Kinetic Mechanism of Fo·F1 Mitochondrial ATPase: Mg2+ Requirement for Mg·ATP Hydrolysis
A. V. Syroeshkin, M. A. Galkin, A. V. Sedlov, and A. D. Vinogradov*
Department of Biochemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (7-095) 939-3955; E-mail: adv@biochem.bio.msu.su* To whom correspondence should be addressed.
Received February 4, 1999; Revision received March 9, 1999
The initial rates of ATP hydrolysis catalyzed by Fo·F1 (bovine heart submitochondrial particles) preincubated in the presence of Pi for complete activation of the oligomycin-sensitive ATPase were measured as a function of ATP, Mg2+, and Mg·ATP concentrations. The results suggest the mechanism in which Mg·ATP complex is the true substrate of the ATPase and the second Mg2+ bound at a specific pH-dependent site is needed for the catalysis. Simple hyperbolic Michaelis--Menten dependences of the reaction rate on the substrate (Mg·ATP) and activating Mg2+ were found. In contrast to the generally accepted view, no inhibition of ATPase by free Mg2+ was found. Inhibition of the reaction by free ATP is due to a decrease of free Mg2+ needed for the catalysis. In the presence of both Ca2+ and Mg2+ the kinetics of ATP hydrolysis suggest that the Ca·ATP complex is neither hydrolyzed nor competes with Mg·ATP, and free Ca2+ does not affect the hydrolysis of Mg·ATP complex. A crucial role of free Mg2+ in the time-dependent inhibition of ATPase by azide is shown. The dependence of apparent Km for Mg·ATP on saturation of the Mg2+-specific site suggests the formal ping-pong mechanism in which bound Mg2+ participates in the overall reaction after dissociation of one product (most likely Pi) thus promoting either release of ADP (catalytic turnover) or slow isomerization of the enzyme--product complex (formation of the dead-end ADP(Mg2+)-inhibited enzyme). The rate of Mg·ATP hydrolysis only slightly depends on pH at saturating Mg2+. In the presence of limited amounts of free Mg2+ the pH dependence of the initial rate corresponds to the titration of a single group with pKa = 7.5. The simple competition between H+ and activating Mg2+ was observed. The specific role of Mg2+ as a coupling cation for energy transduction in Fo·F1-ATPase is discussed.
KEY WORDS: Fo·F1-ATP synthase, F1-ATPase, mitochondria, ATP hydrolysis, oxidative phosphorylation
Abbreviations: MF1, CF1, TF1) solubilized F1s from mitochondria, chloroplasts, and thermophylic bacteria, respectively; CCCP) carbonyl cyanide m-chlorophenylhydrazone; AMP-PNP) 5´-adenylylimidodiphosphate; SMP) submitochondrial particles.
Oxidative phosphorylation and photophosphorylation are catalyzed by
Fo·F1-ATP synthases which are oligomeric
proteins embedded in the coupling membranes of mitochondria,
chloroplasts, and bacteria. The F1 component (a peripheral
part of ATP synthase) has molecular mass of about 380 kD and is
composed of five different polypeptide chains in the stoichiometry
alpha3·beta3·gamma·delta·epsilon
[1-3]. F1 units from
various sources, being energetically uncoupled, rapidly hydrolyze ATP
in the presence of bivalent cations, and their catalytic properties
have been extensively examined for more than 35 years since the enzyme
was first isolated [4]. The 2.8 Å resolution
structure for bovine mitochondrial F1, which is the largest
asymmetric protein structure ever solved, recently become available [5].
The membrane-embedded hydrophobic Fo part contains at least three types of subunits (a, b, and c in stoichiometry a·b2·c9-12 in E. coli [6]) and has more complex structure for bovine heart Fo [7]. The Fo complex contains a proton pathway conducting protons coupled by an unknown mechanism with ATP synthesis at the F1 active site(s) at the expense of an electrochemical H+ gradient (DeltaµH+) across the coupling membrane. Several hypotheses on the mechanism for DeltaµH+-consuming ATP synthesis or DeltaµH+-producing ATP hydrolysis catalyzed by Fo·F1-ATP synthase have been proposed and reviewed [8-13]. None of them is conclusively proved, and the molecular mechanism of coupling between proton flow through Fo and ATP synthesis on F1 seems to be the most challenging problem in bioenergetics.
Three out of six nucleotides which can be cooperatively bound to one F1 molecule are rather rapidly exchangeable with medium ATP or ADP [14, 15], thus suggesting that three sites on F1 are potentially capable of participation in the catalytic steady-state turnovers during ATP hydrolysis or synthesis. The presence of three exchangeable nucleotide-binding sites in addition to one or two Pi-binding site(s) [16] and several Me2+-specific sites [17-21] per F1 molecule gives rise to numerous possible enzyme--substrate (product) complexes which may exist during the steady-state catalysis in either direction. In contrast to several reports in the literature on the substrate--velocity curves for the complex [22-26], we found simple Michaelis--Menten dependence of the reaction rate on ATP concentration in the presence of Mg2+ [27] under the conditions where the true initial azide-insensitive steady-state rates of ATP hydrolysis were measured. Our results suggest that either a single nucleotide-binding site is involved in the catalysis or an extremely strong cooperativity such as a nucleotide binding exchange mechanism [28, 29] exists during the steady-state enzyme turnovers. It should be emphasized that the true initial rates of ATP hydrolysis (when no ADP is present in the assay system) are azide-insensitive, whereas the apparent initial rates (when the enzyme contains tightly bound ADP) are significantly decreased in the presence of azide [27].
