Mini-REVIEW: Translation Termination and Its Regulation in Eukaryotes: Recent Insights Provided by Studies in Yeast
P. Mugnier and M. F. Tuite*
Department of Biosciences, University of Kent, Canterbury, Kent CT2 7NJ, UK; fax: 44 (0) 1227 763912; E-mail: M.F.Tuite@ukc.ac.uk* To whom correspondence should be addressed.
Received July 10, 1999
In protein synthesis, the arrival of one or other of the three stop codons in the ribosomal A-site triggers the binding of a release factor (RF) to the ribosome and subsequent polypeptide chain release. In eukaryotes, the RF is composed of two proteins, eRF1 and eRF3. eRF1 is responsible for the hydrolysis of the peptidyl-tRNA, while eRF3 provides a GTP-dependent function, although its precise role remains to be defined. Recent findings on translation termination and its regulation from studies in the yeast Saccharomyces cerevisiae are reviewed and the potential role of eRF3 is discussed.
KEY WORDS: yeast, translation termination, eukaryote release factor (eRF), nonsense-mediated mRNA decay
The termination of translation is not only the final key step in the translation of an mRNA, it is also the last of the three steps of protein synthesis to be unraveled by a combination of biochemical and genetic studies. Prior to 1994, a considerable body of genetic data had been forthcoming from studies on the yeast Saccharomyces cerevisiae, which implicated the products of several genes in translation termination [1]. The identification by Frolova et al. [2]of one of the key components of the eukaryotic translation termination machinery, namely eukaryotic release factor 1 (eRF1), resulted in renewed interest in translation termination in yeast. These, and parallel studies in higher eukaryotes, quickly established that, in addition to eRF1, a second release factor designated eRF3, was also part of the functional termination complex [3, 4]. In this review, we will discuss the contribution that genetic and biochemical studies of yeast have played in defining both the components and the control of translation termination in eukaryotes. Furthermore, we will explore the link between translation termination in yeast and the pathway by which mRNAs containing premature nonsense codons are degraded, i.e., the nonsense-mediated mRNA decay (NMD) pathway.
GENETIC ANALYSIS OF TRANSLATION TERMINATION IN YEAST
Genetic studies initiated over 30 years ago in yeast identified the genes which were subsequently shown to encode eRF1 and eRF3 (reviewed in [5]). The genetic screens employed were based on the suppression of defined nonsense codons, the rationale being that nonsense suppression would be much more efficient if the termination machinery was defective. The initial screens searched for mutants which resulted in the suppression of nonsense mutations in two or more unlinked genes, the so-called omnipotent suppressor class of mutants. A subsequent development of these assays was to employ strains carrying the weak nonsense (ochre-UAA) suppressor tRNASer encoded by the SUQ5 gene [6]. This suppressor tRNA is unable to compete with the endogenous wild-type termination machinery and thus cannot suppress a number of defined nonsense mutations, e.g., ade2-1. However, any mutation that creates a defective termination step results in the tRNA now being able to suppress the ade2-1 mutation. These simple, yet elegant, screens have allowed us to identify the genes encoding all of the key termination components in yeast.
The two genes first identified, SUP1 (subsequently designated SUP45) and SUP2 (subsequently designated SUP35), came from an omnipotent suppressor screen [7] and were identified as mutants able to suppress all three stop codons--UAA, UAG, and UGA--in the absence of a defined suppressor tRNA. Subsequently, Cox [6] expanded the search by isolating allosuppressor mutants that allowed SUQ5 to efficiently suppress a range of ochre mutations. These mutants mapped to one of five loci designated SAL1-SAL5 while a sixth gene (SAL6) was identified in a different genetic screen [8]. Subsequently, the SAL3 gene was shown to be allelic with the SUP35 gene [9] while the SAL4 gene was allelic with the SUP45 gene [10].
