REVIEW: Targets of Oncogenes and Tumor Suppressors: Key for Understanding Basic Mechanisms of Carcinogenesis
B. P. Kopnin
Institute of Carcinogenesis, Blokhin Russian Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, Moscow, 115478 Russia; fax: (095) 324-1739; E-mail: kopnin@imb.ac.ru
Received September 17, 1999
Changes in expression of protooncogenes and tumor suppressor genes play a key role in oncogenesis. Dysfunction of their protein products leads to abnormal regulation of signaling pathways, which control the cell cycle, apoptosis, genetic stability, cell differentiation, and morphogenetic reactions. Changes in these important physiological processes are obviously responsible both for initial steps of neoplastic cell transformation and for determination of subsequent tumor progression resulting in the development of malignant tumors.
KEY WORDS: oncogene, tumor suppressor, apoptosis, genetic stability, differentiation
Significant progress in understanding of mechanisms of carcinogenesis has arisen from the discovery of oncogenes and protooncogenes and the subsequent discovery of tumor suppressors and mutator genes. Oncogenes are cellular or viral (i.e., inserted into the cell by a virus) genes; their expression can cause the development of a neoplasm. Protooncogenes are normal cellular genes; their conversion to oncogenes can occur via several mechanisms such as amplification or modification. Tumor suppressors (anti-oncogenes, recessive tumor genes) are cellular genes; their inactivation increases the probability of tumor formation, whereas restoration of their functioning may suppress the growth of tumor cells. It should be noted that so-called “mutator” genes, related to tumor suppressors cannot affect growth of tumor cells. However, impairment in their functioning increases the rate of mutations and/or other genetic abnormalities. Inactivation of these genes increases the probability of the appearance of various oncogenic mutations so strongly that tumor formation inevitably occurs sooner or later.
There are a few criteria by which oncogenes and tumor suppressors are determined: a) appropriate changes of structure and/or expression of a desired gene in a certain or various tumor cells; b) appearance of certain tumors in juvenile or young individuals with inherited germinal mutations of a desired gene; c) sharp increase of the tumor rate in transgenic animals which either express an activated form of a desired gene (in the case of oncogenes) or carry knock-out mutations of a desired gene (in the case of tumor suppressors); d) ability to cause morphological transformation and/or unlimited growth (oncogenes) or suppression of cell growth and/or manifestations of transformation (tumor suppressors) in cultivated cells in vitro.
The last two decades are characterized by the discovery of numerous oncogenes and tumor suppressors. Now about one hundred potential oncogenes (cellular and viral) and twenty tumor suppressors have been recognized. Genetic events leading to activation of protooncogenes or inactivation of tumor suppressors have also been described [1-6]. The mechanism of the effect of viral oncogenes includes activation of cellular protooncogenes (retroviruses) or inactivation of tumor suppressors (DNA viruses) [7-11]. Changes in oncogenes and tumor suppressors typical for certain forms of human neoplasms have been recognized and some highly specific abnormalities are employed in diagnostics (Tables 1 and 2) [3, 12].
Table 1. Some changes in protooncogenes
typical for human tumors
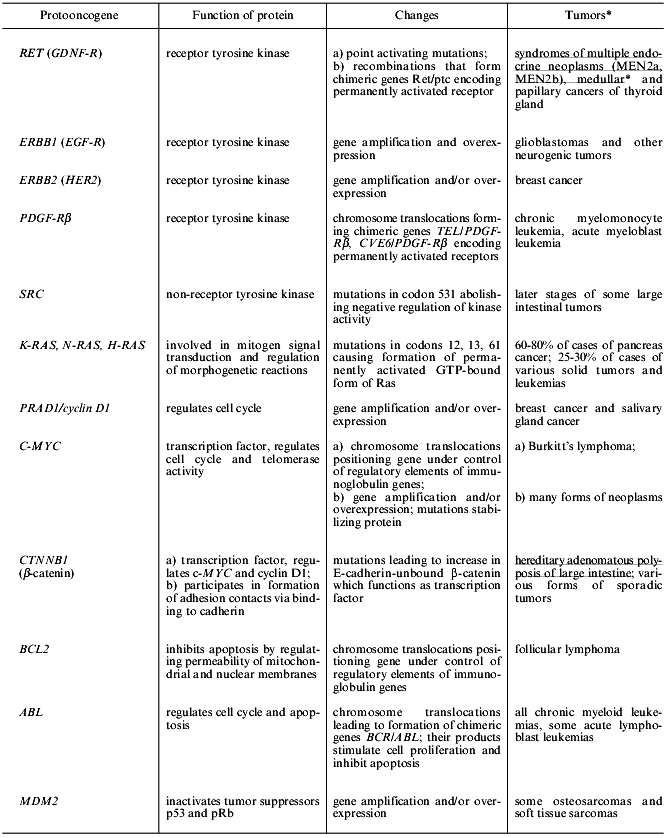
*Hereditary forms of tumors that appear during mutations in sex
cells are underlined. In other cases mutations occur in somatic cells
in which tumors are formed.
Table 2. Forms of human tumors that appear
during inactivation of some tumor suppressors and mutator genes
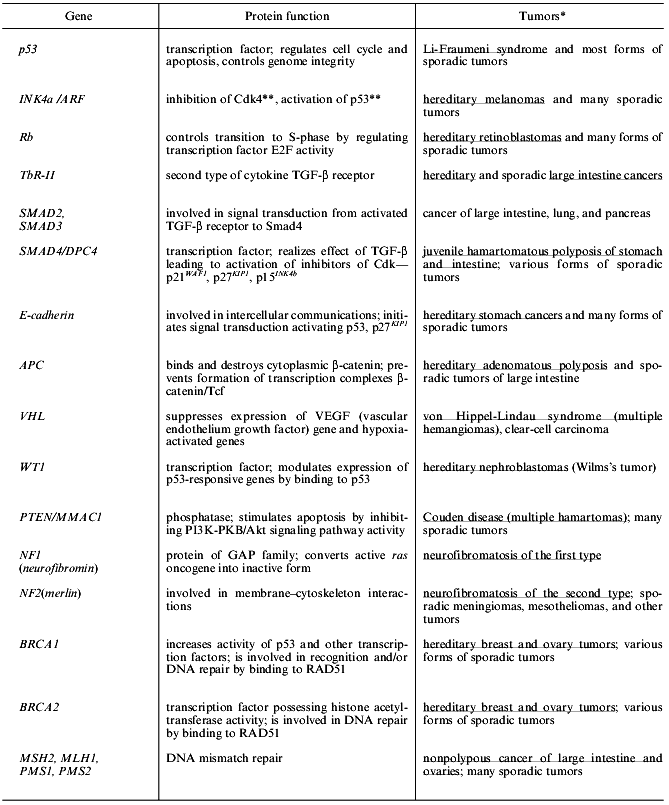
*Hereditary forms of tumors that appear during mutations in sex
cells are underlined.
**Locus INK4a/ARF encodes two proteins: p16INK4a
(inhibitor of cyclin-dependent kinases Cdk4,6) and p19ARF
(product of Alternative Reading Frame, ARF) that binds to p53 and Mdm2
thus blocking their interaction and preventing p53 degradation [13, 14]. Deletions and many
point mutations in locus INK4a/ARF simultaneously cause inactivation of
suppressor activities of these both proteins [15].
However, for a long time our knowledge about each oncogene or tumor suppressor genes remained discrete and poorly related with each other. Only recently it has been recognized that the overwhelming majority of known oncogenes and tumor suppressors are components of a few common signaling pathways that control the cell cycle, apoptosis, genome integrity, morphogenetic reactions, and cell differentiation. Finally, changes in these signaling pathways cause the development of cancer. Here we consider the main targets of oncogenes and tumor suppressor genes.
1. ONCOGENES AND TUMOR SUPPRESSOR GENES IN REGULATION OF THE CELL
CYCLE
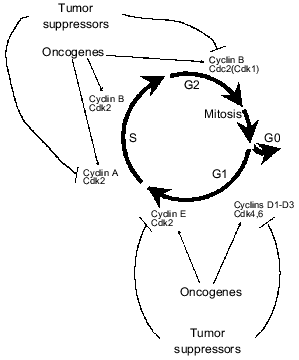
Over-proliferation of certain cells is a basis for tumor formation and therefore impairments of regulation of the cell cycle are inalienable and basic signs of tumor cells. Activities of sequential cyclin-dependent kinases are the “motor” of the cell cycle [16] (Fig. 1). Each cyclin-dependent kinase (Cdk) is a catalytic subunit of the holoenzyme complex that requires the presence of the activating subunit, cyclin, for manifestation of catalytic activity. Regulation of Cdk activity occurs via directed changes of certain cyclins during cell cycle phases. The activity of Cdk is also regulated by phosphorylation. In the active forms cyclin--Cdk complexes phosphorylate regulatory proteins that control running of a given phase.
Many oncogenes and tumor suppressor genes regulate one or another cyclin--Cdk complex. Their protein products increase the activity of cyclin-dependent kinases responsible for initial steps of the presynthetic phase G1 (complexes of cyclins D1-D3 with Cdk4 or Cdk6 depending on cell type) and transition of G1 into S phase of DNA synthesis (cyclin E--Cdk2) (Fig. 1). Some protooncogenes and tumor suppressors regulate activity of complexes cyclin A--Cdk2 (required for DNA replication) and cyclin B--Cdk1 (responsible for the transition of G2 phase to mitosis; another name of Cdk1 is Cdc2).
Tumor suppressor pRb and Rb-like proteins p105 and p130 are the main substrates for cyclin D--Cdk4 and cyclin D--Cdk6 complexes. In non-proliferating cells and in cells at early G1 phase, pRb and its homologs are dephosphorylated [17]. In this state they bind and block transcription complexes E2F--DP (E2F-1, -2, -3, -4, -5 and DP-1, -2, -3) regulating the expression of some genes whose products are required for the beginning and passage of S-phase. In particular E2F--DP regulate expression of genes of thymidine kinase, dihydrofolate reductase, cyclin E, cyclin A, PCNA (proliferating cell nuclear antigen), DNA-polymerase alpha, etc. [18]. Binding of proteins of the E2F family with pRb inhibits their transcription activity. Mitogen signals by growth factors initiate pRb phosphorylation by the cyclin D--Cdk4 complex (or cyclin D--Cdk6) in the middle G1 phase, and this causes release of transcription factors E2F--DP from complexes with pRb and their activation [17]. The latter results in stimulation of transcription of the cyclin E gene and activation of cyclin E--Cdk2 complexes that also phosphorylate pRb. Thus, a regulatory loop is formed. It maintains the activity of transcription factors E2F--DP and their responsive genes that are involved in DNA replication (Fig. 2). After termination of S phase, pRb is dephosphorylated and in this state it blocks the activity of E2F--DP. Initiation of the next S phase requires a new mitogenic stimulus that will activate complexes cyclin D--Cdk4,6. Thus, tumor suppressor pRb plays a key role in the regulation of the transition of the cell into the S phase.Fig. 1. Transition via the cell cycle is determined by sequential activation of various cyclin--Cdk complexes. Most of them are targets of the activating effect of oncogenes or inhibitory effect of tumor suppressors.