The essential requirement of bivalent cations for ATP hydrolysis catalyzed by mitochondrial preparations of different degree of resolution was recognized many years ago [30-34]. However, an adequate answer to the simple question of the actual ATP- and Mg2+-containing species participating as the substrate and activator during ATP hydrolysis has not been found. Numerous studies on this problem have appeared in the literature. It is generally believed that Mg·ATP is the substrate of F1-type ATPase, whereas free ATP and free Mg2+ are inhibitors.
The standard kinetic analysis of enzymatic reactions involving metal--nucleotide complexes must be applied to F1-type ATPase with great precautions for several reasons. When ATP hydrolysis is followed by continuous monitoring methods, the release of product is biphasic or triphasic depending on the "history" of the preparation. It shows a "burst" or "lag" on the minute time scale, and the time dependence for active --> inactive or inactive --> active transitions is a complex function of the nucleotides and Mg2+ concentrations [27, 35, 36]. Thus the possibility of reaching erroneous conclusions from measurements of apparent "initial" rates can hardly be overestimated. Another difficulty in the kinetic analysis of F1-type ATPase is the possible presence of a small inhibitory peptide [37] which seems to be an intrinsic part of the mitochondrial Fo·F1-ATP synthase. The inhibitory effect of this protein is also time- and ATP-dependent in addition to the strong pH and ionic strength dependences [37, 38]. It appears that the presence of uncontrolled contaminations of the protein inhibitor might be a source of serious errors in the measurements of the actual rates of ATP hydrolysis, especially in early studies including our own [30-33]. The third difficulty is concerned with the strong requirement of Mg2+ for pyruvate kinase [39] which is routinely used for removal of inhibitory ADP in the coupled enzyme assay [40]. Thus variations of free Mg2+ in the continuous coupled assay mixtures are limited.
Our search of the literature reveals no report on the kinetic properties of Fo·F1 (which may or may not be identical to those of energetically uncoupled F1) conclusively answering the question of whether ATP or Mg·ATP or Mg·ATP and Mg2+ serve as the substrate(s) of ATPase. In this paper we present initial rate analysis which shows that the Mg·ATP complex is the substrate for the Fo·F1-ATPase and the second Mg2+ bound in pH-dependent fashion is required for the catalysis. The data are suggestive of an ordered reaction mechanism where free Mg2+ participates in the reaction as an uncompetitive (with Mg·ATP complex) activator.
MATERIALS AND METHODS
Submitochondrial particles (SMP). SMP freed of the ATPase inhibitor were protein were prepared from bovine heart mitochondria as described [41] and stored as a suspension (small samples) in 0.25 M sucrose (30-40 mg/ml) in liquid nitrogen. Before the experiments a sample was thawed and diluted to a protein content of 5-10 mg/ml in a mixture containing (final concentrations) 0.25 M sucrose, 0.1 M KCl, 50 µM EDTA, 10 mM Hepes/KOH, and 5 mM potassium phosphate, pH 7.4. The suspension was incubated at 20°C for 1 h and further stored at room temperature (~20°C) during the experiments. The particles treated as described showed no lag in their initial azide-insensitive ATPase activity [42, 43] which was more than 95% inhibited by oligomycin (0.5 nmole/mg protein).
ATPase assay. ATPase activity was measured at 25°C in the standard reaction mixture containing (final concentrations) 0.25 M sucrose, 0.1 M KCl, 10 mM Hepes/KOH buffer, pH 7.4, 3 µM rotenone, 4 µM CCCP, and various concentrations of ATP and MgCl2. The pH of the reaction mixture was adjusted after ATP and MgCl2 were added. The pH was 8.0 when La·ATP was used as the substrate.
When the calculated free Mg2+ concentration was higher than 10-4 M, the activity was followed as a decrease in the absorption of NADH (160 µM) at 340 nm; 1.5 mM phosphoenolpyruvate (tricyclohexylammonium salt), pyruvate kinase (12 units/ml), and lactate dehydrogenase (10 units/ml) were added to the standard mixture, and the reaction was initiated by the addition of SMP (15-25 µg/ml).
When the free Mg2+ concentration was lower than 10-4 M, ATP hydrolysis was followed by "stoichiometric" H+ release [44] as a decrease in the absorption of Phenol Red (DeltaA557-620 nm). Phenol Red (30 µM) was added to the standard reaction mixture and the reaction was initiated by the addition of SMP (30-40 µg/ml). The stoichiometry of ADP formed/H+ released which is dependent on pH and Mg2+ concentration [44] was determined as follows. After the reaction followed at 557-620 nm proceeded for 1-2 min, the ATP hydrolysis was stopped by addition of oligomycin (12 nmoles/ml) and the instrument sensitivity was calibrated by the additions of titrated HCl solution. The amount of ADP formed was then determined in the same sample by measuring of the absorption decrease at 340 nm after the addition of 160 µM NADH, 5 mM MgCl2, 1.5 mM phosphoenolpyruvate, pyruvate kinase (12 units/ml), and lactate dehydrogenase (12 units/ml). It was shown in separate experiments that the pH indicator responded linearly to HCl concentration added and no absorption change of Phenol Red was observed at 340 nm when a pH shift of 0.05 unit was induced by HCl addition. All the activity measurements were performed using a Hitachi-557 spectrophotometer.
It should be noted that all the data and conclusions reported in this paper are valid only for the initial, constant (<30 sec) azide-insensitive rates [42].
Preparation of La·ATP. Equal volumes of 5 mM solutions of LaCl3 and ATP (pH 3.0) were mixed, and the supernatant after centrifugation (to separate a precipitate formed) was used. The concentration of La·ATP was determined by the absorption at 259 nm. No turbidity was observed when the complex thus prepared was added to the assay mixture.
Estimation of ATP, Mg2+, and Mg·ATP concentrations.
