. Cell Motil., 17, 216.
27.Nikolaeva, O. P., Golitsina, N. L., Levitsky, D.
I., Moiseeva, L. N., and Kurganov, B. I. (1994) Biochem. Mol. Biol.
Int., 33, 553-560.
28.Golitsina, N. L., Levitsky, D. I., Moiseeva, L.
N., Nikolaeva, O. P., and Kurganov, B. I. (1991) J. Chem. Biochem.
Kinetics, 1, 311-319.