The SUP35 gene [11, 12] and the SUP45 gene [13, 14] are both essential genes in S. cerevisiae, suggesting that they encode factors that play a crucial and essential role in translation termination. The products of these two genes, originally defined as Sup35p and Sup45p, are in fact eRF3 and eRF1 [2-4] and this nomenclature will be used below.
eRF1 IS THE PRIMARY EFFECTOR OF TRANSLATION TERMINATION IN EUKARYOTES
The first direct evidence that eRF1 is a release factor came from the studies of Frolova et al. [2] using human and Xenopus laevis proteins that showed significant homology to Sup45p. Both proteins were able to promote the in vitro hydrolysis of fMet-tRNAMet in the presence of ribosomes and any of the three stop codons. Thus, unlike in prokaryotes where release factors show codon specificity, the eukaryotic release factor was not nonsense codon specific. This biochemical finding is in keeping with the observed genetic behavior of SUP45 mutants in yeast, i.e., omnipotent suppression.
eRF1 functionally interacts with eRF3, both in vivo and in vitro, to promote translation termination at all three stop codons [3, 4] and both the structure and function of eRF1 are conserved among eukaryotes, from yeast to man [2, 4, 15]. For example, expression of the human and Xenopus eRF1 can complement the otherwise lethal phenotype association with a SUP45 gene deletion in yeast [15]. Furthermore, overexpression of either the human or Xenopus eRF1, together with overexpression of yeast eRF3, will significantly reduce the efficiency of tRNA-mediated nonsense suppression in vivo in yeast [4] indicating a functional interaction between the heterologous factors.
The elongation factor eEF1alpha-like C-terminal domain of eRF3 is also evolutionarily conserved and a thermosensitive variant of yeast eRF3 can be complemented by its Xenopus homologue in vivo [3]. eRF3 is a guanosine triphosphatase which is unable to promote in vitro termination on its own, although it can enhance polypeptide chain release, promoted by eRF1, in a GTPase-dependant manner [3, 16]. Both eRF1 and the ribosome are required for the GTPase activity of eRF3, suggesting that they may play the role of a composite GTPase Activating Protein [16].
Deletions of the C-terminal 28 amino acids of Schizosaccharomyces pombe eRF1 [17] or amino acids of the S. cerevisiae eRF1 protein [18] result in cell death. Using the two-hybrid system, Stansfield et al. [4] showed that eRF1 and eRF3 can interact in vivo. Subsequently, different groups have studied the effect of C-terminal truncations of yeast and human eRF1 on their ability to bind eRF3 using either an in vitro pull-down assay or the two-hybrid system in vivo [17-20]. Although the methodology used is different in each study and the C-terminal length of eRF1 is relatively species specific, these analyses have revealed that deletion of the last 19 (S. cerevisiae [18]) to 27 (H. sapiens [19]) amino acids of eRF1 results in the loss of its eRF3-binding properties both in vivo and in vitro (Fig. 1). Such deletions alter a conserved GFGGIGxxxRY motif present at the C-terminus of eRF1 [18] which suggests that it might play a key role in eRF1 binding to eRF3. Also, the high number of acidic residues present in the eRF1 C-terminal 20 amino acids are removed in such truncated mutants. To study the importance of such acidic residues in the binding to eRF3, Ebihara et al. [20] mutated five of these residues into alanine and shown reduction of binding to eRF3. By analyzing N-terminally truncated eRF1 proteins in the two-hybrid assay, two groups have also identified amino acid residues around position 300 as an eRF3-binding site [17, 19] (Fig. 1).
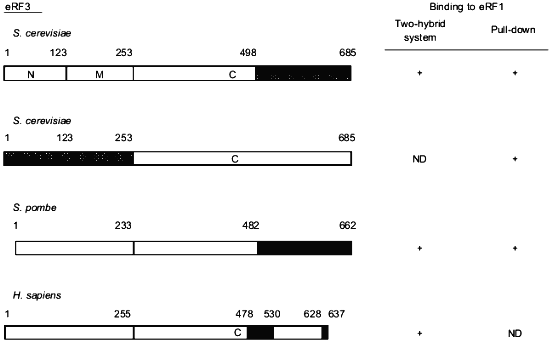
The eRF1-binding domain of eRF3 has also been delineated in different species (Fig. 2). Taken together, the results show that the eRF1-binding domain of eRF3 is distinct from its GTP-binding domain (amino acids residues ca. 250 to 400) in S. cerevisiae [3] which is in agreement with the fact that GTP-binding is not a pre-requisite for eRF3 to bind to eRF1 [17, 18, 20].Fig. 1. Critical regions in eRF1 for the binding to eRF3. Data are for Saccharomyces cerevisiae [18], Schizosaccharomyces pombe [17], and Homo sapiens [19]. Binding properties of eRF1 were tested either in vivo in yeast using the two-hybrid system or in vitro using eRF1 domains tagged with 6 histidines or the glutathione S-transferase protein pull down. The tagged proteins were bound to beads coated with nickel or glutathione, respectively. The capacity of these proteins to "pull down" distinctly overexpressed or purified eRF3 was tested. This comparison shows that, in all three species, the eRF3 binding domain of eRF1 is confined to the C-terminal third of the protein. The domains involved are represented by hatched boxes.
