Overexpression and Purification of Recombinant eRF1 Proteins of Rabbit and Tetrahymena thermophila
A. L. Karamyshev1, Z. N. Karamysheva1,2, K. Ito1, S. Matsufuji2, and Y. Nakamura1*
1Department of Tumor Biology, Institute of Medical Science, University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8630, Japan; fax: +81 3 5449 5415; E-mail: nak@ims.u-tokyo.ac.jp2Department of Biochemistry II, Jikei University, School of Medicine, 3-25-8 Nishishinbashi, Minato-ku, Tokyo 105-8461, Japan; fax: +81 3 3436-3897; E-mail: senya@jikei.ac.jp
* To whom correspondence should be addressed.
Received June 10, 1999
The polypeptide release factor (eRF1) gene was cloned from rabbit and its overexpression and purification system was established in parallel with that of the eRF1 gene of Tetrahymena thermophila that has been cloned recently in this laboratory. The rabbit eRF1 (Ra-eRF1) is composed of 437 amino acids and is completely identical to human eRF1 though 3% distinct in the nucleotide sequence. This is in sharp contrast to Tetrahymena eRF1 (Tt-eRF1) that is only 57% identical to human eRF1. The recombinant Ra-eRF1 was marked with a histidine tag, overexpressed, and purified to homogeneity by two-step chromatography using Ni-NTA-agarose and Mono Q columns. In contrast to Ra-eRF1, Tt-eRF1 formed aggregates upon overexpression in Escherichia coli, hence it was purified under denaturing conditions, and used to raise rabbit antibody. The resulting anti-Tt-eRF1 antibody proved useful for examining conditions for soluble Tt-eRF1 in test cells. Finally, a soluble Tt-eRF1 fraction was purified from Saccharomyces cerevisiae transformed with the Tt-eRF1 expression plasmid by three steps of affinity and anion exchange chromatography. The cloned Ra-eRF1 gene complemented a temperature-sensitive allele in the eRF1 gene, sup45 (ts), of S. cerevisiae, though the complementation activity was significantly impaired by the histidine tag, whereas Tt-eRF1 failed to complement the sup45 (ts) allele.
KEY WORDS: translation termination, polypeptide release factors, rabbit eRF1, Tetrahymena eRF1
Termination of translation during protein biosynthesis takes place on the ribosome in response to a stop, rather than a sense, codon in the decoding site. This process requires two classes of polypeptide release factors (RFs): a class I factor, codon-specific RF (RF1 and RF2 in prokaryotes; eRF1 in eukaryotes), and a class II factor, nonspecific RF (RF3 in prokaryotes; eRF3 in eukaryotes) that binds guanine nucleotides and stimulates class I RF activity [1, 2]. Prokaryotic RF1 and RF2 recognize UAG/UAA and UGA/UAA, respectively [3], and known eukaryotic eRF1s recognize all three stop codons [4].
Although the termination process was first discovered in the late 1960s, much of the mechanism has remained obscure. One of the reasons for relatively slow progresses in the study of translation termination mechanisms was the lack of a complete in vitro system that catalyzes initiation, elongation, and termination of protein synthesis equivalent to the in vivo process. The classical, but still useful, in vitro system of translation termination was to monitor the rate of N-formyl-methionine (fMet) release from the ribosome--fMet--tRNA complex at the stop codon [5, 6]. Obviously, this is not sufficient to investigate the whole mechanism of protein termination or to clarify the interplay between translation termination and the elongation or initiation step of protein synthesis. It was only recently in eukaryotes that a competition against suppressor tRNAs of exogenous eRF1 was investigated in the rabbit reticulocyte lysate system by monitoring terminated and readthrough products in polyacrylamide gel electrophoresis [7]. In prokaryotes the in vitro system is more advanced, but it was only recently that the complete and recycling translation system was reconstituted with initiation, elongation, and termination factors and used to analyze termination and recycling steps of protein synthesis in vitro [8, 9].
In this study, we cloned the rabbit eRF1 gene to facilitate the in vitro translation system composed of rabbit reticulocyte lysate under homologous conditions. One of the assay systems that we are engaged in the construction of is to measure competition between translation termination and frameshifting using the mammalian antizyme frameshift construct of mRNA (unpublished). Cloning and purification of rabbit eRF1, designated Ra-eRF1, is along this line.