Products of many oncogenes are components of signaling pathways responsible for activation of cyclin D--Cdk4,6 and cyclin E--Cdk2 complexes by growth factors and/or cell adhesion to extracellular matrix proteins (Fig. 2). For example, growth factor receptor binding induces dimerization and autophosphorylation of these receptors (one dimer subunit phosphorylates tyrosine residues of its counterpart). This links receptor tyrosine kinases with many signaling proteins that contain SH2 domains and bind phosphotyrosine. For example, activated receptors of platelet-derived growth factor Rbeta (PDGF-Rbeta) interact with SH2 domains of such proteins as phosphatidylinositol-3´-kinase (PI3K) (see review by M. A. Krasilnikov in this issue), phospholipase C (PLC)-gamma1, latent forms of STAT transcriptional factors and adapter protein Grb2 which is involved in signal transduction to Ras proteins [19-21]. Binding of each of these proteins with receptor phosphotyrosines causes activation of intersectional signaling pathways, which terminates in the nucleus by activation of transcriptional factors and specific gene expression (Fig. 2). In particular, Grb2-induced transition of Ras proteins into their activated (GTP-bound) state leads to stimulation of a number of its effectors including serine-threonine type kinases Raf* and MEKK triggering MAP (mitogen activated protein) kinase cascades [20, 22]. End product of these cascades, ERK (MAPK), p38, and JNK (SAPK) are translocated from the cytoplasm into the nucleus where they phosphorylate and activate many substrates including such transcriptional factors as Elk1, Ets1*, Ets2*, Jun*, ATF2, Tcf, etc. This causes activation of other transcription factors. For example, formation of complex between Elk1 and SRF (serum response factor) initiates transcription of genes containing in their promoter SRE elements (e.g., transcription of FOS* gene) [Footnote: Here (see asterisks) and further in this paper modern conventional principles for denomination of protooncogenes and their protein products have been employed. Genes are printed in italics (human and animal genes are written in capital and small letters, respectively) and their protein products are written in small standard letters with the first letter capitalized].Fig. 2. Products of many protooncogenes and tumor suppressor genes regulate the activity of cyclin-dependent kinases phosphorylating pRb. Phosphorylation of pRb and its binding to a number of viral oncoproteins induces release and activation of transcription complexes E2F--DP. They increase expression of genes whose products are necessary for passage of the S phase.
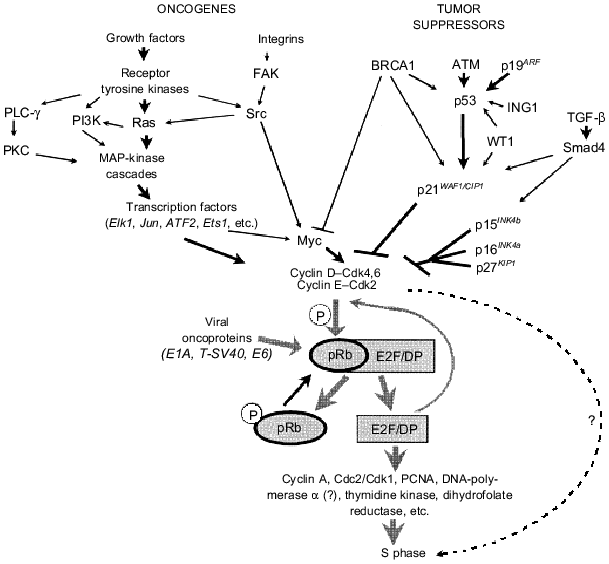
Similar reactions are also observed on binding of integrins (receptors mediating cell adhesion) with extracellular matrix proteins. Such interaction causes activation of autophosphorylation of FAK (focal adhesion kinase). This results in the binding of FAK to the SH2 domain of the Src protooncoprotein followed by recruiting of adapter Grb2 protein, and activation of Ras and MAP kinase cascades (Fig. 2) (see also review by A. G. Tatosyan and O. A. Mizenina in this issue).
Increase of cyclin D1 gene expression (probably mediated by proteins Jun, Ets1, Ets2 [21]) is the major consequence induced by MAP kinase-activated transcriptional factors. Mitogenic signals increase Myc expression that also results in an increase in the activity of cyclin-dependent kinases operating in G1 phase (cyclin D--Cdk4 and cyclin E--Cdk2). This is realized by the following mechanisms: 1) Myc transactivates the Cdc25a-phosphatase gene and this abolishes the inhibitory phosphorylation of Cdk2 and Cdk4 at Thr-14 and Tyr-15; 2) Myc decreases expression of Cdk2 inhibitor, p27KIP1 [23-26]. Mechanisms of Myc activation by growth factors are poorly understood. It is suggested that this activation may be realized via Ras-independent signaling pathways activated by Src oncoprotein and via Ras--Raf--MAP kinase cascades inducing activation of Ets1 and/or E2F (the promoter of the MYC gene contains responsive elements for these transcriptional factors [22, 26]).
Many components of signaling pathways realizing activation of cyclin-dependent kinases (and consequently stimulation of cell division) in response to effects of growth factors are protooncogenes. Changes in their structure (mutations) leading to a loss of control by negative regulatory factors and/or permanent increase in expression convert these protooncogenes into oncogenes [1, 3, 5]. Products of identified oncogenes represent all hierarchical levels of mitogenic signal regulation [5]: 1) growth factors--PDGF-beta (Sis), FGF1, etc.; 2) receptor tyrosine kinases--EGF-R (ErbB), HGF-R (Met), Ret, etc.; 3) proteins of the Ras family--K-Ras, H-Ras, and N-Ras; 4) Ras effectors--serine-threonine kinases Raf and Mos; 5) transcriptional factors--Jun, Ets1, Myc, etc.; 6) cyclin D1 (Prad1). It seems that detailed analysis of each neoplasm reveals changes of at least one of the signaling pathway components (protooncogenes) causing permanent stimulation of cyclin-dependent kinases and initiation of cell division irrespectively to the effect of growth factors.
Interestingly, the Cdk--Rb--E2F signaling pathway is controlled not only by pRb, but also by many other suppressor proteins (Fig. 2). Some of them are inhibitory subunits of Cdk (CKIs, Cdk inhibitors) realizing arrest of the cell cycle in response to various extra- and intracellular signals [16]. Two CKIs families, Ink4 and Cip/Kip, have been recognized. The former consists of four members including tumor suppressors p15INK4b and p16INK4a. Ink4 proteins possess relatively narrow specificity and bind Cdk4 and Cdk6; this prevents complex formation between Cdk4,6 and D cyclins [16, 27]. The Cip/Kip family consists of three members: p21WAF1/CIP1, p27KIP1, and p57KIP2. These proteins bind to (and inhibit) completely formed complexes cyclin D--Cdk4,6, cyclin E--Cdk2, and cyclin A--Cdk2. Protein p21WAF1/CIP1 can also block the complex cyclin B--Cdc2 responsible for proceeding of the G2-phase and entrance into mitosis [16, 27]. Proteins p21WAF1/CIP1 and p27KIP1 also realize effects of other suppressor proteins. Protein p21WAF1/CIP1 is one of the main targets for the transactivating effect of p53 and, consequently, for suppressors involved in regulation of stability/activity of p53 (p19ARF, ATM, WT1) ([13, 14, 28, 29], see also paper by P. M. Chumakov in this issue) or its transcriptional activity (BRCA1 and p33ING1 [30-32]). BRCA1 and WT1 can also activate p21WAF1/CIP1 via unknown p53-independent mechanisms [31, 33].
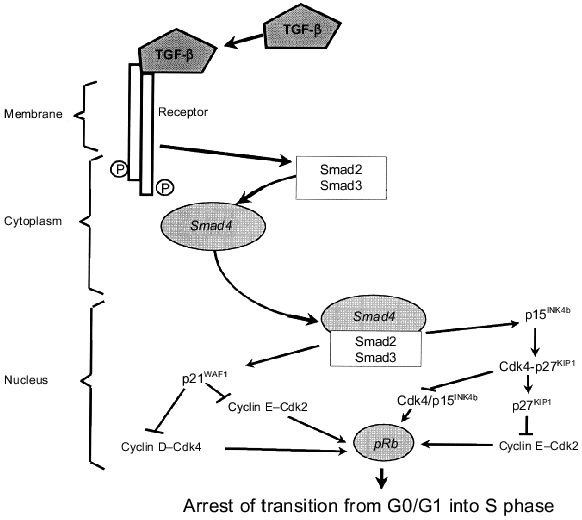
Protein p27KIP1 (as well as p15INK4b) is a key component of inhibitory signal transduction induced by TGF-beta binding to its receptors (Fig. 2). Recently, activated TGF-beta receptors have been shown to phosphorylate specific signaling effectors, proteins Smad2 and Smad3, and this causes their binding with tumor suppressor Smad4. The forming complexes are translocated from the cytoplasm into the nucleus where they regulate transcription of specific genes, in particular genes of Cdk inhibitors. This results in activation of both p21WAF1/CIP1 and p15INK4b [34-37]. The latter displaces p27KIP1 from its complexes with Cdk4,6 and inhibits formation of their complexes with D cyclins required for the proceeding of the G1 phase (Fig. 1). Released p27KIP1 in its turn binds and inhibits cyclin E--Cdk2 complexes responsible for the beginning of the S phase. Increase of p21WAF1/CIP1 expression also results in inhibition of the activity of cyclin D--Cdk4,6 and cyclin E--Cdk2 complexes. This results in arrest of the cell cycle at the G0/G1 phase, and the S phase does not begin (Fig. 3).
Overexpression of MYC or MDM2 oncogenes overcomes the inhibitory effect of TGF-beta [38]. If the effect of Myc is related to activation of various Cdk via the increase of Cdc25A expression [23] and stimulation of p27KIP1 degradation [24], protein Mdm2 besides p53 degradation [13, 14] also causes pRb inactivation [39] releasing active transcriptional complexes E2F--DP. Thus, overexpression of MYC or MDM2 protooncogenes and inactivating mutations in tumor suppressors Smad4, p15INK4b, and pRb have one common consequence: cells rescue from the inhibitory effect of TGF-beta that is very important for development of epithelial tumors, in particular cancers of the intestine and pancreas [40, 41].Fig. 3. TGF-beta binding to its receptor initiates formation of transcriptional complexes Smad4--Smad2,3 which are translocated from the cytoplasm into the nucleus. This results in activation of such targets as inhibitors of cyclin-dependent kinases p21WAF1/CIP1, p15INK4b, and p27KIP1 that inhibits Cdk4,6 and Cdk2 responsible for the proceeding of the G1 phase and entrance into the S phase (see explanations in the text).
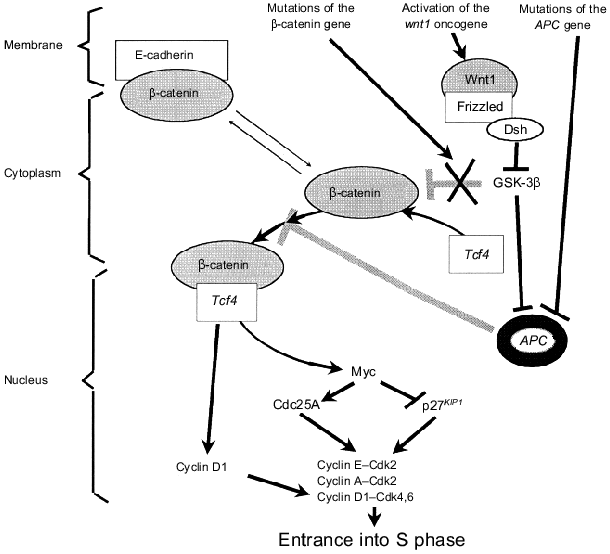
Identification of another important signaling pathway which probably regulates the cell cycle in dependence on the conditions of the membrane and submembranous structures and which is frequently altered in various human tumors [42] is one of brightest achievements of the two last years (Fig. 4). It was found that E-cadherin-unbound beta-catenin can function as a transcription factor. In the cytoplasm it binds to another transcriptional factor, Tcf4, and beta-catenin--Tcf4 complexes are translocated into the nucleus and activate genes possessing special responsive elements. Genes of cyclin D1 [43] and MYC [44] are main targets for the transactivating effect of the beta-catenin--Tcf4 complex. Tumor suppressor APC (its mutations cause the development of adenomatous polyposis of the intestine) binds free cytoplasmic beta-catenin; this is accompanied by beta-catenin degradation [45, 46]. Thus, APC inactivation stimulating formation of beta-catenin--Tcf4 complexes increases transcription of genes of cyclin D1 and MYC; this results in activation of cyclin-dependent kinases responsible for the proceeding of the G1 phase and entrance into the S phase (Fig. 4). Mutations of beta-catenin increasing its stability in the cytoplasm result in the same consequences (such mutations were recognized in patients with familial polyposis without mutations of APC [43, 47]; most mutations were found in beta-catenin sites phosphorylated by glycogen synthase kinase-3beta, GSK-3beta). A similar situation was also observed during activation of WNT1 (Wingless/INT1) protooncogene (Fig. 4). Binding of its product Wnt1 (a member of a family of cysteine-rich glycosylated signaling proteins) with receptor (Frizzled) induces translocation of cytoplasmic Dsh protein to the membrane where it inhibits GSK-3beta kinase activity; the latter phosphorylates beta-catenin and APC and stimulates their binding and beta-catenin degradation. Thus, Wnt1-induced inhibition of GSK-3beta activity is accompanied by stabilization and increase of the intracellular concentration of cytoplasmic beta-catenin; this increases the probability of formation of active transcriptional complexes of beta-catenin with factors of the Tcf/Lef1 family [48]. It is possible that mutations in the E-cadherin gene can be responsible for stimulation of signaling pathways realized by transcriptional activities of beta-catenin.