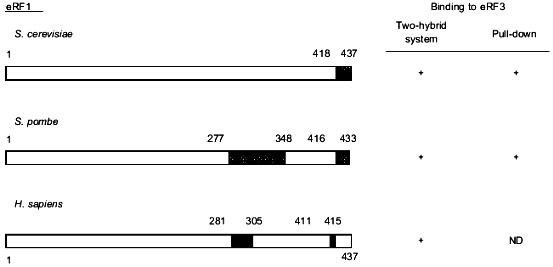
Fig. 2. Critical regions in eRF3 for the binding to eRF1. Data are for Saccharomyces cerevisiae [20, 29], Schizosaccharomyces pombe [20], and Homo sapiens [19]. The methods used for these assignments are similar to those described in the legend to Fig. 1. Data from Paushkin et al. [29] have been obtained using eRF3 domains overproduced in yeast and confirmed by using the NM domain of yeast eRF3 purified from a [PSI+] strain. Comparative data presented here also show that the eRF1 binding domain of eRF3 is situated in the C-terminal part of the eEF1alpha-like domain of eRF3.
WHAT IS THE ROLE OF eRF3 IN TRANSLATION TERMINATION?
eRF1 alone can promote the release of the elongated polypeptide from the ribosomal P-site when a stop codon is present in the A-site in vitro [2, 3]. The addition of eRF3 only renders the reaction GTP-dependant since the addition of eRF3 alone blocks the polypeptide release [3]. However, eRF3 and GTP are required for the reaction when lower mRNA concentrations are used in the assay. Using an in vitro translation assay, Drugeon et al. [21] showed that elevated levels of eRF1 alone can outcompete, i.e., antisuppress a suppressor tRNA at a stop codon and that eRF3 only weakly potentiates this effect. In agreement with these results it has been shown that the overexpression of eRF1 in human cells has an antisuppression effect, but that the concomitant expression of eRF3 does not enhance this effect [22]. However, results in yeast clearly indicate that overexpression of both eRF1 and eRF3 is required to promote antisuppression [4]. These contradictory data from this type of in vivo assay may simply reflect that different species are used and require specific tRNAs and specific reporter genes. Thus, a number of technical differences may be responsible for the discrepancy, including the efficacy and amount of suppressor tRNA, the level of over-expression of the release factors and the resulting ratios of tRNASUP/eRF1. It remains to be established whether or not there is a fundamental difference in the function of eRF3 in yeast and higher eukaryotes.
Studies in yeast have shown that the level of eRF1 can significantly affect translation termination efficiency [23]. An excess of eRF1 with respect to the suppressor tRNA and its strength could be sufficient to detect an antisuppressor phenotype, as suggested previously [22, 24], while a low relative amount of eRF1 would require eRF3 to promote antisuppression as suggested earlier [25]. Intriguingly, although Le Goff et al. checked the stability of the nonsense cat mRNA used in their antisuppression assay when eRF3 was overexpressed, the stability of the nonsense cat mRNA was not monitored during eRF1 overexpression [22]. Considering the connection which exists between translation termination and nonsense-mediated mRNA decay (NMD)--as shown by elegant studies in yeast [26]--one cannot exclude that an NMD-like pathway is triggered by overexpression of eRF1 in human cells and thus results in degradation of the nonsense mRNA, resulting in a low CAT expression that was wrongly interpreted as antisuppression.