Moreover, once an efficient and sensitive termination assay is available in the complete in vitro translation system, it should provide us with a powerful tool to investigate the mechanism of the stop codon reassignment discovered in Tetrahymena, where UAA/UAG codons are decoded as glutamine [10-13]. It is not fully understood how UAA/UAG termination is suppressed efficiently by two glutaminyl-tRNAs cognate to UAA and/or UAG in the presence of eRF1 that is generally omnipotent to three stop codons in eukaryotes. Toward this aim, we recently cloned the eRF1 cDNA from Tetrahymena thermophila [14]. Here, we developed a comparative study of overexpression and purification of recombinant eRF1 proteins of both rabbit and Tetrahymena.
MATERIALS AND METHODS
Strains, plasmids, and media. Strains and plasmids used are listed in Table 1. Yeast cells were grown in YPD or synthetic complete (SC) media [18], and Escherichia coli cells were grown in LB medium supplemented with relevant antibiotics for selection (50 µg/ml ampicillin, 15 µg/ml tetracycline, or 50 µg/ml kanamycin) [19].
Table 1. Strains and plasmids used in this
study
Cloning of rabbit eRF1 cDNA. The primers used for reverse transcription and polymerase chain reaction (PCR) are listed in Table 2. The hybridization screening of the lambda ZapR II rabbit cDNA library (Stratagene, USA) for Ra-eRF1 cDNA was carried out using the human eRF1 cDNA probe (a gift from Dr. S. Hoshino) according to standard procedures [19]. The ECL kit (Amersham, UK) was used for detection of positive clones according to the manufacturer's instructions. For positive clones, in vivo excision of the pBluescript phagemid from the lambda ZapR II vector was done using ExAssist/SOLR system (Stratagene, USA). Hybridization screening was combined with the 5´ and 3´ RACE method essentially as described [20] using the 5´-RACE System for Rapid Amplification of cDNA Ends (GibcoBRL, USA). Reverse-transcribed total cDNA was prepared from rabbit muscle mRNA. For 3´ RACE, primers Z17 and A51 were used. Two 3´ RACE products (1.4 and 1 kb) that differ in 3´-UTR length were cloned into EcoRI/SmaI sites of pUC118. 5´ RACE products were amplified using 5´-RACE Abridged Anchor Primer and primer Z23. The complete Ra-eRF1 cDNA was amplified by PCR from total cDNA using primers Z1 and Z25. PCR product (1.3 kb) was subcloned into EcoRI/NotI sites of the pET-30a(+) expression vector, resulting in pR-eRF1-25/1. The Ra-eRF1 cDNA sequence was confirmed by DNA sequencing on both strands using ABI PRISMTM dye terminator cycle sequencing ready reaction kit (Perkin Elmer, USA).
Overexpression of recombinant eRF1s. For Ra-eRF1 expression, E. coli BL21 (DE3) strain was transformed with plasmid pR-eRF1-25/1 containing eRF1 cDNA with a histidine (His6) tag at the N-terminus under T7 promoter. Transformants were grown to OD600 = 0.5 and induced with 1 mM isopropyl beta-D-thiogalactoside (IPTG) for 4 h at 37°C. For Tetrahymena eRF1 (designated Tt-eRF1) expression in E. coli BL21 (DE3), the coding part of Tt-eRF1 cDNA was recloned into pET-30b(+) vector under the T7 promoter, giving rise to two plasmid constructs (pTT-eRF1-38/1 and pTT-eRF1-39/1) with or without the His6 tag at the N-terminus, respectively. Cells were grown to OD600 = 0.5 and induced with 0.1 mM IPTG for 10 h at 25°C. For Tt-eRF1 expression in the budding yeast Saccharomyces cerevisiae the coding part of Tt-eRF1 cDNA marked with the N-terminal His6 tag was recloned into pYES2 vector under the PGAL1 promoter, resulting in pTT-eRF1-4/12. S. cerevisiae YPH499 cells transformed with pTT-eRF1-4/12 were grown at 25°C in SC medium lacking uracil and supplemented with galactose (for induction of PGAL1 promoter).