The concentrations of the variable substrate (or activator or
inhibitor) were calculated by solving for the positive roots of the
polynomial equations derived from the multiple equilibria:
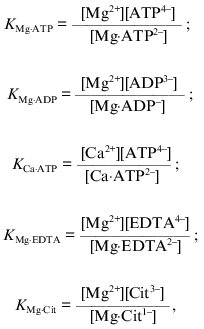
where Cit stands for citrate, and the conservation equations for the total (analytical) concentrations of added Mg2+ ([Mg]t), ATP4- ([ATP]t), EDTA4- ([EDTA]t), ADP3- ([ADP]t), and citrate ([Cit]t) were used. The dissociation constants KMg·ATP (1·10-4 M), KMg·EDTA (1.6·10-6 M), KMg·ADP (1·10-3 M), KMg·Cit (2.5·10-4 M), and KCa·ATP (2.5·10-4 M) corrected for pH 7.4 [45, 46] were used for the calculations. All calculations were performed using the GIM version 2 computer program (copyrighted by A. Drachev). It should be noted that slight variations in the absolute values for the dissociation constants found in the literature do not change our conclusions, although proportional variations in the values of Km(i) for substrate (or activator or inhibitor) are expected.
Reagents. ATP (sodium salt), lactate dehydrogenase, Hepes, EDTA, NADP+, CCCP, oligomycin, and glucose 6-phosphate dehydrogenase were obtained from Sigma Chemical Co. (USA). Phosphoenolpyruvate (tricyclohexylammonium salt), pyruvate kinase, and NADH were from Reanal (Hungary). Hexokinase and rotenone were from Ferak (Germany), Chelex-100 was from Bio-Rad (USA). Other chemicals were of the purest grade commercially available.
The stock solutions of ATP and standard reaction mixture (when no EDTA or citrate were added) were treated with Chelex-100 ion-exchange resin (potassium form) to remove contaminating bivalent cations. The concentration of ATP in stock solutions was determined enzymatically (NADP+, glucose, glucose-6-phosphate dehydrogenase). The concentration of Mg2+ in stock solutions was determined by titration with the standard EDTA in the presence of Eriochrome Black T in NH4OH/NH4Cl buffer (pH 10). The enzymes used for the analyses and assays were extensively dialyzed before use. The protein content was determined by the biuret method [47].
RESULTS
Mg·ATP as the substrate for Fo·F1-ATPase. No measurable ATP hydrolysis was observed in the absence of added Mg2+. At fixed added ATP concentrations the ATPase activity (initial rates) increased as the concentration of added Mg2+ increased, and no inhibition of the reaction was seen up to the ratio [Mg]t/[ATP]t of ~ 30 (Fig. 1a). The dependence of the initial rate on the added ATP concentration at constant [Mg2+]t is shown in Fig. 1b. When the data were plotted as a function of calculated Mg·ATP complex, a single straight line was obtained (Fig. 1c), suggesting simple Michaelis--Menten kinetics of the reaction where Mg·ATP complex is the true substrate (KmMg·ATP = 0.15 mM; Vmax = 6.7 µmoles/min per mg protein). These results show that free ATP4- (in the presence of Mg2+) is neither a substrate or inhibitor of ATP hydrolysis. In agreement with our previous finding [27], we were unable to demonstrate any deviation from a simple hyperbolic dependence of the initial rate on the substrate concentration. No hydrolysis of ATP was observed in the presence of Ca2+. At limited concentration of Mg·ATP (~KmMg·ATP), Ca2+ inhibited the reaction as it would be quantitatively expected if the inhibition were due to redistribution between catalytically inert Ca·ATP and active Mg·ATP complexes (table).
Apparent inhibition of Fo·F1-ATPase by Ca2+Fig. 1. Dependence of the initial rate of ATP hydrolysis on Mg2+ and ATP concentrations. a) Variations of [Mg2+]t at constant concentrations of added ATP (indicated by the figures on the curves). The ATPase activity was assayed with ATP-regenerating system when [Mg2+]t > [ATP]t and by H+ release when [Mg2+]t < [ATP]t. b) Varying [ATP]t at 5 (1) and 10 mM [Mg2+]t (2). c) The double reciprocal plot of the combined data presented in (a, Delta) and (b, {} and {}) recalculated assuming that Mg·ATP is the substrate for hydrolysis.
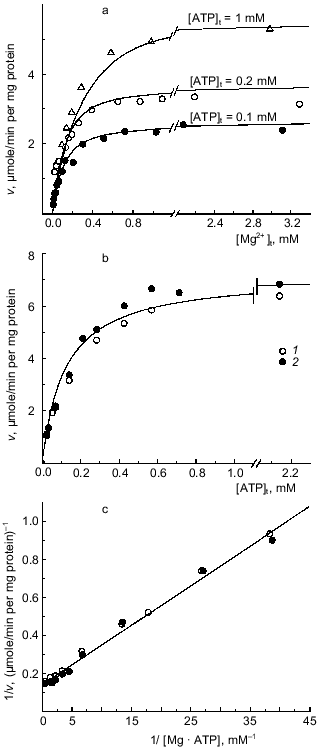
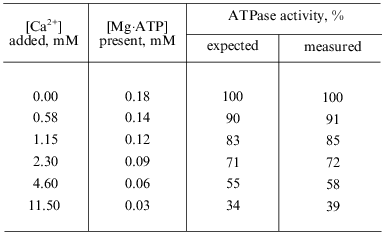
*ATPase activity was measured as H+-release (see "Materials and Methods" section). 100% correspond to the activity calculated using the parameters: Vmax = 6.7 µmoles/min per mg protein and KmMg·ATP = 0.15 mM. The concentrations of Mg·ATP were calculated by solving the cubic equation derived from the equilibria between ATP, Mg2+, and Ca2+ at pH 7.4. The expected rates were calculated assuming a simple Michaelis--Menten dependence of the velocity on [Mg·ATP] and inability of Ca·ATP binding and hydrolysis at the catalytic site. [ATP]t and [Mg2+]t were 0.2 and 1 mM, respectively.