In conclusion we should note that most known protooncogenes and tumor suppressors somehow regulate activity of cyclin-dependent kinases responsible for entrance to the S phase of the cell cycle. Products of some cellular (Mdm2) or viral oncogenes (e.g., T-antigen of SV40 virus, E1A adenovirus, E7 HPV, etc.) bind and inactivate the main substrate of such Cdk--pRb. Apparently, impairments in signaling pathways --> Cdk2,4,6 --> pRb --> E2F/DP are necessary precondition for the appearance of constantly proliferating neoplastic cells.Fig. 4. Mutations in tumor suppressors APC and beta-catenin and activation of the wnt1 gene stimulate formation of beta-catenin--Tcf4 transcriptional complexes regulating the cyclin D1 and MYC genes. This increases the activity of cyclin--Cdk complexes (see explanations in text).
2. ONCOGENES AND TUMOR SUPPRESSORS IN REGULATION OF
APOPTOSIS
Involvement in control of apoptosis (programmed cell death) is another important point for regulatory activities of oncogenes and tumor suppressors. Apoptosis can be induced by various signals such as receptor binding of specific killer ligands, a deficit of growth/rescue factors, damage to DNA and cytoskeleton, hypoxia, and other unfavorable conditions (see reviews [49-52]). Two main phases have been recognized in apoptosis: induction and execution. The latter is realized via activation of caspases, a family of cysteine proteases that cleave their substrates at aspartate residues. The so-called “effector” or “executing” caspases 3, 6, 7 catalyze degradation of key substrates such as DFF45/ICAD, nuclease DFF40/CAD inhibitor (caspase 3 substrate), lamins, nuclear cytoskeleton proteins (caspase 6 substrates), etc. and this results in DNA fragmentation and cell destruction [52]. Caspases exist in the cytoplasm as proenzymes; their activation into functional proteases occurs via proenzyme cleavage into small and large subunits and their subsequent cleavage of N-terminal domains. Mature subunits form tetramers with two active sites [49, 52]. Various proteases including other caspases are involved in proteolytic activation of procaspases.
Activation of caspases 3, 6, 7 is thought to be realized via at least two completely different signaling pathways [49, 52] (Fig. 5). The first is initiated by specific receptor binding of killer molecules (Fas-ligand, TNF-alpha, etc.) that results in recruiting of adapter proteins and procaspases, in particular procaspase 8. Aggregation of procaspase 8 molecules can initiate their autoprocessing (self-cleavage) and formation of active caspase 8 that in turn processes “executing” caspases. An alternative mechanism of cleavage of caspases 3, 6, 7 involves caspase 9 that is activated during release of AIF (apoptosis inducing factor) protease and/or cytochrome c from mitochondria. Cytochrome c stimulates procaspase binding to Apaf1 protein (a homolog of CED-4 protein in C. elegans) and consequent formation of procaspase 9 aggregates and their autoprocessing to the active forms. Proteins of the Bcl2 family regulate the permeability of the mitochondrial membrane for AIF and cytochrome c. This family of structurally related proteins consists of more than twenty members including bcl2 and bcl-x protooncogene products that can block apoptosis and tumor suppressor Bax that on the contrary can induce apoptosis [53-55]. Antiapoptogenic molecules Bcl2 and Bcl-x are suggested to be localized on mitochondrial membranes and block channels responsible for the release of cytochrome c and/or AIF from mitochondria. Apoptogenic signals initiate translocation of Bax from cytoplasmic compartments to mitochondrial membranes, where it interacts with integral protein of the outer mitochondrial membrane, VDAC. This stimulates opening of a channel secreting cytochrome c. Bax also forms complexes with proteins Bcl2 and Bcl-x, and this probably opens the closed channels [54, 55]. Other proapoptotic proteins of the Bcl2 family (Bak, Bad, Bid, etc.) possibly exert similar effects [53, 55].
If Bcl2, Bcl-x, and Bax directly control release of apoptogenic molecules from mitochondria, other protooncogenes and tumor suppressors regulate activity of these and other proteins of Bcl2 (Fig. 5). Tumor suppressor p53 is one of the most potent regulators. Being activated in response to various unfavorable treatments (DNA damage, hypoxia, loss of cell contacts with substrate, permanent uncontrolled stimulation of mitogenic signal and many others [13, 14, 56-58]) (see also paper by P. M. Chumakov in this issue), p53 simultaneously activates BAX gene expression and BCL2 gene repression at the transcriptional level [57, 59]. p53 also increases expression of the PIG genes; their products induce oxidative stress that can cause impairment in permeability of mitochondrial and nuclear membranes [60]. p53 can transactivate some killer receptors, in particular Fas and KILLER/DR5 [57, 61, 62]. Thus, p53 activation provides a potent apoptogenic signal and various mechanisms of induction of “executive” caspases are involved in its realization. It is important to emphasize that p53-dependent apoptosis eliminates from the organism no only damaged cells, but also cells with uncontrolled stimulation of proliferation induced, for example, by constitutive activation of the MYC oncogene and/or transcriptional factor E2F. Stabilization of p53 during oncogene activation is related to E2F-induced increase in transcription of the p19ARF gene and its protein product prevents Mdm2-dependent degradation of p53 [13, 14] (see also paper by P. M. Chumakov in this issue). It is clear that inactivating mutations of p53 or p19ARF that impair operation of this protective mechanism will increase the probability of appearance of constantly proliferating cell clones and, therefore, the probability of subsequent development of tumors.Fig. 5. Involvement of oncogenes and tumor suppressors in regulation of apoptosis (see explanations in the text).
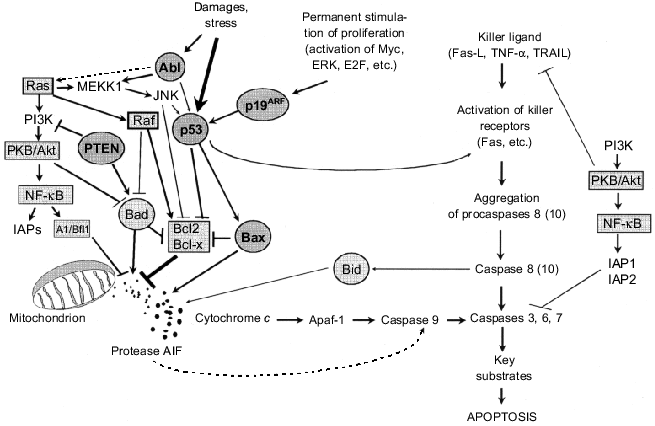
Constitutive expression of Ras oncogenes simultaneously initiates apoptogenic and antiapoptogenic signals. The former are due to the activation of the Ras--Raf--MAPK--E2F--p19ARF--p53 signaling pathway [63]. The latter are related to the action of Ras effectors: Raf protein (which phosphorylates and inactivates the proapoptotic protein Bad, a member of Bcl2 family) and PI3K (phosphoinositol-3-kinase) [21, 51, 63, 64] (see also review by M. A. Krasilnikov in this issue). Antiapoptotic effects of PI3K are due to its activation of serine-threonine protein kinase PKB/Akt (which was originally identified as an oncogene of retrovirus AKT8 inducing T-cell lymphomas in AKR mice). This kinase blocks apoptosis via several mechanisms [65] (Fig. 5). A PKB/Akt like Raf protein can phosphorylate and inactivate Bad protein. By suppressing the function of DAF-16 protein, a transcriptional factor of the Forkhead family, PKB/Akt can inhibit production of certain killer molecules (e.g., Fas-ligand). PKB/Akt has recently been shown to activate transcriptional factors of the Rel/NF-kB family [65] that can inhibit apoptosis via several pathways (Fig. 5). (These factors are homologs of viral v-Rel oncoprotein; amplifications and rearrangements of their genes are typical for many human tumors [66]). In particular PKB/Akt transactivated gene encoding A1/Bfl1 protein (a member of Bcl2 protein family) that inhibits release of cytochrome c and/or AIF from mitochondria [67]. NF-kB also increases expression of inhibitors of apoptosis, IAP1 and IAP2, which are members of the IAP (inhibitors of apoptosis) family of proteins blocking functions of caspases 3, 6, 7, 8, 9 [66]. This clarifies one of the protective functions of the PTEN tumor suppressor (its inactivation has been recognized in gliomas, breast and prostate cancers, and inborn mutations cause the development of syndrome of multiples hamartomas [68], Table 2): PTEN protein possessing tyrosine phosphatase activity suppresses the antiapoptogenic effects of the PI3K--PKB/Akt signal [69].
Neoplastic cells are characterized by impairments in functioning of other tumor suppressors that exert positive regulation on apoptosis. For example, the development of chronic myeloid leukemia is caused by chromosome translocation t(9; 22) that results in formation of the chimeric BCR/ABL gene. Such rearrangement simultaneously results in two important consequences: 1) a sharp increase in tyrosine kinase activity of Abl protein leading to stimulation of mitogenic and anti-apoptotic signals realized via Ras-signaling pathways [70, 71] and an increase in integrin synthesis providing better cell adhesion to the extracellular matrix [72]; 2) inactivation of apoptogenic activities of Abl [73-75] apparently due to its involvement in positive regulation of JNK kinase (another name SAPK, stress activated protein kinase) which can suppress activity of Blc2 and activate p53 (Fig. 5). There are some indications that protein Abl can directly bind p53 and modify its proapoptotic function [57, 65, 75].
Chromosome translocation t(15; 17) observed in most cases of acute promyelocytic leukemia results in connection of the gene of the retinoic acid receptor (RAR-alpha) with the tumor suppressor PML gene [3, 12, 76] (its product forms specific matrix-associated bodies in the nucleus). The chimeric protein product of the PML/RAR-alpha gene is suggested to inactivate apoptogenic functions of normal PML protein via a dominant negative mechanism by forming heterodimers with it. The mechanisms of induction of apoptosis during PML overexpression are not completely understood. PML is involved in the activation of caspases 1, 3 and in recruiting of protein Bax at apoptosis induced by TNF-alpha, interferons 1 and 2, Fas activation, and also by DNA damage [77, 78]. Besides regulation of apoptosis, PML also controls proliferation and differentiation of myeloid precursors. Activation of p21WAF1/CIP1 responsible for retinoic acid-induced arrest of the cell cycle was shown to be realized by PML [79]. Thus, expression of the chimeric protein PML/RAR-alpha induces inactivation of normal functioning of PML protein and like rearrangement of the BCR/ABL gene it simultaneously results in changes in cell cycle control and in partial blockade of induction of apoptosis. However, it should be noted that in contrast to BCR/ABL, rearrangement of PML/RAR-alpha also blocks differentiation (see section 8). Multidirectional effects of hybrid molecules result in the appearance of cells with increased proliferative potential and resistance to negative regulatory signals and/or unfavorable environmental conditions. Such changes are suggested to be sufficient for the development of some forms of hemoblastoses. Actually, rearrangements BCR/ABL or PML/RAR-alpha are the only genetic changes that are frequently recognized in chronic myeloid and acute promyelocytic leukemias, respectively [3, 12].