Two reports have described an interaction between the N terminal domain of eRF3 and the full length eRF1 (Fig. 2). In the first study, Hoshino et al. [27] used the two-hybrid system, but results that contradict their finding have subsequently been published [28] without a clear explanation for the difference in the findings. In a previous report, Paushkin et al. [29] showed that a GST-eRF1 fused protein, expressed in E. coli, can pull down the (N + M)-domain of eRF3 (eRF3NM) overexpressed in S. cerevisiae, or precipitate semi-purified eRF3NM from a yeast lysate. Such an interaction between the (N + M)-domain of S. cerevisiae eRF3 prepared from E. coli and eRF1 was not detected [20, 29]. Ebihara et al. [20] proposed that an adapter protein such as Upf1p, a protein implicated in the NMD pathway, could have been pulled-down from the yeast lysate to allow the indirect interaction (via the formation of a ternary complex) between the eRF3 N-terminal domain and eRF1. However, such a ternary complex has never been detected using the two-hybrid system [19, 20] and the addition of yeast lysate to the eRF1--eRF3NM binding reaction did not allow these two proteins to interact when eRF3NM was purified from E. coli [29]. Although it cannot be ruled out that indeed a third factor played the role of an adapter to give this unexpected result, it is likely, as suggested earlier [29], that a conformational difference due to the different origins of the two eRF3NM could explain this behavior.
eRF3 IN YEAST IS A PRION PROTEIN
In addition to its native structure, eRF3 can also take up an alternative structural form in yeast [30]. This altered protein structure, and the high-molecular-weight aggregates that they form, are inherited in a non-Mendelian manner through cytoplasmic exchange during cell division in a manner that parallels the behavior of the mammalian prion protein, PrP [31, 32]. This prion-associated phenotype, called the [PSI+] factor, results in a depletion of the soluble fraction of eRF3 [31, 33] and a subsequent termination defect as detected by a nonsense suppressor-based assay [34]. Decoding of a stop codon by a nonsense suppressor tRNA is hence facilitated, giving [PSI+] strains an allosuppressor phenotype.
The N-terminal domain of eRF3 in S. cerevisiae is responsible for [PSI+] maintenance [35] but it is not required for cell viability or termination efficiency [25]. The function of the protein is conferred by the essential eEF1alpha-like C-terminal domain (Fig. 2) [25]. Since the exact function of eRF3 in translation termination is unknown, it is not yet clear why the [PSI+] phenotype results in a termination defect. eRF1 and Upf1p could be sequestered in the eRF3 particles [26, 29] resulting in a concomitant depletion of these two other termination factors. In this case the remaining eRF1 and Upf1p available in the soluble fraction [26, 29, 33] might account for the "basal" and sufficient-for-viability termination activity observed in a [PSI+] strain. However, this model is weakened by the fact that eRF1 overexpression does not reverse the allosuppressor phenotype of a [PSI+] strain but rather enhances it [24].
It is most likely that the termination defect observed in a [PSI+] strain results from the loss of the main termination function of eRF3 itself, which is located in its C-terminal domain and which also defines the essential protein. Since there is approximately one eRF1 molecule per 20 ribosomes [36], it has already been speculated that the essential C terminal domain of eRF3 is required to recycle eRF1 to the ribosome and to allow efficient termination [29]. Indeed, eRF3 is required together with GTP to promote efficient polypeptide chain release in vitro when the mRNA is added at a non-saturating concentration [3].
The fact that the eRF3 NM domain produced from a [PSI+] strain can interact with eRF1 in vitro, and the finding that eRF1 can co-precipitate with eRF3NM aggregates in vivo [29] indicate that eRF1 might interact with the NM domain of eRF3 when it is in its prion conformation. In a recent model, Kisselev and Frolova [37] proposed that the N-terminal domains of eRF1 and eRF3 are free in the heterodimeric eRF complex, while C-terminal domains interact. Since the N-terminal domain of RF2 in E. coli is believed to carry the peptidyl-tRNA hydrolysis activity [38] such an interaction between the N-domain of eRF1 and the N-domain and eRF3[PSI+] could alter eRF1 release activity. Although this model needs to be confirmed experimentally, it would provide an additional explanation for the allosuppressor phenotype observed in a [PSI+] strain.
WHAT IS THE PHYSIOLOGICAL SIGNIFICANCE OF REDUCED TERMINATION EFFICIENCY
IN [PSI+] STRAINS?