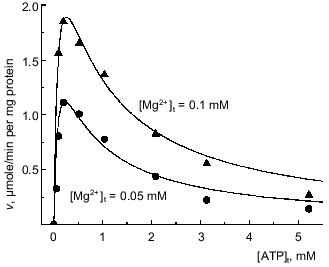
Binding of free Mg2+ is needed for hydrolysis of Mg·ATP. When added ATP was a variable parameter with a limited concentration of Mg2+, the ATPase activity reached a maximum at the ratio [ATP]t/[Mg2+]t of approximately 1 and decreased upon further increase of free ATP (Fig. 2). The ascending parts of the curves are consistent with the results presented in the previous section suggesting that Mg·ATP is the substrate for the hydrolysis. At least two reasons for the descending parts of the curves may be considered. The possibility that free ATP4- is an inhibitor of the reaction seemed to be unlikely in light of the results presented in Fig. 1c. An inhibition of the reaction at [ATP]t/[Mg2+]t >> 1 would be also expected if the presence of free Mg2+ is needed for the hydrolysis of Mg·ATP. To test the latter explanation the buffer system containing Mg2+, ATP, and other anions capable of Mg2+ binding was used to create a system in which concentration of the substrate (Mg·ATP) would be maintained at a constant level upon variation of free Mg2+. For instance, if the reaction mixture contained 1.65 mM EDTA and 1 mM Mg2+ (total concentrations) the variation in [ATP]t from 0.2 to 1 mM results in variation of [Mg·ATP] from 4.7 to 23 µM, whereas the concentration of free Mg2+ remains almost constant (~2.4 µM). This complex buffer system allow varying the substrate concentration at a particular constant free [Mg2+].
Figure 3 presents an example of the dependences of the reaction rate on the substrate concentrations ([Mg·ATP]) at different constant [Mg2+]. When apparent maximal rates extrapolated to infinite [Mg·ATP] were plotted as a function of free Mg2+ concentrations, a simple hyperbolae was obtained with the parameters: KmMg = 0.02 mM and Vmax = 6.3 µmoles/min per mg protein (Fig. 3a). It appears that a single activating Mg2+-specific site is operating during the steady-state catalysis. The kinetic behavior of the reaction (Fig. 3b) suggests the formal ping-pong mechanism, where the substrate (Mg·ATP) transformation at the enzyme active site (hydrolysis of Mg·ATP and/or release of one product (ADP or Pi)) precedes the involvement of free Mg2+, which is needed to complete the catalytic cycle. The kinetic equation for such a mechanism (see scheme shown in Fig. 8 in the "Discussion" section) appears as:Fig. 2. Dependence of the initial rate of ATP hydrolysis on [ATP]t at fixed added Mg2+ concentrations. Concentrations of added Mg2+ are indicated on the curves. Symbols (o, Delta), the experimental points; continuous lines, the theoretical values calculated according Eq. (1) with the parameters: Vmax = 6.1 µmoles/min per mg protein; KsMg·ATP = 0.1 mM; and KsMg = 0.025 mM.
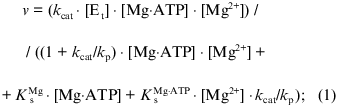
and it is expected that an inhibition of the reaction would occur at high concentrations of added ATP at any given Mg2+ concentration. Figure 2 shows that the experimentally determined initial rates fit the theoretical prediction when the approximated values of KsMg·ATP and KsMg were used. Taken together, the results presented in this and previous section strongly suggest that Mg·ATP is a true substrate for Fo·F1-ATPase and free Mg2+ is needed as an obligatory activator for the steady-state ATP hydrolysis.
It should be noted that an alternative interpretation of the data shown in Fig. 2 is possible assuming that Mg·ATP is the substrate and free ATP is an inhibitor. An ambiguity in interpretation is due to the fact that the concentrations of ATP and Mg2+ at equilibrium are certain to obey the mass action low. It was desirable to find a more direct experimental approach to show the requirement of free Mg2+ for the ATPase reaction. The kinetically inert complexes of metals with ATP [48, 49] seemed to be a useful tool to discriminate between the two possible interpretations. This approach was used previously by Pedersen's group for the soluble rat liver F1 [48]. As depicted in Fig. 4, the La·ATP complex is not hydrolyzed unless Mg2+ is added. In the presence of free Mg2+ the rate of ATP hydrolysis is about 15% of that observed for the Mg·ATP maximal rate.Fig. 3. Kinetic parameters of Mg·ATP hydrolysis at different Mg2+ concentrations. a) The reaction rates (v) extrapolated to infinite Mg·ATP were measured as a function of free Mg2+ (double reciprocal plot). Concentrations of constant free Mg2+ were maintained utilizing Mg2+/citrate/ATP buffer (o) or Mg2+/EDTA/ATP buffer (o) (see "Materials and Methods" section). b) Secondary plot (Vmax versus KmMg·ATP) of data obtained as in (a).