However, development of malignant solid tumors (cancers, sarcomas, etc.) also requires other changes which first of include: impaired interactions with adjacent cells and extracellular matrix, loss of contact inhibition of reproduction, increased locomotor activity responsible for invasion into the surrounding and other tissues, tumor neovascularization promoting its nutrition. Thus, it is not surprising that a number of mutations and other genetic abnormalities recognized in solid tumor cells are usually higher than in leukemic cells. The number of genetic reorganizations may reach a few tens. It is clear that the usual rate of mutations cannot account for appearance of numerous mutations within one cell. So, before we start to analyze the role of protooncogenes and tumor suppressors in the regulation of morphogenetic reactions of a cell and neoangiogenesis, let us consider mechanisms underlying the appearance of genetic instability, another important characteristic feature of the neoplastic cell.
3. PROTOONCOGENES AND TUMOR SUPPRESSORS IN CONTROL OF GENETIC
STABILITY
Suppression of induction of apoptosis (observed in neoplastic cells) increases viability of cells exposed to DNA-damaging treatments and therefore increases the probability of preservation of new genetic defects. However, more specialized systems of control of genome integrity exist in the cell, and impairments in their operation are also typical for tumor cells.
Systems controlling genome integrity can be subdivided into two groups: 1) repair systems which recognize and correct errors leading to changes in nucleotide sequence in DNA; 2) systems of cell cycle control which prevent subsequent proliferation in cells in which changes in structure or number of chromosomes occur.
Changes in repair systems are obviously typical for a relatively small proportion of tumors. However, they can play decisive role in the development of certain tumors. For example, inborn defects of genes whose products are responsible for excision repair of DNA cause pigment xeroderma, a syndrome characterized by the development of multiple tumors of skin exposed to solar irradiation [80]. In spite of involvement of excision repair in correction of defects induced not only by UV irradiation but also by various mutagens/carcinogens [81, 82], the frequency of appearance of other tumors remains almost unchanged. Transgenic mice with the same defects of excision repair system are characterized by an increased rate of tumor induction in internal organs by various chemical carcinogens [82]. Preferential appearance of skin tumors in patients with pigment xeroderma may indicate an insignificant role of environmental chemical contaminants in the development of human tumors [83].
Inborn defects of another repair system involved in mismatch repair during DNA replication cause Lynch syndrome. The development of large intestine tumors (so-called hereditary non-polypous colonorectal cancer) and/or ovarian tumors is a characteristic feature of this syndrome [83-86]. (Preferential development of these particular intestinal tumors with defects in this repair system is probably related to extremely high proliferative potential of cells at the bottom of the intestinal crypts, which is inevitably accompanied by increased rate of replication mistakes.) Four genes, MSH2, MLH1, PMS1, and PMS2, in which inactivating mutations cause this syndrome have been identified [84-86]. Easily detectable instability of micro-satellite DNA sequences is a marker of inactivation of any of them [83, 87]. Impairments in mismatch repair system are also typical for some forms of sporadic (non-hereditary) tumors: they are recognized in 13-15% of large intestine tumors and cancers of the stomach and endometrium; in other tumors they are found in only <2% [83].
Impairments in double-strand break repair, which are due to homologous recombinations, are suggested to result in the development of certain tumors as well. Data showing that germinal mutations of suppressor proteins BRCA1 and BRCA2 are responsible for hereditary forms of breast and ovary cancer support this idea [85, 86, 88]. Normal proteins BRCA1 and BRCA2 can form complexes with protein RAD51, a homolog of bacterial protein RecA responsible for homologous recombination, whereas inactivation (knockout) of BRCA1 and BRCA2 genes causes a sharp increase of sensitivity to gamma-irradiation [89-91]. However, it is still unclear whether carcinogenesis is actually due to impairment of these particular functions of BRCA1 and BRCA2 and not by some other activities of these proteins. It should be noted that repair of double-strand DNA breaks occurs at certain periods of the cell cycle, and arrest at these periods sharply increases the efficiency of the repair process. It is possible that the ability of protein BRCA1 to increase expression of p21WAF1/CIP1 via p53-dependent and p53-independent mechanisms [30, 31] and to suppress the transactivation effect of Myc protein [92] is directed toward the arrest of the cell cycle in damaged cells.
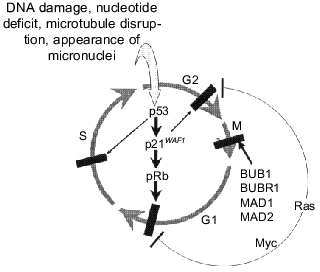
If impairments of repair systems and related “nucleotide instability” are involved in the development of a relatively small number of certain tumors, “chromosome instability” resulting in impairments of normal regulation is typical for the overwhelming majority of solid tumors. The existence of so-called checkpoints has been postulated in the cell cycle; their passing is possible only in the case of normal completion of previous stages and lack of breakage. At least four such points have been distinguished: in G1, S, G2, and also a “spindle assembly checkpoint” in mitosis [27, 93-95].
Checkpoint at G1. The intactness of DNA is a main requirement for the cell to enter the S phase because replication of damaged DNA will lead to transmission of genetic abnormalities to offspring. So a cell exposed to mutagenic treatments inducing DNA breaks (UV and gamma-irradiation) stop in G1 and do not enter S phase [95, 96]. Arrest at G1 is observed not only after DNA damaging treatments but also under other conditions accompanied by changes in chromosome number in the uncompleted previous cell cycle if it did not end by mitosis (chromosome disjunction) [97]. Arrest at G1 is also observed on incorrect chromosome segregation during mitosis which results in micronuclei formation [98] and also on microtubule destruction which may induce subsequent impairments in mitosis [99]. Arrest at G1 may be irreversible (as in the case of gamma-irradiation [100]) or reversible, which terminates with termination of the effect of the stop-inducing factor: on restoration of the normal nucleotide pool [56, 101] or microtubule system [98].
Checkpoint at S phase. This checkpoint monitors the correctness of DNA replication. In particular, arrest at a certain period of the S phase is observed at nucleotide deficit in cells that did not stop for some reason at G1 [102].
Checkpoint at G2. DNA damage and other impairments induce arrest of cells not only at G1 and S, but also at G2 phase of the cell cycle. This allows revealing damages that were either missed during passage through previous checkpoints or acquired during previous stages of the cell cycle. Arrest at G2 phase also allows detection of the completeness of DNA replication, and cells in which DNA are under-replicated do not enter mitosis [103].
Spindle assembly checkpoint. To avoid incorrect chromosome distribution, cells stop at metaphase until all kinetochores are attached to microtubules. Disruption of unattached kinetochores by laser pencil initiates the beginning of anaphase [104] when delay of chromosomes that are not attached to the spindle occurs and micronuclei are formed from them. Changes of interactions between kinetochore associated proteins, BUB1, BUBR1, MAD1, and MAD2 play a certain role in induction of this stop in metaphase [105, 106].
Tumor cells are characterized by changes in the cell cycle checkpoints that are either sensors of changes or effectors realizing cell cycle arrest. For example, inactivation of spindle-assembly checkpoint due to impaired functions of MAD1 or MAD2 is observed in some cases of breast cancer and T-cell leukemias induced by HTLV-1 virus (MAD1 is the direct target of viral oncoprotein Tax). Mutations of genes BUB1 and BUBR1 have been recognized in some cases of large intestine cancers [83, 105]. However, dysfunction of some tumor suppressors and protooncogenes, in particular p53, pRb, Myc, and Ras, has greater importance for inactivation of cell cycle checkpoints (Fig. 6).
p53 is a key component for some checkpoints. As indicated above (see section 2), it is activated in response to various unfavorable treatments that result in genetic abnormalities: DNA breaks [28, 59], deficit of nucleotide pool [56], disruption of microtubules [98], lack of chromosome segregation at mitosis [97] or its incorrect terminations resulting in micronuclei formation [98]. DNA protein kinase and/or protein ATM (ataxia-telangiectasia mutated) are sensors of DNA damages. They can recognize free DNA ends and also phosphorylate p53 at Ser-15; the latter prevents binding of p53 to Mdm2 and its transport from the nucleus and degradation [14, 28]. Sensors of other abnormalities and pathways for their signaling to p53 are not clear.Fig. 6. Cell cycle checkpoints and involvement of some tumor suppressors and oncogenes in their regulation (see explanations in text).
Activation of p53 has a few consequences: change in expression of BAX, BCL2, and other genes controlling apoptosis (see previous section) and expression of p21WAF1 and GADD45 (growth arrest and DNA damage-induced) which stop the cell cycle [56, 57, 59]. This will promote elimination of cells with genetic defects by induction of apoptosis or arrest at G1, G2, or sometimes at S phase of the cell cycle. The choice between these two possible reactions of the cell on p53 activation (apoptosis or cell cycle arrest) is determined by many factors: histogenetic type of cell (cell cycle arrest is more typical for normal fibroblasts, whereas apoptosis dominates in lymphocytes), degree of p53 activation (increase of its expression increases probability of apoptosis), functional activity of p21WAF1--pRb--E2F signaling pathway responsible for arrest at G1 (apoptosis is observed in fibroblasts with inactivated p21WAF1 or pRb), etc. [56, 57, 59]. A point of cell cycle arrest is determined by a phase of the cell cycle in which increased expression of p53 occurred in a given cell [107] and a factor provoking this activation [56]. Impaired functions of p53 typical for most human tumors significantly attenuate controlling functions of the cell cycle checkpoints and simultaneously inhibit induction of apoptosis [106, 108]. Together with some other consequences of p53 dysfunction (loss of mechanism limiting formation of additional centrosomes [109]) these impairments sharply increase the probability of appearance of proliferating cells with spontaneous or induced genetic abnormalities: changes of chromosome number [110-112], breaks and recombination of chromosomes [110, 112, 113], amplification of certain genes [112, 114-116]. Restoration of normal functioning of p53 in cells with its insufficiency reduces the rate of appearance of genetic abnormalities [111].
Genome destabilization is also observed during dysfunction of other tumor suppressors, in particular pRb. However, in this case the rate of appearance of genetic changes and their spectrum in proliferating cells are significantly lower than in cells with p53 dysfunction because pRb inactivation attenuates only the operation of the checkpoint at G1 (Fig. 6) and insignificantly influences the checkpoint at G2. Inactivation of pRb also does not block p53-dependent apoptosis in abnormal cells.
Activation of some protooncogenes may also attenuate the operation of the cell cycle checkpoints (Fig. 6) and consequently increase genetic instability. For example, Myc overexpression allows overcoming the inhibiting effect of p21WAF1 on the cyclin D--CdK4 and cyclin E--Cdk2 complexes, thus abolishing arrest at G1 induced by p53 activation. Ras hyperfunction can also attenuate operation checkpoints at G1 and G2 and induced genetic instability. However such effects are realized only in cells characterized by certain abnormalities in p53-regulated signaling pathways [117].
Thus, changes of tumor suppressors (inactivation of p53, pRb, and, possibly, p16INK4a-p19ARF) and/or protooncogenes (activation of Myc, Ras, and others), which are often observed in human tumors to result in dysfunction of the cell cycle checkpoints and genome instability. Tumor cells are also characterized by changes in some other genes responsible for maintenance of genome integrity. Moreover, inborn inactivating mutations not only of p53 or pRb but also some other genes of repair systems always result in the development of certain tumors. This suggests an important role of genetic instability in the genesis of tumors and/or their subsequent progression. Although increased genome instability is not ultimately required for oncogenesis, it is ultimately required for the appearance of a sufficient number of mutations in one cell that determine malignant growth of solid tumors. Creating heterogeneity of the cellular population of genetic instability constantly provides material for selection of more and more autonomous and aggressive cells.