The advantage conferred on a cell by the [PSI+] prion has been proposed to be the synthesis of protein which would not have been produced in a [psi-] background, i.e., translation of mRNAs containing a nonsense codon within their sequence or by-passing of native stop codons to produce extended proteins [39]. Phenotypically, [PSI+] confers upon cells resistance to heat and ethanol stress [33] and, indeed, it allows synthesis of a heat shock transcription factor which contains a nonsense codon within its coding sequence [40]. This proposed mechanism could be extended to other mRNAs whose translation is indeed increased in stress conditions. For example, genes like GCN4 [41] or YAP1 and YAP2 [42] are regulatory genes used by the cells to protect themselves against stress (namely, amino acid starvation and chemicals stress, respectively). The 5´-UTR of the corresponding mRNAs contain short upstream open reading frames (uORF) which act to block the ribosome scanning down to the main ORF [43] or to accelerate decay of the mRNA [44]. In strains with a [PSI+]-associated termination defect, low-level read-through of the termination codon of these uORFs might allow for more efficient translation of such stress response mRNAs. The validity of this model remains to be tested.
WHAT IS THE FUNCTION OF THE eRF3 NM DOMAIN?
All eRF3 proteins described so far contain a variable N terminal domain [37]. Its primary function is unlikely to be to catalyze prion formation, since such a property has not been described for eRF3 in species other than S. cerevisiae. However, this domain is not essential for cell viability in yeast [25] and its deletion results in a nonsense suppression phenotype only in Podospora anserina [45]. The effect of this truncation might actually be more general and, as suggested above, the fact that suppression assays used in different species cannot be standardized [4, 22, 45], could account for the apparent discrepancies between the findings in these different fungal species.
eRF3 binds to Upf1p more efficiently in vitro than eRF1 and Upf1p is sequestered together with eRF1 in eRF3 aggregates in vivo in [PSI+] strains [26]. It is, therefore, possible that this interaction allows a functional link between termination and the termination-associated nonsense-mediated mRNA decay (NMD) pathway involving the Upfp proteins--Upf1p, Upf2p, and Upf3p [46]. Upf1p is an RNA binding protein displaying an RNA-dependant ATPase activity. In vitro, it has 5´--3´ helicase properties which are ATPase-dependent and are believed to resolve mRNA structures in the NMD pathway before they are degraded by exonucleases [47]. Disruption of the non-essential UPF1 gene results in the stabilization of nonsense-containing mRNA transcripts and suppression of mutant nonsense codons [48]. To verify that the latter was not simply a consequence of the former, Weng et al. [48] used truncated and mutant forms of Upf1p and showed that some mutations in the N-terminal cysteine- and histidine-rich region specifically led to nonsense suppression without altering mRNA decay. These results suggest that Upf1p is responsible for an important step in pre-termination. However, prior efficient termination is required for NMD to occur [49]. Since eRF3 and RNA compete for interaction with Upf1p [26], it has been proposed that the interaction of Upf1p with the termination complex on the ribosome is required for the subsequent binding of the RNA helicase Upf1p to the mRNA once the termination complex has left [46].
eRF3 MAY LINK TERMINATION AND INITIATION STEPS IN PROTEIN SYNTHESIS
It has been shown recently that the N-terminal domain of human eRF3 can interact with the C-terminal domain of the human poly(A)-binding protein (PABP) [28]. The interaction of this latter protein with eIF4G and the CAP-binding complex stabilizes 5´-capped poly(A)-tailed mRNAs and thus stimulates their translation [50]. PABP acts as a homopolymer to stabilize the 3´-poly(A) tail of mRNAs. Its interaction with mRNA is mediated by the four RNA recognizing motifs (RRM) whereas its C-terminal domain is responsible for homodimerization [51]. If this finding is confirmed in other species, the PABP--eRF3 interaction might play a role in destabilizing PABP dimers and subsequent poly(A) protection and interaction with the CAP-binding complex. Such a destabilization of the mRNA would increase its susceptibility to exonucleases and participate in sense or nonsense mRNA decay pathways [28, 51]. The interaction of the human PABP with the variable N-terminus of eRF3 raises the question of the existence of such an interaction in other species including yeast. This remains to be established.
It is now believed that the full-length eRF3 protein has four associated activities: eRF1 binding, GTPase activity, Upf1p binding, and PABP binding.