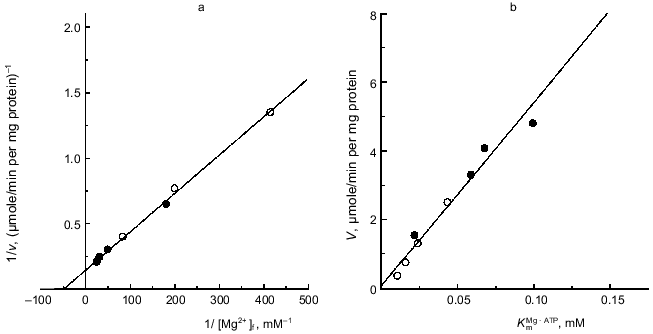
Since the affinity of the binding site for a cation is expected to be pH dependent, it was of interest to determine how the pH dependence of the enzyme activity (if it exists) is related to the Mg2+ requirement. Figure 5 demonstrates that the initial rate of ATP hydrolysis was only slightly changed within the pH interval from 6.8 to 8.0 when free Mg2+ was in excess (~10 KmMg) and becomes strongly pH dependent when free Mg2+ was limiting. An apparent pKa of 7.4 for a group or groups involved in Mg2+ binding was estimated from the titration curve, and simple competition was observed between activating free Mg2+ and H+ (Fig. 6).Fig. 4. Hydrolysis of La·ATP by Fo·F1-ATPase. The reaction was followed as H+ release measured with Phenol Red (see "Materials and Methods" section). La·ATP (0.2 mM), MgCl2 (0.2 mM), SMP (0.4 mg/ml), and oligomycin (12 nmoles/ml) were added where indicated by the arrows. The rate of La·ATP hydrolysis initiated by the addition of Mg2+ was 0.7 µmole/min per mg protein.
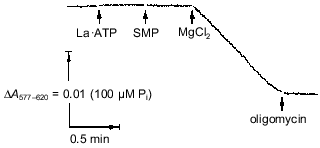
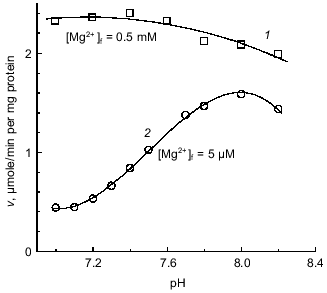
Fig. 5. pH dependence of ATPase activity. 1) Mg2+ concentration was 0.5 mM; ATP-regenerating system was used for ATPase assay; 2) Mg2+ concentration was 5 µM; ATPase activity was assayed as H+ release. H+ released/ADP formed ratio was varied from 0.68 at pH 7.0 to 0.96 at pH 8.2 and the rates were corrected as described in "Materials and Methods" section.
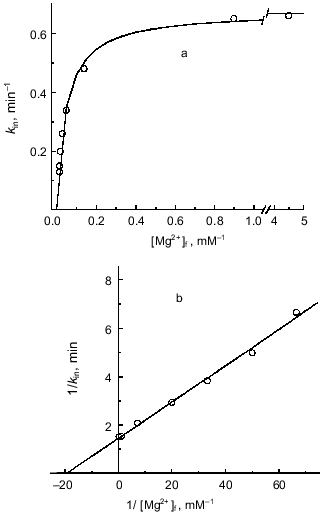
Mg2+(ADP)-specific time-dependent inhibition and requirement of free Mg2+ for ATP hydrolysis. It has been shown that ADP-loaded Fo·F1-ATPase is sensitive to Mg2+-induced slow isomerization of the ADP--enzyme complex, producing inactive ATPase which is stabilized by azide [42, 50]. Since the binding of free Mg2+ at the enzyme specific site was shown to be required for the steady-state ATP hydrolysis (see previous section), it was of interest to get deeper insight into the relations between the role of Mg2+ in the catalysis and in the deactivation process. The parameters of ATP hydrolysis activation by Mg2+ (Fig. 3a) and those determined as the first-order rate constant for the azide-induced deactivation [42] (Fig. 7) were quantitatively estimated. Both dependences were found to fit a simple hyperbolic function with the dissociation constant for Mg2+ of about 4·10-5 M. These results are in accord with the proposal on a single free Mg2+ binding site.Fig. 6. Competitive inhibition of ATPase by H+. [Mg·ATP] was 0.1 mM; calculated reciprocal free concentrations of Mg2+ are plotted on the abscissa.
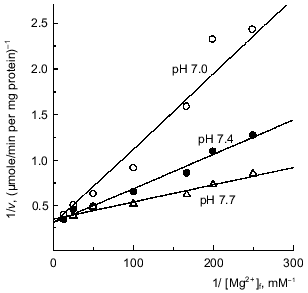
Fig. 7. Mg2+ dependence of inhibition of ATPase by azide. a) The standard reaction mixture contained 200 µM sodium azide and increased amount of pyruvate kinase (300 units/ml) to avoid rate limitation at low free [Mg2+]. The concentration of Mg·ATP was 1 mM. The reaction was initiated by the addition of SMP and followed continuously. The first-order rate constant of azide-induced inhibition was calculated [42] and plotted as a function of free [Mg2+]. Concentrations of free Mg2+ were varied by the excess of ATP added. b) Double reciprocal plot of the data shown in (a).
DISCUSSION
Many years ago we reported that depending on the "history" of the preparation the mitochondrial F1- or Fo·F1-ATPase shows either lag-phase or burst in the activity assayed in the presence of an ATP-regenerating system [27]. The interplay of Mg2+-dependent deactivation of the enzyme--ADP intermediate [27, 50] and ATP-dependent reactivation of the inhibited enzyme [27] (both reactions are slow compared with the enzyme turnover) could lead to erroneous interpretation of the velocity versus substrate curves. Moreover, the meaning of the "initial rate", which is a standard parameter used for steady-state kinetic analysis, becomes ambiguous when applied to a system where the amount of catalytically competent enzyme is changed within the time scale comparable with the resolution limit of the assay. These considerations prompted us to reinvestigate a long standing problem: what are the true species which interact with Fo·F1-ATPase during steady-state ATP hydrolysis? Our data confirm and reinforce the generally accepted belief (for exceptions, see [51, 52]) that Mg·ATP complex is the specie taken up from solution as the substrate for the reaction. However, our results reject another generally accepted view, that free Mg2+ is an inhibitor of the reaction [34]. The most likely reason for the latter is that no precautions for complete activation of ATPase (removal of tightly bound ADP [27]) were taken in the previously published works. When the concentration of free Mg2+ in the assay system is high, a fraction of the enzyme preparation becomes rapidly deactivated, and the apparent "initial rate" is underestimated.