4. ONCOGENES, TUMOR SUPPRESSORS AND IMPAIRMENTS OF MORPHOGENETIC
REACTIONS OF CELLS
“Asocial behavior” is a characteristic feature of neoplastic cells. First of all this is related to dysfunction of normal morphogenetic reactions: loss of contact inhibition of reproduction, acquisition of proliferative ability irrespectively to substrate adhesion, changes of adhesive interactions, shape and motility of cells, etc. These impairments together with other properties (ability to secrete proteolytic enzymes and angiogenic factors) predetermine the invasive character of growth (penetration into surrounding normal tissues) and subsequent metastasizing (formation of secondary foci of tumor growth) [118]. Changes in protooncogene and/or tumor suppressor functioning play a primary role in impairments of these morphogenetic reactions (Figs. 7 and 8).
Fig. 7. Changes in activity of suppressor proteins and protooncogene-regulated signaling pathways determining the dependence of cells on substrate adhesion and contact inhibition of proliferation (see explanation in text).
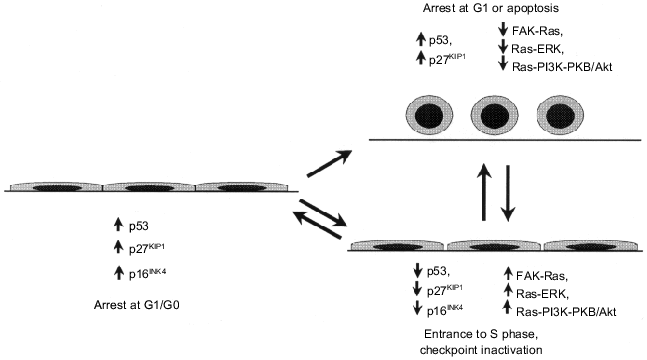
Contact inhibition of proliferation (establishment of contacts with adjacent cells halts proliferation), a property of normal cells, is related to an increase in expression of tumor suppressors p16INK4a and p27KIP1 [37, 119, 120] and subsequent pRb dephosphorylation and blockade of entrance to S phase [17] (see section 1). Signal transduction pathways from plasma membrane to inhibitors of cyclin-dependent kinases are not clear. It is known that the increase in E-cadherin expression in epithelial cells induced by transduction of its gene is accompanied by accumulation of p27KIP1 and growth arrest [121]. Recently the existence of another pathway of blockade of the cell cycle in response to formation of intercellular contacts has been demonstrated. Formation of an epithelial layer induces accumulation of p53, whereas mutations of E-cadherin and/or uncoupling of intercellular contacts have the opposite effect: they cause destabilization of p53 followed by abolishing inhibitory effects of p21WAF1 on cyclin--Cdk complexes. It is possible that the oncogenic potential of E-cadherin mutations responsible for the development of hereditary forms of stomach cancer and other tumors [122] is at least partially due to changes of the cell cycle, apoptosis, and genetic stability control [123].Fig. 8. Oncogene-regulated signaling pathways responsible for changes in morphology and locomotor phenotype of neoplastic cells (see explanations in text).
Besides inactivation of tumor suppressors (E-cadherin, p53, p27KIP1, pRb) caused by mutations or binding to viral oncoproteins (pRb with E1A, E7, T-SV40; p53 with E1B, E6, T-SV40; p27KIP1 with E1A, etc. [7, 10, 124]), hyperfunction of protooncogenes modifying the activity of the Cdk--pRb--E2F signaling pathway can also result in the loss of contact inhibition of reproduction. This may be caused by increased Myc expression or Ras protooncogene activation (the former induces degradation of p27KIP1 and transactivation of Cdc25a, the latter causes the degradation of p27KIP1 and increase of cyclin D1 expression, see section 1).
Anchorage independence. Survival and proliferation of most types of normal cells require anchorage to extracellular matrix. In detached cells growth factors cannot activate cyclin E--Cdk2 complexes responsible for entrance to the S phase [125]. Lack of adhesion interactions induces apoptosis in most cell types (this type of apoptosis has a special term, anoikis) [126]. Suppression of proliferation and induction of apoptosis in detached cells may be related to p53 activation induced by lack of both cell anchoring to substrate and signals from integrin receptors [58, 127]. Besides activation of p53--p21WAF1 pathway, the accumulation of p27KIP1 observed during lack of cell contacts with matrix [127, 128] is also responsible for blockade of cell entrance to S phase. However, besides triggering of mechanisms of negative proliferation control (blockade of entrance to S phase and induction of apoptosis) in response to cell detachment from matrix, independent mechanisms of positive regulation and cell proliferation also exist. They are initiated by integrin binding to extracellular matrix proteins and subsequent activation of non-receptor FAK tyrosine kinase (focal adhesion kinase is a key component of signal transduction from integrin receptors which physically interacts with the cytoplasmic domain of the beta-subunit of integrin [118, 126]). Integrin binding to matrix and FAK activation are necessary for mitogenic signal transduction from growth factor (EGF, PDGF) receptors to terminal MAP kinases, ERK1/2. (In detached cell growth factor signals are blocked at the intermediary MAP kinase, MEK1, and by some unknown reasons the latter does not phosphorylate its targets, ERK1/2 [64]). The integrin receptors not only transduce mitogenic signal (by Ras activation via adapter protein Shc) but also suppress anoikis (apoptosis) via activation of the Ras--PI3K--PKB/Akt signaling pathway [126] (see section 2). The existence of a few mechanisms determining the dependence of viability and/or cell proliferation on binding to matrix may well explain why acquisition of independence from adhesion interactions typical for tumor cells requires several events. Such events would allow overcoming suppressor effects of p53 (mutations/deletions of this gene, MDM2 oncogene overexpression, etc.) and/or p27KIP1 (mutations/deletions of this gene, RAS and MYC oncogene overexpression resulting in degradation of this protein) and bypass inhibition of mitogenic signal at MEK1 kinase (for example, due to activation of proteins Src or Myc, which induce activation of cyclin E--Cdk2 complexes) and also block anoikis via Ras--PI3K--PKB/Akt.
Changes of form and motility of cells. Changes in cell morphology are a typical property of tumor cells that is used for microscopic diagnostics of malignant transformation. They are due to interrelated changes of cytoskeleton and cell--cell and cell--extracellular matrix interactions. They are expressed as impairments in formations of focal contacts, impaired attachment of cells to matrix, disorganization of actin-microfilament system. This results in changes in the activity of pseudopodia and cell motility. The whole scenario is reminiscent of changes that appear in normal cells during the action of motogenic cytokines, factors stimulating cell motility. However, so-called locomotor phenotype of neoplastic cells is usually so exaggerated that it allows distinguishing tumor cell morphology and a normal moving cell.
Molecular mechanisms underlying the appearance of locomotor phenotype both in normal cells under mitogenic stimuli and in neoplastic transformation are not clear. Only some key points in cross-linking connections of signal transduction pathways responsible for the appearance of these changes have been recognized. Many cytokines (e.g., HGF/SF, EGF, FGF, PDGF, IGF-1, etc.) are mitogens and motogens simultaneously [118]. For example, HGF/SF (hepatocyte growth factor/scatter factor) is a potent mitogen for hepatocytes and a motogen for various epithelial cells (mammary gland cells, endotheliocytes, etc.). The motogenic effect of HGF/SF is determined by HGF/SF-induced stimulation of Ras--Raf--MAPK signaling pathways; inhibition of MEK1 functioning and blockade of signal transduction to ERK1/2 abolishes the motogenic effect [129-131]. However, besides Raf activation, disconnection of E-cadherin intercellular contacts in epithelial cells and stimulation of their locomotion also requires simultaneous PI3K activation, which can be induced via Ras-dependent and Ras-independent pathways [130, 132]. Effectors of PI3K responsible for realization of the motogenic effect remain unknown. Although a basal level of Rac activity is necessary for cytoskeleton reorganization and disconnection of intercellular contacts required for locomotion [132], neither PKB/Akt nor Rac can cause the motogenic effect [130]. Decrease of fibronectin gene expression is one of the most important effects induced by HGF/SF in soft tissue sarcoma cells; this can alter adhesion interactions with matrix and locomotion [133]. Mutations in HGF/SF receptor gene (protooncogene Met) leading to permanent stimulation of its tyrosine kinase activity are oncogenic: they can induced morphologic transformation of cultivated cells in vitro [134]. These mutations are also responsible for the development of hereditary papillar kidney cancer and some other human tumors (Table 1). Molecular mechanisms of motogenic effects of other cytokines are less studied; however, as in the case of HGF/SF they are suggested to be related to stimulation of Ras--PI3K and Ras--MAPK signaling pathways [135-139].
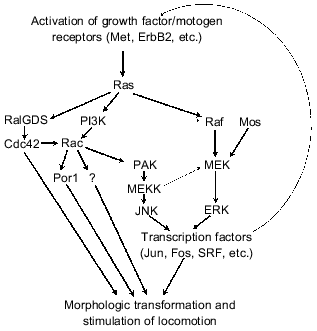
Constitutive expression of activated Ras oncogenes in fibroblasts and epithelial cells induces a sharp increase of locomotion activity and stable morphological changes typical for neoplastic cells [140, 141]. (It should be noted that these were recognized only in cells with abnormalities in p53- and/or p16INK4a-regulated signaling pathways; in other cases apoptosis or arrest at G1 reducing manifestations of Ras-induced morphological changes [117, 142] can be induced in response to permanent Ras overexpression (see also paper by P. M. Chumakov in this issue)). Several Ras effectors, first of all Raf and GTPases of the Rho family (Rac, Cdc42, Rho), are suggested to be responsible for manifestation of morphological transformation and stimulation of locomotion activity of neoplastic cells [20, 143] (Fig. 8). As in the case of cytokine motogenic effects activation of two signaling cascades, Ras--PI3K--Rac and Ras--Raf--ERK probably play a key role [20, 143-145]. Constitutive activation of either pathway can induce fibroblast transformation. In fact, transduction of activated Rac1, Raf, Mos (which exerts Ras-like stimulation of MEK1) or MEK1 can induce morphological transformation of rodent fibroblasts [146-150]. Blockade of MEK1 or Rac functioning in Ras-transformed cells results in partial reversion of morphological changes but does not prevent transformation [148, 151]. However, in epithelial cells which are well transformed by Ras oncogene the activation of Raf--ERK cascades is not sufficient for induction of morphologic transformation [149]. Expression of activated Rac acquires some signs of the transformed phenotype to epithelial cells (formation of lamellopodia and membrane ruffling [143, 152]); however, strong morphological transformation is apparently achieved only during simultaneous activation of Rac-dependent and Raf--ERK signaling pathways [144, 153].
Limited information about molecular events responsible for morphological transformation during activation of Rac and Raf--ERK signaling pathways is now available. Stimulation of MAP kinases ERK1/2 results in the activation of some transcriptional factors, such as Elk1, Fos, and SRF. The stimulation of another MAP kinase cascade (Fig. 8) resulting in activation of transcriptional factors Jun and ATF2 by its end product JNK is one of the most important consequences of Rac hyperfunction. Thus, the Raf--ERK and Rac--JNK signaling pathways regulate activity of transcriptional complexes AP-1 (they consist of homodimers Jun/Jun or heterodimers Jun/Fos) that are important for induction of morphological changes. Oncogene Jun can induce cell transformation [154, 155], whereas suppression of Jun functioning is accompanied by reversion of transformed phenotype [156, 157]. It is possible that the transforming potential of Jun (AP-1) is related not to direct effect on targets regulating adhesion interactions, organization of cytoskeleton, and locomotion but to formation of an autocrine loop due to stimulation of production of growth factors/motogens (EGF, etc.) which activate Ras and its effectors, in particular, Rac and other GTPases of the Rho family (Cdc42, RhoA) responsible for morphological changes [155, 157] (Fig. 8). For example, it is suggested that Rac provides formation of lamellopodia and ruffling, and Cdc42 is involved in formation of filopodia, whereas RhoA participates in formation of focal contacts and stress fibers [143, 152].