Purification of recombinant rabbit eRF1. Recombinant Ra-eRF1 was isolated from the soluble fraction of bacterial lysate using Ni-NTA-agarose (Qiagen, Germany) according to the manufacturer's instruction and purified on Mono Q column (Pharmacia, Sweden) by linear gradient 0.15-1.00 M KCl in buffer containing 50 mM Tris-HCl (pH 7.5) and 2 mM DTT. Ra-eRF1 fractions were combined, dialyzed against 50 mM Tris-HCl (pH 7.5), 50 mM KCl, 2 mM DTT, and 5% glycerol, and stored at -80°C.
Purification of the recombinant Tetrahymena eRF1 under denaturing conditions. Overexpressed Tt-eRF1 formed inclusion bodies in E. coli and was isolated under denaturing conditions from E. coli BL21(DE3) cells transformed with plasmid pTT-eRF1-39/1. The inclusion bodies were collected by low speed centrifugation after cell lysis, washed with solution containing 2% Triton X-100, 10 mM EDTA, and 10 mM Tris-HCl (pH 8.0), and solubilized in 10 mM Tris-HCl (pH 8.0) solution containing 8 M urea. eRF1 was further purified by FPLC anion exchange chromatography using Mono Q column with linear gradient of 0-0.75 M NaCl in buffer containing 10 mM Tris-HCl (pH 8.0) and 8 M urea.
Purification of the recombinant Tetrahymena eRF1 under non-denaturing conditions. The native recombinant Tt-eRF1 protein was purified from S. cerevisiae YPH499 cells transformed with plasmid pTT-eRF1-4/12. Cells were disrupted by French Press in buffer containing 50 mM NaH2PO4 (pH 7.7), 300 mM NaCl, 10 mM MgCl2, 20 mM imidazole, 5 mM beta-mercaptoethanol, and 1 mM phenylmethylsulfonyl fluoride. Cell debris was removed by low speed centrifugation, and ribosomes were collected by centrifugation at 130,000g, and the resulting post-ribosomal supernatant was applied to chromatography in Ni-NTA-agarose (Qiagen) according to the manufacturer's instruction. Fractions containing Tt-eRF1 were subjected to chromatography in AF-Heparin Toyopearl 650 M (Tosoh Co.) with a linear gradient of 0.05-1.0 M NaCl in buffer containing 50 mM Tris-HCl (pH 7.6), 10 mM MgCl2, and 5 mM beta-mercaptoethanol. Tt-eRF1 was finally purified by anion exchange chromatography on a Mono Q column by linear gradient of 0-0.8 M NaCl in buffer containing 20 mM Tris-HCl (pH 8.0) and 1 mM DTT. Purified protein was dialyzed against 20 mM Tris-HCl (pH 7.6), 25 mM KCl, and 1 mM DTT.
Other DNA techniques. Site-directed mutagenesis was performed using Altered Sites II in vitro Mutagenesis System (Promega, USA) and primers A23 through A32 (see Table 2). The DNA fragments of the coding region of Tt-eRF1 cDNA for expression constructs were amplified by PCR using sets of primers A33 and A34 or A34 and A35 (Table 2). The preparation of rabbit RNA was conducted using QuickPrep (Pharmacia) according to the manufacturer's instruction. Yeast transformation with plasmid DNA was done using the lithium acetate method [21]. Other DNA manipulations were conducted according to standard methods [19].
Table 2. List of oligonucleotides
RESULTS AND DISCUSSION
Cloning of rabbit eRF1. A rabbit cDNA library was screened using a human eRF1 cDNA as probe, and one positive clone was isolated. It contained a 327-bp insert, and the deduced coding sequence was completely identical to human eRF1 sequence [4]. Based on this sequence information, 5´ and 3´ RACE were conducted, giving rise to one 5´ RACE product (0.9 kb) and two 3´ RACE products with sizes of 1.4 and 1.0 kb. Sequences of both 3´ RACE products were identical except for the 3´ UTR length; i.e., 861 and 514 bp sequences preceding poly(A), suggesting alternative polyadenylation at distinct sites (see Fig. 1). The 5´ and 3´ UTR sequences of Ra-eRF1 mRNA were highly homologous to those of human eRF1 mRNA (Fig. 1). A cDNA encoding full-length Ra-eRF1 was synthesized by PCR from rabbit muscle mRNA. The cloned Ra-eRF1 cDNA encoded a 1,314-bp open reading frame (ORF) flanked by the AUG initiator codon and the UAG terminator codon (Fig. 1). The deduced protein is composed of 437 amino acids (49 kD), and its protein sequence was completely identical to human eRF1 regardless of being 3% distinct in the nucleotide sequence, showing the high conservative structural and functional feature of the mammalian eRF1 family. The nucleotide sequence data reported in this paper will appear in the DDBJ/EMBL/GenBank nucleotide sequence databases with the accession number AB029089.