Our data show that free Mg2+ (in addition to Mg·ATP) is needed for the catalysis. This is in accord with the conclusion reached by other authors for F1 from rat liver [48] and from Halobacterium saccharovorum [53]. In addition to providing new information about the participation of free Mg2+ in Mg·ATP hydrolysis by Fo·F1-ATPase, the results presented here are relevant to the sequence of the catalytic steps. The extended kinetic scheme of the reaction is shown in Fig. 8. As shown in this study, the activating effect of free Mg2+ is described by the formal ping-pong mechanism. Mechanistically this would mean that activating Mg2+ participates in the overall reaction after one of the products (ADP or Pi) is released. Several findings suggest that Pi leaves the ATPase active site before ADP. First, it is well documented that Pi is not required for ADP binding. Moreover, Pi reverses ADP-induced (in the presence of Mg2+) deactivation of the ATPase [43] or Mg2+-induced deactivation of the ADP-preloaded ATPase [50]. Second, both the steady-state rate of ATP hydrolysis (routes (1)-(5)) and the deactivation rate (routes (1)-(6)) are changed proportionally when either Mg·ATP [27] or free Mg2+ (this paper) are variable parameters, independently of the presence of Pi. The absence of any inhibitory effect of Pi on Fo·F1-ATPase [43] suggests that Pi leaves the enzyme during uncoupled ATP hydrolysis from a very low-affinity site. The low affinity for leaving Pi compared to the high affinity for ADP [35] can provide a thermodynamic basis for the kinetic sequence of the release of the products during ATP hydrolysis. It worth noting that the apparent Km value for Pi during oxidative phosphorylation under conditions similar to those used in this work for ATPase assay is about 1 mM (our unpublished observation). The absence of the inhibitory effect of Pi on the ATPase is hard to reconcile with simple reversibility of the reaction steps during hydrolysis and synthesis of ATP.
Strong data for the presence of a Mg2+-specific site on mitochondrial Fo·F1-ATPase [50] and CF1 [55] which is responsible for Mg2+-induced deactivation of ATPase have been reported. A two-step mechanism for Mg2+-induced deactivation of ADP-loaded enzyme was proposed, which includes rapid, pH and nucleotide-dependent binding of Mg2+ (Ks value of 1·10-4 M at pH 7.4) and subsequent isomerization of the complex resulting in the inactive enzyme [50]. The apparent affinities for free Mg2+ determined from quantitation of: (1) Mg2+-induced deactivation of ADP-preloaded Fo·F1 [50], (2) Mg2+ requirement of Mg·ATP hydrolysis (this paper), and (3) Mg2+ dependence of azide-induced inhibition (this paper) at pH 7.4 are 1·10-4, 2·10-5, and 4·10-5 M, respectively. The latter two values are similar, and it is not unexpected that the value determined for (1) is considerably higher: a strong dependence of the apparent affinity for deactivating Mg2+ on occupation of nucleotide-binding sites has been documented [21]. The actual occupancy of three rapidly exchangeable nucleotide-binding sites during steady-state ATP hydrolysis is not known. Thus, we believe that the same Mg2+-specific site is responsible for all three processes, and the reaction sequences (3)-(4)-(5) and (3)-(4)-(6) (Fig. 8) describe the final steps of ATP hydrolysis and deactivation, respectively. The simple competitive inhibition of Mg·ATP hydrolysis by free ADP (our results which are not included in this paper) is expected if the reaction sequence proceeds as depicted in the reaction scheme whereby free ADP leaves the ATPase catalytic site via a Mg2+-dependent mechanism. The tight binding of ADP and some nucleotide derivatives to TF1 and MF1 in the absence of Mg2+ and major rearrangements of the nucleotide-binding sites after subsequent addition of Mg2+ have been documented [56, 57]. On the other hand, Mg·ADP serves as the substrate for photo- [58] or oxidative phosphorylation (our unpublished results). The difference in the reactive species leaving the catalytic site during steady-state ATP hydrolysis and entering the catalytic site during steady-state ATP synthesis is in accord with our hypothesis of different catalytic mechanisms for operation of Fo·F1 in the hydrolytic and synthetic directions [59, 60].Fig. 8. Reaction scheme for hydrolysis of ATP catalyzed by the mitochondrial Fo·F1-ATPase. E, T, D, Pi, M, and Ma stand for the enzyme, ATP, ADP, inorganic phosphate, Mg2+ originated from Mg·ATP, and free activating Mg2+, respectively. The scheme does not attempt to portray the interactions between the enzyme subunits during the reaction. The hydrolytic steps (2)-(5) may certainly depend on the cooperative interactions within multisubunit enzyme (binding change mechanism [12]) possibly including rotation of the gamma-subunit [54]. The scheme is rather intended to describe the fate of the substrate during catalytic turnover. It should be noted that steps (4) and (5) are shown as binding and release of Mg2+ for the sake of clarity; no difference in the kinetic behavior of the system (Eq. (1)) would be seen if the activating Mg2+ (Ma) remains permanently bound to the enzyme during the catalysis, participating in the reaction cycle after Pi release. For simplicity, a combination of equilibrium (steps (1)-(2) and (4)) and steady-state (steps (3) and (5)) approximations were applied to derive Eq. (1) describing the reaction scheme.