The existing models consider formation of lamellopodia as a consequence of Rac-induced increase in activity of Por1 protein [126, 152] and the appearance of stress fibers as the result of Rho kinase-induced activation of myosin phosphatase. Focal contact formation is considered to be the result of actin binding with cytoskeleton proteins, prophylin and gelsolin, which is stimulated by phosphatidylinositol bisphosphate (one of the targets of Rho) [126, 152]. However, its remains unclear whether all these reactions are due to signal transduction solely among cytoplasmic proteins, or whether changes in the activity of numerous transcriptional factors also contribute to these processes.
Thus, protooncogenes play the major role in morphological transformation and acquisition of locomotor phenotype. Changes in their activity result in activation of proteins of the Ras family and/or its effectors, PI3K, Raf, and, perhaps, RalGDS. Apparently, only the combination of protooncogene-induced changes in the regulation of activity of pseudopodia and assembly/disassembly of cytoskeleton and focal contacts with simultaneous change in activity of many transcriptional factors finally provides acquisition of so-called “completely transformed” phenotype causing aggressive cell proliferation. Manifestation of these changes also requires inactivation of tumor suppressors (p53, p19ARF, and/or p16INK4a) protecting the organism against the appearance of cell clones with constantly activated Ras--MAP kinase signaling pathways. Increased expression of some other tumor suppressors, E-cadherin [118] and pRb [158], also reduces manifestation of morphological transformation and locomotion ability. Moreover, two other most important signs of the neoplastic cell, attenuation of contact inhibition of proliferation and acquisition of substrate-independence depends on inactivation of certain tumor suppressors: p53, p27KIP, Rb, and E-cadherin. So, it is clear that the appearance of changes of morphogenetic reactions typical for tumor cells requires a few genetic events (mutations) involving both tumor suppressors and protooncogenes.
5. ONCOGENES AND TUMOR SUPPRESSORS IN NEOANGIOGENESIS
Neoangiogenesis, capillary network formation from endothelial cells lining small venules, is a necessary precondition for subsequent growth of the tumor nidus to 2-4 mm in diameter [118, 159]. The ability of neoplastic cells to stimulate proliferation and migration of endothelial cells is apparently related to two main events, termination of secretion of angiogenesis inhibiting factors (thrombospondins, etc.) and increase in cytokine production. Cytokines are growth factors and motogens for endotheliocytes (VEGF and also FGF, EGF, TGF-alpha); their production is accompanied by an increase in secretion and/or activity of proteases providing proteolysis of extracellular matrix and endotheliocyte invasion to the neoplastic tissue.
Inactivation of tumor suppressor p53 controlling expression of some inhibitors and stimulators of angiogenesis plays a key role in formation of angiogenic phenotype of neoplastic cells. For example, genes of thrombospondins 1 and 2 are targets for the transactivating effect of p53 [160, 161]; the latter also suppresses gene transcription of VEGF [162, 163]. Together with p53 activation in response to hypoxia [164], this represents the mechanism by which normal p53 functioning protects the organism against tumor growth. The development of hypoxia in the center of the neoplastic nidus induces p53 and as a consequence apoptosis or cell cycle arrest. This is accompanied by increased secretion of thrombospondins and decreased VEGF expression that must prevent nidus neovascularization. Thus, p53 inactivation may be an important step in acquisition of angiogenesis stimulating activity. In fact, analyzing the mechanisms of appearance of angiogenic phenotype in human fibroblasts, it was found that in most cases p53 inactivation was the initiating event [165].
Expression of oncogenes may result in subsequent increase in angiogenesis stimulation. In particular, expression of the RAS oncogene family induces activation of transcriptional complex AP-1 and increase in VEGF secretion; the gene of the latter contains AP-1-responsive elements [165-169]. Activation of AP-1 transcriptional complex also causes production of matrix metalloproteases (MMP-9/collagenase IV, MMP-1, etc.) [169-171]. Their genes are also regulated by AP-1 [172, 173] and other Ras-inducible transcriptional factors [172, 173].
Another sequence of events resulting in the development of angiogenic phenotype is also possible. The appearance of angiogenic phenotype in fibrosarcomas of transgenic mice was associated with increased expression of JunB and c-Jun components of AP-1 complex accompanied by subsequent FGF-beta secretion from tumor cells rather than with mutations of p53 [174]. Some evidence exists that other oncogenes and tumor suppressors are also involved in regulation of angiogenesis. For example, Myc suppresses transcription of thrombospondin 1 [26]. Mutations of tumor suppressor VHL causes von Hippel--Lindau syndrome (the development of multiple hemangiomas) and kidney carcinoma [175] (Table 2); tumor suppressor VHL is involved in negative regulation of VEGF gene expression in stromal cells of hemangioma [176] and in kidney epithelial cells [177]. Thus, there is evidence that changes in the activity of certain tumor suppressors and oncogenes play decisive roles in stimulation of angiogenesis.
6. THE ROLE OF ONCOGENES AND TUMOR SUPPRESSORS IN ACQUISITION OF
METASTASIZING ABILITY
Metastasizing, the formation of secondary nidi of tumor growth, is the most dangerous manifestation of tumor progression. It is the main reason for the deaths of oncological patients. For metastasizing the cell must acquire a number of properties. It must penetrate deep inside normal surrounding tissues (including blood or lymphatic vessels), survive in vessels and then penetrate through the vascular wall and proliferate in unusual (for the given cell type) microenvironments giving a new nidus of tumor growth [118]. Thus, metastasizing ability represents a complex of simpler signs: acquisition of locomotor phenotype, increase in proteolytic activity, and the ability to stimulate angiogenesis. They are responsible for tumor cell evacuation from the primary nidus and appearance of substrate-independence and inhibition of apoptosis (these were considered in the previous sections). Manifestation of either sign increases the probability of increase in metastasizing potential. However, such proteins (and genes which encode them) as p53, Ras, and Src are of the major importance because changes in their activity cause simultaneous appearance of a few components of metastatic phenotype and genetic instability that promotes the appearance of additional signs required for metastasis. Abnormalities of p53 functioning significantly increase metastasizing ability of model cellular systems in vivo [58, 178, 179]. Transcription of a recently recognized gene of transmembrane protein KAI1 that forms complexes with E-cadherin is directly activated by p53. Loss of protein KAI1 expression due to various reasons including p53 inactivation [180] was recognized in later stages of various human tumors (60-90% of cases of cancers of the prostate, pancreas, mammary glands, small cell type lung cancer, hepatocellular cancer, etc.) [181-185] whereas restoration of its expression causes inhibition of the metastasizing process [186-188]. Overexpression of calcium binding protein S100A4/MTS1/CAPL (metastasin) occurs at later stages of human tumors [189], and this increases the invading and metastasizing potential of the cells [190-192]. Expression of this protein also exerts pleiotropic effects: inhibition of E-cadherin content [193], suppression of metalloprotease inhibitor, TIMP-1 [194], changes in the regulation of cytoskeleton reorganization (as the result of inhibition of myosin heavy chains phosphorylation) [195], and possibly to sequestration and functional inactivation of p53 [189].
Of course, all of these changes do not exhaust the list of signs controlled by tumor suppressors and/or protooncogenes that may play a significant role in the acquisition of metastasizing ability. For example, G. I. Deichman described so-called “H2O2CA + PGES” phenotype which consists of an increase in antioxidant activity, prostaglandin E2 secretion that increases tolerance to factors of natural resistance and acquired immunity [196, 197]. This phenotype was induced in vitro in cultivated cells via transduction of certain isoforms of v-src oncogene but not other studied genes (activated H-ras, myc, bcl2, mutant p53, E1A, LT SV40). It may also appear during tumor growth in vivo, and metastasizing ability of cells correlate with the appearance of this phenotype (see review by G. I. Deichman in this issue). In the near future we can expect important new discoveries that will clarify metastasizing mechanisms and the role of tumor suppressors and oncogenes in these processes.
7. ROLE OF ONCOGENES AND TUMOR SUPPRESSORS IN NEOPLASTIC CELL
IMMORTALIZATION
Tumor formation from a single precursor cell and subsequent metastasizing requires a large number of cell divisions. However, the number of division of most normal cells (with the exception of stem cells) is limited. For example, cultivated in vitro fibroblasts and human epithelial cells irreversibly halt after 50-60 divisions (the so-called Hayflick number) at G1 or G2 phases of the cell cycle [198]. This phenomenon has been called “replicative aging” (see reviews [199-202]). Such a limiting mechanism is due to progressive shortening of the telomere which results from incomplete replication of chromosome ends during each mitotic cycle [203-205]. According to modern concepts, the arrest of the cell cycle is due to formation of chromosome sticky ends and their subsequent sticking triggers reactions that are similar to those observed during effects of DNA damaging agents [200]. However, the presence of active telomerase, an enzyme responsible for de novo elongation of DNA telomere repeats, or activation of so-called “alternative mechanisms of telomere elongation” (for example, based on non-reciprocal recombination of their sites [206, 207]) may result in cell immortalization, which means cancellation of the limitation of cell division number [201, 202, 208, 209]. The following data support this concept: 1) in contrast to normal human cells, tumor cells as well as stem cells contain active telomerase [201, 208-210]; 2) transduction of vectors expressing the catalytic subunit of telomerase (TERT) increases life span of some normal human cell lines by at least 20 divisions [211, 212].
Recently it has been shown that telomerase activity is controlled by Myc oncoprotein, which increases transcription of the gene encoding the TERT subunit [212]; the level of its expression determines telomerase activity in normal cells [211, 212]. Other cellular and viral oncoproteins (activated Ras, Mdm2, cyclin D1, Cdc25A, E7 HPV) do not activate telomerase [212], whereas E6 HPV16 does activate it [213]. The latter effect is realized via Myc expression [212]. It is possible that telomerase activation in mitogen-stimulated lymphocytes and in proliferative zones of hair follicles and intestinal crypts (see review [209]) is also due to expression of Myc protein in them [212]. (It should be noted that in cells of proliferating normal cells, telomerase is usually inactive [201, 210].)
However, the activation of mechanisms preventing telomere shortening is not the only precondition for immortalization of cells. For example, transduction of TERT or E6 abolishing limitation of a number of cell divisions in some cell lines did not result in immortalization of IMR-90 fibroblasts [212], keratinocytes, and mammary gland epithelial cells [214] in spite of telomerase activation and telomere elongation in them. Immortalization of such cells requires additional inactivation of certain tumor suppressors [201]. Different cell types require inactivation of different suppressors [208]. For example, immortalization of human keratinocytes and mammary gland epitheliocytes was observed during TERT transduction and simultaneous inactivation of either pRb or p16INK4a, whereas elimination of p53 or p19ARF did not cause such effect [214]. In contrast to human cells, mouse cells contain constitutively activated telomerase [215] and as a rule the inactivation of p53 or p19ARF results in immortalization (see for review [13]). However, premature aging of the mouse cells (as well as human ones) can be induced by increasing the activity of either of the above-mentioned tumor suppressors, p53, p19ARF, p16INK4a, pRb, and also p21WAF1 [216-219].
Constitutive telomerase activation in many types of normal mouse cells and long initial length of their telomeres (20 kb, whereas in human cells telomere length varies from 5 to 15 kb) may well explain the increased ability of mouse cells for immortalization and transformation in response to the effect of various carcinogens. This may also explain why proliferating stem cells (characterized by constitutive telomerase activation) are more susceptible to neoplastic transformation than differentiated cells.