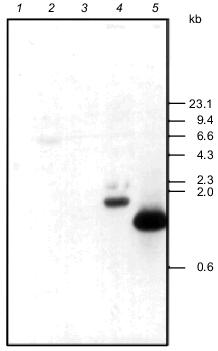
Primary structure of Tetrahymena thermophila eRF1 cDNA. Tt-eRF1 cDNA has been cloned recently from Tetrahymena thermophila using the 5´- and 3´-RACE method [14]. The deduced Tt-eRF1 ORF is composed of a 1,308-bp sequence starting at the AUG initiator codon and ending at the UGA termination codon (Fig. 2). This ORF was interrupted by nine UAA and one UAG codons that are known to be reassigned to the glutamine codon in Tetrahymena [10-13] (Fig. 2). The predicted Tt-eRF1 protein contains 435 amino acids (molecular mass 49.5 kD) and shares 57% identity and 66% similarity with rabbit, human, and Xenopus eRF1s. Southern hybridization using the cloned Tt-eRF1 probe detected a unique signal in Tetrahymena DNA while not in DNAs from E. coli, S. cerevisiae, and Schizosaccharomyces pombe (Fig. 3), showing that the cloned Tt-eRF1 sequence is derived from T. thermophila and its sequence is not highly conservative compared with other eRF1s.Fig. 1. Sequence alignment of rabbit and human eRF1 cDNAs. Human eRF1 cDNA sequence (GenBank accession No. X81625), referred to as “H”, was aligned with Ra-eRF1 cDNA (this work), referred to as “R”, using the Genetyx Mac 10.1 program. The initiating codon (ATG) and termination codon (TGA) are boxed. Asterisks denote nucleotides identical to the Ra-eRF1 cDNA. The position of the end of the smaller 3´ UTR is marked by an arrow. The location of the two putative polyadenylation signals in the 3´ UTR is underlined.

Fig. 2. Nucleotide sequence of Tt-eRF1 cDNA. Initiation (ATG) and termination (TGA) codons are boxed. The underlined TAA and TAG codons are reassigned to glutamine (shown by asterisks). Deduced amino acid sequence is also shown.

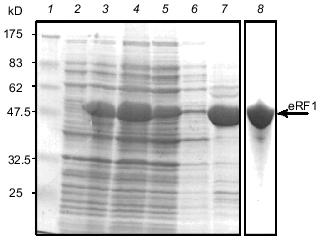
Overproduction and purification of rabbit eRF1. Ra-eRF1 cDNA was cloned into pET-30a(+) expression vector. This expression plasmid yielded a large amount of soluble His6-tagged Ra-eRF1 protein in E. coli BL21 strain. Presence of the N-terminal His6 tag enabled quick isolation of eRF1 using Ni-NTA resin. Recombinant Ra-eRF1 was purified from the soluble fraction of bacterial lysate using Ni-NTA-agarose and Mono Q column chromatographies to homogeneity (Fig. 4).Fig. 3. Southern hybridization analyses using PCR fragment of Tt-eRF1 cDNA as a probe. Total genomic DNA (7-10 µg) of E. coli (lane 1), S. cerevisiae (lane 2), S. pombe (lane 3), and T. thermophila (lane 4) were digested with HindIII, separated by electrophoresis in 0.9% agarose gel, transferred to nylon filter, and hybridized with the ECL-labeled PCR fragment of Tt-eRF1 according to the manufacturer's instruction. The PCR fragment of Tt-eRF1 cDNA (lane 5) was used as a positive control of hybridization. DNA size markers of HindIII digests of lambda DNA are shown.