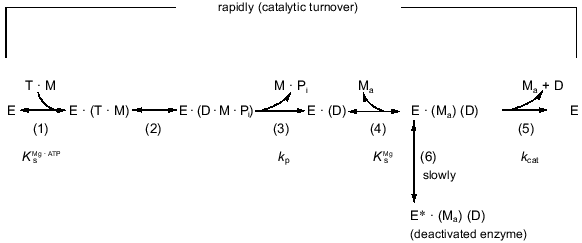
Since the structure at 2.8 Å resolution of bovine heart mitochondrial F1-ATPase became available [5], an obvious question arises: where is the single Mg2+-specific site located? Structures of the nucleotide-binding sites of asymmetric F1 containing Mg2+--nucleotide complexes with a slight difference in the ligands to Mg2+ in the alpha- and beta-subunits were identified [5]. Surprisingly, no Mg2+ other than that originating from the Mg2+--nucleotide complexes was visualized despite the presence of 250 µM AMP-PNP, 5 µM ADP, 6 mM Mg2+, and 3 mM sodium azide and the absence of Pi in the crystallization medium [61], i.e., conditions which must produce the deactivated Mg2+-containing enzyme. Several possibilities for an explanation of the absence of visualized Mg2+ can be offered. Major rearrangement of F1 subunits and the substrate/activator binding sites induced by interaction of F1 and Fo is one possible reason [62]. Another possibility is that the Mg2+-specific site is located on a disordered "invisible" part of the gamma-subunit. Such speculation may be supported by the findings that ATP hydrolysis catalyzed by the reconstituted 3alpha·3beta TF1 is not sensitive to azide [63, 64], which is known to inhibit ATPase by a Mg2+-dependent mechanism ([21, 42, 50] and this paper). If this hypothesis is correct, the structural arrangements of the events shown in the reaction scheme (Fig. 8) are that free ADP formed at the catalytic site after (Pi·Mg) release is removed to the solution via the interaction between the Mg2+-liganded arm of the gamma-subunit and beta-subunit. A key role of Mg2+ in the energy transducing enzymes has long been suggested [65, 66]. The large pH dependence of the Mg2+-specific site evidenced from the Mg2+-dependent steady-state hydrolysis of Mg·ATP (Figs. 5 and 6) is in line with this proposal. The prediction of the hypothesis on the key role of Mg2+ in coupling between transmembrane proton or sodium [67] flows and binding/release of nucleotides at the catalytic site(s) is that the binding of Mg2+ to the F1 part of Fo·F1-ATPase/synthase must be DeltaµH+-dependent. Experiments aimed to clarify this possibility are currently underway in our laboratory.
We thank Drs. E. Maklashina, V. Grivennikova, and E. Gavrikova for their help in the preparation of SMP.
This work was supported by the Russian Foundation for Basic Research (grants 96-04-48185 and 99-04-48082) and by the Program for Advanced Schools in Science (grant 96-15-97822).
REFERENCES
1. Catteral, W. A., and Pedersen, P. L. (1971) J.
Biol. Chem., 246, 4987-4994.
2. Kagawa, Y., Sone, N., Yoshida, M., Hirata, H., and
Okamoto, H. (1976) J. Biochem. (Tokyo), 80, 141-151.
3. Foster, D. L., and Fillingame, R. H. (1979) J.
Biol. Chem., 254, 8230-8236.
4. Penefsky, H. S., Pullman, M. E., Datta, A., and
Racker, E. (1960) J. Biol. Chem., 235, 3330-3336.
5. Abrahams, J. P., Leslie, A. G. W., Lutter, R., and
Walker, J. E. (1994) Nature (London), 370, 621-628.
6. Foster, D. L., and Fillingame, R. H. (1982) J.
Biol. Chem., 257, 2009-2015.
7. Collinson, I. R., Runswick, M. J., Buchanan, S.
K., Fearnley, I. M., Skehel, J. M., van Raaij, M. J., Griffiths, D. E.,
and Walker, J. E. (1994) Biochemistry, 33, 7971-7978.
8. Mitchell, P. (1961) Nature (London),
191, 144-148.
9. Mitchell, P. (1974) FEBS Lett., 43,
189-194.
10. Senior, A. E. (1988) Physiol. Rev.,
68, 177-231.
11. Penefsky, H. S., and Cross, R. L. (1991) Adv.
Enzymol., 64, 173-214.
12. Boyer, P. D. (1993) Biochim. Biophys.
Acta, 1140, 215-250.
13. Pedersen, P. L., and Amzel, L. M. (1993) J.
Biol. Chem., 268, 9937-9940.
14. Cross, R. L., and Nalin, G. M. (1982) J.
Biol. Chem., 257, 2874-2881.
15. Kironde, F. A. S., and Cross, R. L. (1986) J.
Biol. Chem., 261, 12544-12549.
16. Kasahara, M., and Penefsky, H. S. (1978) J.
Biol. Chem., 253, 4180-4187.
17. Senior, A. E. (1979) J. Biol. Chem.,
254, 11319-11322.
18. Williams, N., Hullihen, J., and Pedersen, P. L.
(1987) Biochemistry, 26, 162-169.
19. Haddy, A. E., Frasch, W. D., and Sharp, R. R.
(1989) Biochemistry, 28, 3664-3669.
20. Pedersen, P. L., Williams, N., and Hullihen, J.
(1987) Biochemistry, 26, 8631-8637.
21. Bulygin, V. V., Syroeshkin, A. V., and
Vinogradov, A. D. (1993) FEBS Lett., 328, 193-196.
22. Ebel, R. E., and Lardy, H. A. (1975) J. Biol.
Chem., 250, 190-196.
23. Schuster, S. M., Ebel, R. F., and Lardy, H. A.
(1975) J. Biol. Chem., 250, 7848-7853.
24. Roveri, O. A., and Calcaterra, N. B. (1985)
FEBS Lett., 192, 123-127.
25. Wong, S. Y., Matsuno-Yagi, A., and Hatefi, Y.
(1984) Biochemistry, 23, 5004-5010.
26. Harris, D. A. (1989) Biochim. Biophys.
Acta, 974, 156-162.