The possible use of potential telomerase inhibitors for tumor therapy has recently been discussed in the literature. However, studies on transgenic mice with gene knockout of the RNA-subunit of telomerase gave unexpected results that question such therapeutic treatment (see review [219]). Two phases, called “early crisis” and “genetic catastrophe”, exist in replicative aging. The former is related to either activation of p53 and/or Cdk inhibitors in response to telomere shortening to some critical size or some other signals. The latter (“genetic catastrophe”) is obviously due to total telomere dysfunction and chromosome sticking. It was found that during p53 inactivation (typical for most tumors) “early crisis” was not observed in spite of telomerase knockout and telomere shortening. Moreover, telomerase blockade in such cells even increases genetic instability. In these cells (at least in mouse cells) “genetic catastrophe” was not observed, possibly due to triggering of alternative mechanisms of telomere elongation. Thus, inactivation of the telomerase gene in mice (with or without additional knockout of p53 gene) does not prevent the formation of tumors; moreover it actually increases the rate of their appearance [219]. However, knockout of tumor suppressor Ink4a with additional knockout of the telomerase gene reduced the rate of tumor formation in mice. In this connection it is possible that telomerase blockade may exert a therapeutic effect with respect of some tumors that preserve functional p53 [219].
8. ONCOGENES, TUMOR SUPPRESSORS, AND ABNORMALITIES OF CELL
DIFFERENTIATION
Abnormalities of cell differentiation are a characteristic feature of tumor cells that is widely used in diagnostics. They are especially demonstrative in hemoblastoses when cell clones are fixed (“frozen”) at a certain stage of maturation. According to a conventional concept, lower maturation of leukemic cells reflects their origin from immature cells (with blocked processes of subsequent differentiation) rather than de-differentiation of mature cells that underwent neoplastic transformation (Fig. 9) (see also paper by G. I. Abelev in this issue). This concept has strong experimental support: transduction of chimeric gene PML/RAR-alpha (its formation is responsible for the development of acute promyelocytic leukemia, Table 1) and some other oncogenes (MYC, MYB, v-erbA) is accompanied by a loss of differentiation of immature cell recipients which is normally induced by retinoic acid, specific cytokines, and other maturation inducers [76, 220-222]. Expression of PML/RAR-alpha protein prevents not only myeloid but also megakaryocyte differentiation induced in corresponding precursor cells by thrombopoietin [222].
It should be noted that arrest of differentiation is not sufficient for the development of leukemia. For example, binding of v-erbA oncogene (which encodes reconstructed nuclear thyroid hormone receptor possessing a dominant negative effect) to specific responsive elements of some genes completely blocks erythrocyte formation from erythroblasts but does not cause the development of erythroblastosis. This disease appears only in the case of simultaneous stimulation of erythroblast proliferation induced by additional expression of oncogenes that trigger Ras--Raf--MAP kinase cascades and/or activation of transcription AP-1 complexes [220].Fig. 9. Models explaining neoplastic origin from immature cells of a certain differentiation stage with preserved or blocked mechanisms responsible for subsequent maturation (see explanation in text).
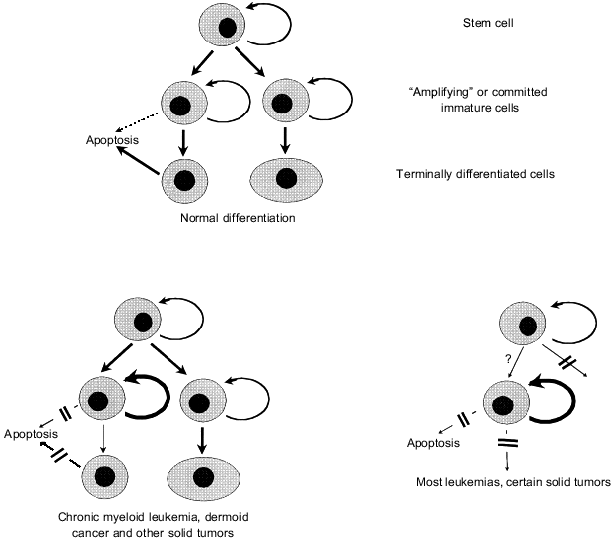
Moreover, blockade of cell differentiation is not an obligatory precondition for tumor growth even in the case of leukemias. For example, chromosome translocation in immature non-committed cell results in chronic myeloid leukemia. This translocation causes the expression of chimeric protein p210BCR/ABL that stimulates proliferation and inhibits apoptosis (see section 2) accompanied by a significant increase in quite mature myeloid cells. Besides myeloid cells, other mature committed cells, lymphocytes and histiocytes, may be descendants of the progenitor leukemic cell [223, 224]. It should be noted that the course of chronic myeloid leukemia is more or less non-malignant and it becomes malignant only during so-called blast crisis when a cell clone with blocked differentiation appears as a result of additional genetic changes.
Maintenance of differentiation ability is also observed in many solid tumors, but in contrast to leukemias, cell maturation does not prevent acquisition of malignant phenotype. Dermoid horn cancers and highly differentiated colon adenocarcinomas originating from either amplifying cells (stem cell descendants that divide several times and then differentiate) or from committed immature cells [225]. The origin from immature cells does not contradict the de-differentiation concept that in the course of progression the tumor cells can undergo certain de-differentiation and loose differentiation markers; their lack provides some selective advantages to cells (receptors of steroid hormones in breast cancers, etc.). As Abelev and Sell indicate [226], complete loss of tissue specificity was never observed in tumors; this may be explained by the tissue specificity of expression of some oncogenes or other genes required for maintenance of neoplastic transformation.
If oncogene expression can block the differentiation processes, the activation of tumor suppressors, on the contrary, can induce cell maturation. For example, the intensity of B-cell, erythroid, enterocyte, epidermal, muscular differentiations depend on the activity of p53 [227-230], p21WAF1 [231-234], and pRb [235]. The stimulation of cell differentiation by the suppressor proteins is suggested to be related to cell cycle arrest at G0/G1 that is the necessary precondition for maturation of many cell types [236]. Involvement of some other additional mechanisms is also possible. For example, p53 acting as a transcriptional factor may stimulate expression those genes whose products are involved in some special differentiation.
Although it is reasonable to suggest that the influence of oncogenes on cell differentiation is mainly related to changes in the regulation of proliferation, the actual effects of oncogenes are more complex. First of all they strongly depend on the tissue specificity of cells. For example, activated Ras and Myc stimulate proliferation of many cell types. However, their expression in monoblasts blocks proliferation and transition into monocytes [237]. In transgenic animals constitutive Myc expression stimulates terminal keratinocyte differentiation by stimulating division and transition of stem cells into amplifying ones [225].
Second, mechanisms of oncogene effects on cell differentiation are obviously not limited by their influence on proliferation. For example, Spi1 (PU.1), a member of the Ets transcription factor family, belongs to a category of so-called differentiation “master genes” which regulate the activity of a large number of genes that determine committing and subsequent cell maturation in one or another direction. Spi1 activity, also responsible for activation of proliferation suppressor p21WAF1 during the action of various differentiation stimuli [238], predetermines myeloid cell differentiation. Perverted functioning of this protein in non-committed hemopoietic cells results in the development of erythroleukemias [76]. Leukosogenic effect of Myb oncoproteins blocking differentiation of immature myeloid cells is probably related to the uncoupling of regulatory mechanisms of cell proliferation and expression of proteins involved in their differentiation. The product of the MYB protooncogene is a transcriptional factor which directly activates transcription of genes for myeloperoxidase, neutrophil elastase, CD34, CD13, etc. [76, 221]. However, its oncogenic derivatives that looses this function during reorganizations preserve antiapoptotic activities (protein c-Myb transactivates BCL2) and the ability to promote entrance into S phase [221].
In concluding this section it should be noted that mechanisms of regulation of cell differentiation are the less studied aspects of the action of oncogenes and tumor suppressors. In the recent future this problem will certainly attract the attention of researchers.
Thus, carcinogenesis is a multistage process of accumulation of mutations and other genetic changes leading to abnormalities of cells, apoptosis, differentiation, morphogenetic reactions, and also to non-effective functioning of factors determining specific and nonspecific anti-tumor immunity. Only the sum of such changes acquired as a result of relatively long evolution of neoplastic clones (resulting in selection cells with required signs) can provide the development of tumor formation. The probability of the appearance of a few genetic changes in one cell is sharply increased during impairments in systems controlling genome integrity. So, mutations leading to genetic instability are also an inalienable stage of tumor progression. Moreover, some inborn abnormalities of genetic control systems predetermine the inevitable appearance of neoplasms. They increase the probability of the appearance of various oncogenic mutations in each cell so strongly that during selection pressure the whole complex of changes required for tumor formation will be accumulated in some cell(s) of a proliferating clone sooner or later, and a tumor appears.
Abnormal functioning of tumor suppressors and protooncogenes play a key role in the appearance of the above-mentioned properties of neoplastic cell. Recent studies have identified signaling pathways that are under control of most of these genes. Most of them regulate the activity of the same pathways at different levels of signal transduction. Some of these signaling pathways are simultaneously involved in the regulation of several of the most important physiological processes. For example, activation of the Raf--MAP kinase cascade not only stimulates the entrance into S phase, but also causes changes in the shape and motility of cells; in some cells it inhibits apoptosis. Products of some tumor suppressors and protooncogenes are the key points in cross-linking connections of various signaling pathways. For example, p53 activated in response to various damaging, stressor, or normal regulatory effects interacts with various targets and controls apoptosis, cell cycle passage, genome stability, morphogenetic reactions, and cell differentiation. Ras proteins play a key role in the regulation of division, survival, and differentiation of cells and their interaction with extracellular matrix and locomotion; these effects being realized via the activation of Raf, PI3K, and RalGDS. This explains high incidents of changes in RAS and p53 genes recognized in various tumors; mutations in these genes allow overcoming several important stages on the pathway of tumor progression and acquire a few required properties for the neoplastic cell.
Some neoplasms (especially leukemias) are characterized by specific genetic changes typical only for this disease. These are chromosome translocations resulting in the transposition of protooncogenes and/or tumor suppressors in the genome. Specificity of such changes can be explained by the following reasons. 1) Certain cell types are characterized by increased probability of some genetic reorganizations. For example, during B-cell differentiation a programmed reorganization of immunoglobulin genes occur. Chromosome translocations connecting immunoglobulin genes with MYC protooncogene are the typical error of such reorganizations; MYC protooncogene contains specific signal sequences that are recognized by recombinases exerting immunoglobulin gene reorganizations. It is reasonable to suggest that in B-lymphocyte progenitors such translocations impairing normal regulation of Myc protein occur more often than the other mutations leading to similar biological consequences. 2) Expression or effects of certain oncogenes/tumor suppressors may be tissue specific. 3) Various cell types require different sets of biological properties for acquisition of malignant phenotype. For example, in the case of hemopoietic cells the acquisition of such signs as loss of contact inhibition and locomotor phenotype are less important compared with other cells. Stimulation of proliferation and inhibition of apoptosis and blockade of specific differentiation are decisive features that determine malignant transformation of hemopoietic cells. So, PML/RAR-alpha type reorganizations that are able to acquire these three properties for certain cell types have special selection value during the development of hemoblastoses.
In spite of recent significant progress in our understanding of basic mechanisms of carcinogenesis, many questions still remain unanswered. Mechanisms of tissue specific effects of oncogenes and tumor suppressors are among them, and the study of this problems will undoubtedly become one of the most rapidly developing areas of oncology.
Author is grateful to Yu. A. Rovensky and G. I. Abelev for constructive criticism and stimulating discussion.
REFERENCES
1.Bishop, J. M. (1991) Cell, 64,
235-248.
2.Levine, A. J. (1993) Annu. Rev. Biochem.,
62, 623-651.
3.Rabbits, T. H. (1994) Nature, 72,
143-149.
4.Weinberg, R. A. (1995) Ann. N. Y. Acad.
Sci., 758, 331-338.
5.Hunter, T. (1997) Cell, 88,
333-346.
6.Hooper, M. L. (1998) EMBO J., 17,
6783-6789.
7.Dyson, N., Buchkovich, K., Whyte, P., and Harlow,
E. (1989) Princess Takamatsu Symp., 20, 191-198.
8.Sugden, B. (1993) Trends Biochem. Sci.,
18, 233-235.