Overproduction and isolation of Tetrahymena eRF1 from E. coli under denaturing conditions. To establish an overexpression system of Tt-eRF1 in E. coli or in standard eukaryotes, ten internal UAA/UAG codons of Tt-eRF1 cDNA were changed to glutamine codons (CAG and CAA) by site-directed mutagenesis. Overexpression of both His6-tagged and tag-free Tt-eRF1s was achieved by the T7 promoter-driven expression system in E. coli. Although both constructs gave very high level of protein expression in E. coli BL21 strain, the overproduced eRF1, in contrast to Ra-eRF1, was fully insoluble in both cases (data not shown), which greatly impeded the purification of active Tt-eRF1. Hence, we decided to isolate first denatured Tt-eRF1 to prepare antibody against the recombinant Tt-eRF1 for Western immunoblot detection. Tt-eRF1-containing inclusion bodies were isolated by centrifugation and Tt-eRF1 was purified by anion-exchange chromatography on Mono Q to homogeneity under denaturing conditions (Fig. 5). About 5 mg of solubilized Tt-eRF1 was purified and used for rabbit immunization, and high-titer anti-Tt-eRF1 antibody was prepared.Fig. 4. Purification of recombinant Ra-eRF1 under native conditions. Lanes: 1) protein markers; 2, 3) whole cell proteins before and after induction, respectively; 4) cleared cell lysate; 5) flow-through fraction of Ni-NTA column; 6) wash; 7) Ra-eRF1 eluted from Ni-NTA-agarose; 8) Ra-eRF1 purified on Mono Q column.
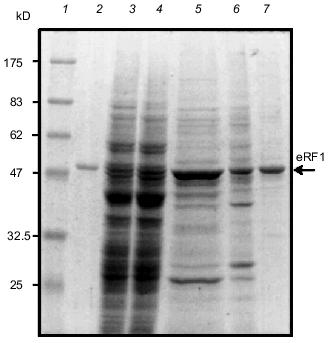
Purification of the soluble Tetrahymena eRF1. Several attempts were made to increase the soluble fraction of Tt-eRF1 or to prevent the aggregate formation in vivo as well as in vitro, though without any improvement: 1) denaturation and renaturation treatment with several solubilization chemicals or gel-filtration; 2) co-expression of E. coli chaperons; 3) fusion with thioredoxin protein; and 4) test of several growth media or growth conditions (data not shown). Finally, we found that Tt-eRF1 remains in a soluble form once expressed in S. cerevisiae although the expressionlevel was very low. Since the first affinity purification of His6-tagged Tt-eRF1 with Ni-NTA-agarose was incomplete in the purity, the fraction was further subjected to chromatography on a Heparin column and on Mono Q column. A single protein was purified to near homogeneity (Fig. 6). The purified protein was reactive to anti-Tt-eRF1 antibody described above (not shown).Fig. 5. Purification of His6-tag free Tt-eRF1 under denaturing conditions. Samples were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and Coomassie Blue staining at different purification steps. Lanes: 1, 2) whole cell proteins before and after induction, respectively; 3) cleared lysate; 4) pellet of cell debris with Tt-eRF1 inclusion bodies; 5) supernatant obtained after treatment of the pellet with 2% Triton X-100, 10 mM EDTA, and 10 mM Tris-HCl (pH 8.0); 6) Tt-eRF1 protein solubilized in 8 M urea prior to Mono Q purification; 7) Tt-eRF1 after Mono Q column chromatography.