27. Vasilyeva, E. A., Minkov, I. B., Fitin, A. F.,
and Vinogradov, A. D. (1982) Biochem. J., 202, 9-14.
28. Repke, K. R. H., and Schön, R. (1973)
Acta Biol. Med. Germ., 31, K-19-K-30.
29. Rosing, J., Kayalar, C., and Boyer, P. D. (1977)
J. Biol. Chem., 252, 2478-2485.
30. Cooper, C., and Lehninger, A. L. (1957) J.
Biol. Chem., 224, 547-560.
31. Selwyn, M. J. (1968) Nature (London),
219, 490-493.
32. Akimenko, V. K., Minkov, I. B., Bakeeva, L. E.,
and Vinogradov, A. D. (1972) Biokhimiya, 37, 285-293.
33. Adolfsen, R., and Moudrianakis, E. (1973)
Biochemistry, 12, 2926-2933.
34. Fleury, B., Di Pietro, A., Godinot, C., and
Gautheron, D. (1980) Biochimie, 62, 733-737.
35. Vasilyeva, E. A., Fitin, A. F., Minkov, I. B.,
and Vinogradov, A. D. (1980) Biochem. J., 188,
807-815.
36. Vinogradov, A. D. (1972) Biokhimiya,
49, 1220-1238.
37. Pullman, E. M., and Monroy, G. G. (1963) J.
Biol. Chem., 238, 3762-3769.
38. Panchenko, M. V., and Vinogradov, A. D. (1985)
FEBS Lett., 184, 226-230.
39. Gupta, R. K., Oesterling, R. M., and Mildvan, A.
S. (1976) Biochemistry, 15, 2881-2887.
40. Pullman, M. E., Penefsky, H. S., Datta, A., and
Racker, E. (1960) J. Biol. Chem., 235, 3322-3329.
41. Kotlyar, A. B., and Vinogradov, A. D. (1990)
Biochim. Biophys. Acta, 1019, 151-158.
42. Vasilyeva, E. A., Minkov, I. B., Fitin, A. F.,
and Vinogradov, A. D. (1982) Biochem. J., 202, 15-23.
43. Yalamova, M. V., Vasilyeva, E. A., and
Vinogradov, A. D. (1982) Biochem. Int., 4, 337-344.
44. Chance, B., and Nishimura, M. (1967) Meth.
Enzymol., 10, 641-650.
45. Shikama, K. (1971) Arch. Biochem.
Biophys., 147, 311-317.
46. Dawson, R. M. C., Elliott, D. C., Elliott, W.
H., and Jones, K. M. (1986) Data for Biochemical Research,
Clarendon Press, Oxford.
47. Gornall, A. G., Bardawill, C. J., and David, M.
M. (1949) J. Biol. Chem., 177, 751-766.
48. Hanley-Trawick, S., Carpen, M. E.,
Dunaway-Mariano, D., Pedersen, P. L., and Hullihen, J. (1989) Arch.
Biochem. Biophys., 268, 116-123.
49. Bossard, M. J., Vik, T. A., and Schuster, S. M.
(1980) J. Biol. Chem., 255, 5342-5346.
50. Bulygin, V. V., and Vinogradov, A. D. (1991)
Biochem. J., 276, 149-156.
51. Berger, G., Girault, G., Galmiche, J.-M., and
Pezennec, S. (1994) J. Bioenerg. Biomembr., 26,
335-346.
52. Pezennec, S., Berger, G., Andrianambinintsoa,
S., Radziszewski, N., Girault, G., Galmiche, J.-M., and Bäuerlein,
E. (1995) Biochim. Biophys. Acta, 1231, 98-110.
53. Schobert, B. (1992) J. Biol. Chem.,
267, 10252-10257.
54. Noji, H., Yasuda, R., Yoshida, M., and Kinosita,
K. (1997) Nature, 386, 299-302.
55. Guerrero, K. J., Xue, Z., and Boyer, P. D.
(1990) J. Biol. Chem., 265, 16280-16287.
56. Yoshida, M., and Allison, W. S. (1983) J.
Biol. Chem., 258, 14407-14412.
57. Burgard, S., Nett, J. H., Sauer, H. S., Kagawa,
Y., Schäfer, H.-J., Wise, J. G., Vogel, P. D., and Tommer, W. E.
(1994) J. Biol. Chem., 269, 17815-17819.
58. Zhou, J.-M., and Boyer, P. D. (1992)
Biochemistry, 31, 3166-3171.
59. Minkov, I. B., Vasilyeva, E. A., Fitin, A. F.,
and Vinogradov, A. D. (1980) Biochem. Int., 1,
478-485.
60. Syroeshkin, A. V., Vasilyeva, E. A., and
Vinogradov, A. D. (1995) FEBS Lett., 366, 29-32.
61. Lutter, R., Abrahams, J. P., van Raaij, M. J.,
Todd, R. J., Lundqvist, T., Buchanan, S. K., Leslie, A. G. W., and
Walker, J. E. (1993) J. Mol. Biol., 229, 787-790.
62. Böttcher, B., Gräber, P., Boekema, E.
J., and Lücken, U. (1995) FEBS Lett., 373,
262-264.
63. Kagawa, Y., Ohta, S., and Otawara-Hamamoto, Y.
(1989) FEBS Lett., 249, 67-69.
64. Miwa, K., and Yoshida, M. (1989) Proc. Natl.
Acad. Sci. USA, 86, 6484-6487.
65. Racker, E. (1977) Annu. Rev. Biochem.,
46, 1006-1014.
66. Jenkins, W. T., Marshall, M. M., and Lewin, A.
S. (1984) Arch. Biochem. Biophys., 232, 496-504.
67. Laubinger, W., and Dimroth, P. (1988)
Biochemistry, 27, 7531-7537.