In addition, this protein may play a role in controlling the G1 to S phase transition of the yeast cell cycle [52] although this effect is most likely to be indirect. The main experimental issue is now to determine if all these features are carried by the eEF1alpha-like C-terminal domain or if the N and/or M domains are also involved. Concerning eRF1, a role has been assigned only to its C-terminal domain and molecular elements involved in the stop codon recognition and the peptidyl-tRNA hydrolysis have not been identified so far. Three dimensional structure determination and structure--function analysis using site-directed mutants should provide us with this information, while further exploitation of genetic analysis in yeast will undoubtedly reveal new insights into eukaryotic translation termination and its regulation.
The work in the author's laboratory on translation termination in yeast is supported by project grants from the BBSRC and The Wellcome Trust.
REFERENCES
1.Stansfield, I., and Tuite, M. F. (1995) Curr.
Genetics, 25, 385-395.
2.Frolova, L., Le Goff, X., Rasmussen, H. H.,
Cheperegin, S., Drugeon, G., Kress, M., Arman, I., Haenni, A.-L.,
Celis, J. E., Philippe, M., Justesen, J., and Kisselev, L. (1994)
Nature, 372, 701-703.
3.Zhouravleva, G., Frolova, L., Le Goff, X., Le
Guellec, R., Inge-Vechtomov, S., Kisselev, L., and Philippe, M. (1995)
EMBO J., 14, 4065-4072.
4.Stansfield, I., Kones, K. M., Kushnirov, V. V.,
Dagkesamanskaya, A. R., Poznyakovski, A. I., Paushkin, S. V., Nierras,
C. R., Cox, B. S., Ter-Avanesyan, M. D., and Tuite, M. F. (1995)
EMBO J., 14, 4365-4373.
5.Hawthorne, D. C., and Leupold, U. (1974) Curr.
Top. Microbiol. Immunol., 64, 1-47.
6.Cox, B. (1977) Genet. Res., 30,
187-205.
7.Inge-Vechtomov, S. G., and Andrianova, V. M. (1970)
Genetika, 6, 103-115.
8.Song, J. M., and Liebman, S. W. (1987)
Genetics, 115, 451-460.
9.Crouzet, M., and Tuite, M. F. (1987) Mol. Gen.
Genet., 210, 581-583.
10.Crouzet, M., Izgu, F., Grant, C. M., and Tuite,
M. F. (1988) Curr. Genet., 14, 537-543.
11.Kushnirov, V. V., Ter-Avanesyan, M. D., Telckov,
M. V., Surguchov, A. P., Smirnov, V. N., and Inge-Vechtomov, S. G.
(1988) Gene, 66, 45-54.
12.Wilson, P. G., and Culbertson, M. R. (1988) J.
Mol. Biol., 199, 559-573.
13.Breining, P., and Piepersberg, W. (1985)
Nucleic Acids Res., 14, 5187-5197.
14.Himmelfarb, H. J., Maicas, E., and Friesen, J. D.
(1985) Mol. Cell. Biol., 5, 816-822.
15.Urbero, B., Eurwilaichitr, L., Stansfield, I.,
Tassan, J. P., Le Goff, X., Kress, M., and Tuite, M. F. (1997)
Biochimie, 79, 27-36.
16.Frolova, L., Le Goff, X., Zhouravleva, G.,
Davydova, E., Philippe, M., and Kisselev, L. (1996) RNA,
2, 334-341.
17.Ito, K., Ebihara, K., and Nakamura, Y. (1998)
RNA, 4, 958-972.
18.Eurwilaichitr, L., Graves, F. M., Stansfield, I.,
and Tuite, M. F. (1999) Mol. Microbiol., 32, 485-496.
19.Merkulova, T. I., Frolova, L. Y., Lazar, M.,
Camonis, J., and Kisselev, L. L. (1999) FEBS Lett., 443,
41-47.
20.Ebihara, K., and Nakamura, Y. (1999) RNA,
5, 739-750.
21.Drugeon, G., Jean-Jean, O., Frolova, L., Le Goff,
X., Philippe, M., Kisselev, L., and Haenni, A.-L. (1997) Nucleic
Acids Res., 25, 2254-2258.
22.Le Goff, X., Philippe, M., and Jean-Jean, O.
(1997) Mol. Cell. Biol., 17, 3164-3172.
23.Stansfield, I., Eurwilaichitr, L., Akhmaloka, and
Tuite, M. F. (1995) Mol. Microbiol., 20, 1135-1143.