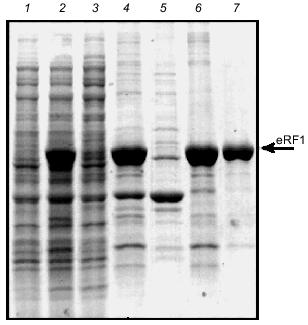
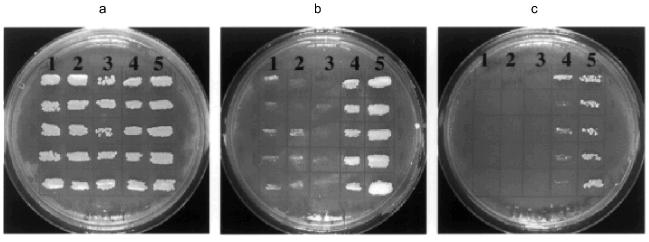
Functional test of recombinant eRF1s in yeast. Strain MT557/4d has a temperature-sensitive mutation in the eRF1 gene of S. cerevisiae (sup45 ts allele; sal4-2 [16]) and was used for heterologous complementation test of recombinant eRF1s. Both His6-tagged and tag-free constructs of Tt-eRF1 and Ra-eRF1 cDNAs were cloned under the PGAL1 promoter in the pYES2 vector (plasmids pR-eRF1-25/2, pR-eRF1-25/3, pTT-eRF1-4/12, and pTT-eRF1-1/3; see Table 1), and transformed into MT557/4d strain. Growth of these transformants were monitored on uracil-free SC medium plates supplemented with galactose for induction of PGAL1 promoter at the permissive and non-permissive temperatures (Fig. 7). The data indicated that Ra-eRF1 complemented efficiently the sup45 ts allele of S. cerevisiae, showing that eRF1 genes of rabbit and yeast are functionally exchangeable. This observation is consistent with the previous results that the eRF1 genes from S. pombe [22], Xenopus [23, 24], human, and hamster [24] are able to complement the lethal sup45 mutation of S. cerevisiae. The recombinant Ra-eRF1, however, became less active for complementation upon tagging with His6 (see Fig. 7), suggesting that the exogenous polypeptide including His6 tag at the N terminus is slightly but significantly harmful to the function of Ra-eRF1.Fig. 6. Purification of native Tt-eRF1 protein synthesized in S. cerevisiae.Samples were analyzed by SDS-PAGE and Coomassie Blue staining at different purification steps. Lanes: 1) protein markers; 2) denatured Tt-eRF1 protein isolated from inclusion body in E. coli (control); 3) total lysate; 4) lysate after removal of ribosomes; 5) Tt-eRF1 fraction after chromatography in Ni-NTA-agarose; 6) Tt-eRF1 fraction after chromatography in Heparin Toyopearl; 7) Tt-eRF1 purified by FPLC chromatography on a Mono Q column.
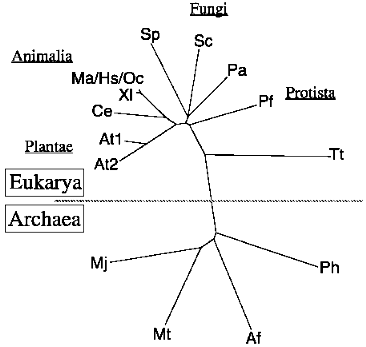
In contrast to Ra-eRF1 and most other eRF1s so far tested, Tt-eRF1 (with or without His6-tag) failed to complement the sup45 ts mutation (Fig. 7). The disability of Tt-eRF1 for the complementation could be explained by assuming that Tt-eRF1 recognizes only UGA but not the other two codons, unlike yeast eRF1, or by assuming that Tt-eRF1 cannot interact properly with the heterologous ribosome derived from S. cerevisiae. The latter possibility should not be the sole reason for the disability but could well be supported by the large phylogenetic distance of Tt-eRF1 from yeast eRF1 (Fig. 8). It is quite remarkable that Tt-eRF1 is very isolated from most other eRF1s in the phylogenetic tree, showing only 57% identity to mammalian and yeast eRF1. Therefore, one could argue that Tetrahymena eRF1 might have evolved rapidly compared with other eRF1s. It remains to be examined whether eRF1 genes from other ciliates are also distantly related with the mammalian release factor genes.Fig. 7. Complementation test of eRF1 clones in sup45 ts mutant of S. cerevisiae. S. cerevisiae strain MT 557/4d transformants (in quintet) were grown on the solid SC media lacking uracil and supplemented with galactose at 25 (a), 35 (b), or 37°C (c). eRF1 genes transformed: 1) Tt-eRF1 (with His6 tag); 2) Tt-eRF1 (without His6 tag); 3) pYES2 vector (control); 4) Ra-eRF1 (with His6 tag); 5) Ra-eRF1 (without His6 tag).
We thank Dr. Mick F. Tuite for S. cerevisiae MT557/4d strain and Dr. Shinichi Hoshino for human eRF1 cDNA clone. This work was supported in part by grants from the Ministry of Education, Science, Sports and Culture, Japan; the Human Frontier Science Program (awarded in 1993 and 1997); and the Basic Research for Innovation Biosciences Program of Bio-oriented Technology Research Advancement Institution (BRAIN). A. L. K. was a postdoctoral fellow of the Japan Society for the Promotion of Science.Fig. 8. Phylogenetic tree of the eukaryotic and archaebacterial release factors. Ph, Pyrococcus horikoshii (GenBank accession No. AP000006); Af, Archaeoglobus fulgidus (AE001020); Mt, Methanobacterium thermoautotrophicus (AE000864); Mj, Methanococcus jannaschii (U67526); Sp, Schizosaccharomyces pombe (D63883); Pa, Podospora anserina (AF053983); Sc, S. cerevisiae (X04082); At, Arabidopsis thaliana (AT81KBGEN, AB016886); Ce, Caenorhabditis elegans (CELT05H4); Xl, Xenopus laevis (Z14253); Ma, Mesocricetus auratus (MAC114); Hs, Homo sapiens (X81625); Oc, Oryctolagus cuniculus (rabbit, this work); Pf, Plasmodium falciparum (AE001402); Tt, Tetrahymena thermophila (AB026195).
REFERENCES
1.Nakamura, Y., Ito, K., and Isaksson, L. A. (1996)
Cell, 87, 147-150.
2.Buckingham, R. H., Grentzmann, G., and Kisselev, L.
(1997) Mol. Microbiol., 24, 449-456.
3.Scolnick, E., Tompkins, R., Caskey, T., and
Nirenberg, M. (1968) Proc. Natl. Acad. Sci. USA, 61,
768-774.
4.Frolova, L., Le Goff, X., Rasmussen, H. H.,
Cheperegin, S., Drugeon, G., Kress, M., Arman, I., Haenni, A. L.,
Celis, J. E., Philippe, M., Justesen, K., and Kisselev, L. (1994)
Nature, 372, 701-703.
5.Caskey, C. T., Tompkins, R., Scolnick, E., Caryk,
T., and Nirenberg, M. (1968) Science, 162, 135-138.
6.Caskey, C. T., Beaudet, A. L., and Tate, W. P.
(1974) Meth. Enzymol., 30, 293-303.
7.Drugeon, G., Jean-Jean, O., Frolova, L., Le Goff,
X., Philippe, M., Kisselev, L., and Haenni, A. L. (1997) Nucleic
Acids Res., 25, 2254-2258.
8.Freistroffer, D. V., Pavlov, M. Y., MacDougall, J.,
Buckingham, R. H., and Ehrenberg, M. (1997) EMBO J., 16,
4126-4133.
9.Pavlov, M. Y., Freistroffer, D. V., MacDougall, J.,
Buckingham, R. H., and Ehrenberg, M. (1997) EMBO J., 16,
4134-4141.
10.Horowitz, S., and Gorovsky, M. A. (1985) Proc.
Natl. Acad. Sci. USA, 82, 2452-2455.
11.Cupples, C., and Pearlman, R. E. (1986) Proc.
Natl. Acad. Sci. USA, 83, 5160-5164.
12.Kuchino, Y., Hanyu, N., Tashiro, F., and
Nishimura, S. (1985) Proc. Natl. Acad. Sci. USA, 82,
4758-4762.
13.Hanyu, N., Kuchino, Y., and Nishimura, S. (1986)
EMBO J., 5, 1307-1311.
14.Karamyshev, A. L., Ito, K., and Nakamura, Y.
(1999) FEBS Lett., submitted.
15.Sikorski, R. S., and Hieter, P. (1989)
Genetics, 122, 19-27.
16.Stansfield, I., Akhmaloka, and Tuite, M. F.
(1995) Curr. Genet., 27, 417-426.
17.Vieira, J., and Messing, J. (1987) Meth.
Enzymol., 153, 3-11.
18.Sherman, F. (1991) Meth. Enzymol.,
194, 3-20.
19.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, New York.
20.Frohman, M. A., Dush, M. K., and Martin, G. R.
(1988) Proc. Natl. Acad. Sci. USA, 85, 8998-9002.
21.Rose, M. D., Winston, F., and Hieter, P. (1990)
Methods in Yeast Genetics. A Laboratory Course Manual, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
22.Ito, K., Ebihara, K., Uno, M., and Nakamura, Y.
(1996) Proc. Natl. Acad. Sci. USA, 93, 5443-5448.
23.Tassan, J. P., Le Guellec, K., Kress, M., Faure,
M., Camonis, J., Jacquet, M., and Philippe, M. (1993) Mol. Cell.
Biol., 13, 2815-2821.
24.Urbero, B., Eurwilaichitr, L., Stansfield, I.,
Tassan, J. P., Le Goff, X., Kress, M., and Tuite, M. F. (1997)
Biochimie, 79, 27-36.