24.Derkatch, I. L., Bradley, M. E., and Liebman, S.
W. (1998) Proc. Natl. Acad. Sci. USA, 95, 2400-2405.
25.Ter-Avanesyan, M. D., Kushnirov, V. V.,
Dagkesamanskaya, A. R., Didichenko, S. A., Chernoff, Y. O.,
Inge-Vechtonov, S. G., and Smirnov, V. N. (1993) Mol.
Microbiol., 7, 683-692.
26.Czaplinski, K., Ruiz-Echevarria, M. J., Paushkin,
S. V., Han, X., Weng, Y., Perlick, H. A., Dietz, H. C., Ter-Avanesyan,
M. D., and Peltz, S. W. (1998) Genes Dev., 12,
1665-1677.
27.Hoshino, S., Imai, M., Mizutani, M., Kikuchi, Y.,
Hanaoka, F., and Katada, T. (1998) J. Biol. Chem., 273,
22254-22259.
28.Hoshino, S., Imai, M., Kobayashi, T., Uchida, N.,
and Katada, T. (1999) J. Biol. Chem., 274,
16677-16680.
29.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, M. D. (1997) Mol. Cell. Biol., 17,
2798-2805.
30.Glover, J. R., Kowal, A. S., Schirmer, E. C.,
Patino, M. M., Liu, J. J., and Lindquist, S. (1997) Cell,
89, 811-819.
31.Patino, M. M., Liu, J. J., Glover, J. R., and
Lindquist, S. (1995) Science, 273, 622-626.
32.Wickner, R. B. (1994) Science, 264,
566-569.
33.Eaglestone, S. S., Cox, B. S., and Tuite, M. F.
(1999) EMBO J., 18, 1974-1981.
34.Cox, B. S. (1965) Heredity, 20,
505-521.
35.Ter-Avanesyan, M. D., Dagkesamanskaya, A. R.,
Kushnirov, V. V., and Smirnov, V. N. (1994) Genetics,
137, 671-676.
36.Stansfield, I., Grant, C. M., Akhmaloka, and
Tuite, M. F. (1992) Mol. Microbiol., 6, 3469-3478.
37.Kisselev, L. L., and Frolova, L. (1999)
Biochemistry (Moscow), 64, 8-16.
38.Moffat, J. G., and Tate, W. P. (1994) J. Biol.
Chem., 269, 18899-18903.
39.Lindquist, S. (1997) Cell, 89,
495-498.
40.Lindquist, S., and Kim, G. (1995) Proc. Natl.
Acad. Sci. USA, 93, 5301-5306.
41.Hinnebusch, A. G. (1995) in Translational
Control (Hershey, J. W. B., Mathews, M. B., and Sonenberg, N.,
eds.) Cold Spring Harbour Laboratory Press, pp. 199-244.
42.Bossier, P., Fernandes, L., Rocha, D., and
Rodrigues-Pousada, C. (1993) J. Biol. Chem., 268,
23640-23645.
43.Mueller, P. P., and Hinnebusch, A. G. (1985)
Cell, 45, 201-207.
44.Vilela, C., Velasco Ramirez, C., Linz, B.,
Rodrigues-Pousada, C., and McCarthy, J. E. G. (1999) EMBO J.,
18, 3139-3152.
45.Gagny, B., and Silar, P. (1998) Genetics,
149, 1763-1775.
46.Culbertson, M. R. (1999) Trends Genet.,
15, 74-80.
47.Czaplinski, K., Weng, Y., Hagan, K. W., and
Peltz, S. W. (1995) RNA, 1, 610-623.
48.Weng, Y., Czaplinski, K., and Peltz, S. W. (1995)
Mol. Cell. Biol., 16, 5491-5506.
49.Losson, R., and Lacroute, F. (1979) Proc.
Natl. Acad. Sci. USA, 76, 5134-5137.
50.Otero, L. J., Ashe, M. P., and Sachs, A. B.
(1999) EMBO J., 18, 3153-3163.
51.Kuhn, U., and Pieler, T. (1995) J. Mol.
Biol., 256, 20-30.
52.Kikuchi, Y., Shimatake, H., and Kikuchi, A.
(1988) EMBO J., 7, 1175-1172.