9.Hoppe-Seyler, F., and Butz, K. (1995) J. Mol.
Med., 73, 529-538.
10.Flint, J., and Shenk, T. (1997) Annu. Rev.
Genet., 31, 177-212.
11.Scarpa, A., and Tognon, M. (1998) Int. J. Mol.
Med., 1, 1011-1023.
12.Sanchez-Garcia, I. (1997) Annu. Rev.
Genet., 31, 429-453.
13.Sherr, C. J. (1998) Genes Dev., 12,
2984-2991.
14.Prives, C. (1998) Cell, 95,
5-8.
15.Ruas, M., and Peters, G. (1998) Biochim.
Biophys. Acta, 1378, F115-F177.
16.Morgan, D. O. (1997) Annu. Rev. Cell Dev.
Biol., 13, 261-291.
17.Mittnacht, S. (1998) Curr. Opin. Genet.
Dev., 8, 21-27.
18.Helin, K. (1998) Curr. Opin. Genet. Dev.,
8, 28-35.
19.Porter, A. C., and Vaillancourt, R. R. (1998)
Oncogene, 17, 1343-1352.
20.Campbell, S. L., Khosravi-Far, R., Rossman, K.
L., Clark, G. J., and Der, C. J. (1998) Oncogene, 17,
1395-1414.
21.Pawson, T., and Saxton, T. (1999) Cell,
97, 675-678.
22.Dhanasekaran, N., and Reddy, E. P. (1998)
Oncogene, 17, 144-145.
23.Galaktionov, K., Chen, X., and Beach, D. (1996)
Nature, 382, 511-517.
24.Alevizopoulos, K., Vlach, J., Hennecke, S., and
Amati, B. (1997) EMBO J., 17, 5322-5333.
25.Fashini, L. M., and Penn, L. Z. (1998) FASEB
J., 12, 633-651.
26.Dang, C. V. (1999) Mol. Cell. Biol.,
19, 1-11.
27.Sherr, C. J. (1996) Science, 274,
1672-1677.
28.Giaccia, A. J., and Kastan, M. B. (1998) Genes
Dev., 12, 2973-2983.
29.Maheswaran, S., Englert, C., Bennett, P.,
Heinrich, G., and Haber, D. A. (1995) Genes Dev., 9,
2143-2156.
30.Ouchi, T., Monteiro, A. N. A., August, A.,
Aaronson, S. A., and Hanafusa, H. (1998) Proc. Natl. Acad. Sci.
USA, 95, 2302-2306.
31.Zhang, H., Somasundaram, K., Peng, Y., Tian, H.,
Zhang, H., Bi, D., Weber, B. L., and El-Deiry, W. S. (1998)
Oncogene, 16, 1713-1721.
32.Garkavtsev, I., Grigorian, I. A., Ossovskaya, V.
S., Chernov, M. V., and Gudkov, A. V. (1998) Nature, 391,
295-298.
33.Englert, C., Maheswaran, S., Garvin, A. J.,
Kreidberg, J., and Haber, D. A. (1997) Cancer Res., 57,
1429-1434.
34.Datto, M. B., Hu, P. P., Kowalik, T. F.,
Yingling, J., and Wang, X. F. (1997) Mol. Cell. Biol.,
17, 2030-2037.
35.Grau, A. M., Zhang, L., Wang, W., Ruan, S.,
Evans, D. B., Abbruzzese, J. L., Zhang, W., and Chiao, P. J. (1997)
Cancer Res., 57, 3929-3934.
36.Miyazaki, M., Ohashi, R., Tsuji, T., Mihara, K.,
Gohda, E., and Namba, M. (1997) Biochem. Biophys. Res. Commun.,
246, 873-880.
37.Massague, J., and Polyak, K. (1995) Curr.
Opin. Genet. Dev., 5, 91-96.
38.Sun, P., Dong, P., Dai, K., Hannon, G. J., and
Beach, D. (1998) Science, 282, 2270-2272.
39.Xiao, Z. X., Chen, J., Levine, A. J., Modjtahedi,
N., Xing, J., Sellers, W. R., and Livingston, D. M. (1995)
Nature, 375, 694-698.
40.Derynck, R., Zhang, Y., and Feng, X.-H. (1998)
Cell, 95, 737-740.
41.Kretzschmar, M., and Massague, J. (1998) Curr.
Opin. Genet. Dev., 8, 103-111.
42.Bullions, L. C., and Levine, A. J. (1998)
Curr. Opin. Oncol., 10, 81-87.
43.Tetsu, O., and McCormick, F. (1999)
Nature, 398, 422-426.
44.He, T.-C., Sparks, A. B., Rago, C., Hermeking,
H., Zawel, L., da Costa, L. T., Morin, P. J., Vogestein, B., and
Kinzler, K. W. (1998) Science, 281, 1509-1512.
45.Rubinfeld, B., Robbins, P., El-Gamil, M., Albert,
I., Porfiri, E., and Polakis, P. (1997) Science, 275,
1790-1792.
46.Polakis, P. (1997) Biochim. Biophys. Acta,
1332, F127-F147.
47.Morin, P. J., Sparks, A. B., Korinek, V., Barker,
N., Clevers, H., Vogelstein, B., and Kinzler, K. W. (1997)
Science, 275, 1787-1790.
48.Willert, K., and Nusse, R. (1998) Curr. Opin.
Genet. Dev., 8, 95-102.
49.Green, D. R. (1998) Cell, 94,
695-698.
50.Evan, G., and Littlewood, T. (1998)
Science, 281, 1317-1322.
51.Dragovich, T., Rudin, C. M., and Thompson, C. B.
(1998) Oncogene, 17, 3207-3213.
52.Nunez, G., Benedict, M. A., Hu, Y., and Inohara,
N. (1998) Oncogene, 17, 3237-3245.
53.Adams, J. M., and Cory, S. (1998) Science,
281, 1322-1326.
54.Chao, D. T., and Korsmeyer, S. J. (1998) Annu.
Rev. Immunol., 16, 395-419.
55.Reed, J. C. (1998) Oncogene, 17,
3225-3236.
56.Agarwal, M. L., Taylor, W. R., Chernov, M. V.,
Chernova, O. B., and Stark, G. R. (1998) J. Biol. Chem.,
273, 1-4.
57.Amundson, S. A., Myers, T. G., and Fornace, A.
J., Jr. (1998) Oncogene, 17, 3287-3299.
58.Nikiforov, M. A., Hagen, K., Ossovskaya, V. S.,
Connor, T. M. F., Lowe, S. W., Deichman, G. I., and Gudkov, A. (1996)
Oncogene, 13, 1709-1719.
59.Ko, L. J., and Prives, C. (1996) Genes
Dev., 10, 1054-1072.
60.Polyak, K., Xia, Y., Zweller, J. L., Kinzler, K.
W., and Vogelstein, B. (1997) Nature, 389, 300-306.
61.Wu, G. S., Burns, T. F., McDonald, E. R., III,
Jiang, W., Meng, R., Krantz, I. D., Kao, G., Gan, D. D., Zhou, J. Y.,
Muschel, R., Hamilton, S. R., Spinner, N. B., Markowitz, S., Wu, G.,
and El-Deiry, W. S. (1997) Nat. Genet., 17, 141-143.
62.Sheikh, M. S., Burns, T. F., Huang, Y., Wu, G.
S., Amundson, S., Brooks, K. S., Fornace, A. J., Jr., and El-Deiry, W.
S. (1998) Cancer Res., 58, 1593-1598.
63.Downward,J. (1998) Curr. Opin. Genet.
Dev., 8, 49-54.
64.Jarpe, M. B., Widmann, C., Knall, C.,
Schlesinger, T. K., Gibson, S., Yujiri, T., Fanger, G. R., Gelfand, E.
W., and Johnson, G. L. (1998) Oncogene, 17,
1475-1482.
65.Khwaja, A. (1999) Nature, 401,
33-34.
66.LaCasse, E. C., Baird, S., Korneluk, R. G., and
MacKenzie, A. E. (1998) Oncogene, 17, 3247-3259.
67.Wang, C. Y., Guttridge, D. C., Mayo, M. W., and
Baldwin, A. S., Jr. (1999) Mol. Cell. Biol., 19,
5923-5929.
68.Marsh, D. J., Coulon, V., Lunetta, K. L.,
Rocca-Serra, P., Dahia, P. L., Zheng, Z., Liaw, D., Caron, S., Duboue,
B., Lin, A. Y., Richardson, A. L., Bonnetblanc, J. M., Bressieux, J.
M., Cabarrot-Moreau, A., Chompret, A., Demange, L., Eeles, R. A.,
Yahanda, A. M., Fearon, E. R., Fricker, J. P., Gorlin, R. J., Hodgson,
S. V., Huson, S., Lacombe, D., and Eng, C. (1998) Hum. Mol.
Genet., 7, 507-515.
69.Stambolic, V., Suzuki, A., de la Pompa, J. L.,
Brothers, G. M., Mirtsos, C., Sasaki, T., Ruland, J., Penninger, J. M.,
Siderovski, D. P., and Mak, T. W. (1998) Cell, 95,
29-39.
70.Gotoh, A., and Broxmeyer, H. E. (1997) Curr.
Opin. Hematol., 4, 3-11.
71.Amarante-Mendes, G. P., McGahon, A. J., Nishioka,
W. K., Afar, D. E., Witte, O. N., and Green, D. R. (1998)
Oncogene, 16, 1383-1390.
72.Kramer, A., Horner, S., Willer, A., Fruehauf, S.,
Hochhaus, A., Hallek, M., and Hehlmann, R. (1999) Proc. Natl. Acad.
Sci. USA, 96, 2087-2092.
73.Yuan, Z. M., Huang, Y., Ishiko, T., Kharbanda,
S., Weichselbaum, R., and Kufe, D. (1997) Proc. Natl. Acad. Sci.
USA, 94, 1437-1440.
74.Huang, Y., Yuan, Z. M., Ishiko, T., Nakada, S.,
Utsugisawa, T., Kato, T., Kharbanda, S., and Kufe, D. W. (1997)
Oncogene, 15, 1947-1952.
75.Kharbanda, S., Yuan, Z.-M., Weichselbaum, R., and
Kufe, D. (1998) Oncogene, 17, 3309-3318.
76.Tenen, D. G., Hromas, R., Licht, J. D., and
Zhang, D.-E. (1997) Blood, 90, 489-519.
77.Quignon, F., de Bels, F., Koken, M., Feunteun,
J., Ameisen, J. C., and de The, H. (1998) Nat. Genet.,
20, 259-265.
78.Wang, Z. G., Ruggero, D., Ronchetti, S., Zhong,
S., Gaboli, M., Rivi, R., and Pandolfi, P. P. (1998) Nat.
Genet., 20, 266-272.
79.Wang, Z. G., Delva, L., Gaboli, M., Rivi, R.,
Giorgio, M., Cordon-Cardo, C., Grosveld, F., and Pandolfi, P. P. (1998)
Science, 279, 1547-1551.
80.Cleaver, J. E. (1968) Nature, 218,
652-656.
81.Bol, S. A., van Steeg, H., Jansen, J. G., van
Oostrom, C., de Vries, A., de Groot, A. J., Tates, A. D., Vrieling, H.,
van Zeeland, A. A., and Mullenders, L. H. (1998) Cancer Res.,
58, 2850-2856.
82.De Vries, A., Dolle, M. E., Broekhof, J. L.,
Muller, J. J., Kroese, E. D., van Kreijl, C. F., Capel, P. J., Vijg,
J., and van Steeg, H. (1997) Carcinogenesis, 18,
2327-2332.
83.Lengauer, C., Kinzler, K. W., and Vogelstein, B.
(1997) Nature, 396, 643-649.
84.Lynch, H. T., Lemon, S. J., Karr, B., Franklin,
B., Lynch, J. F., Watson, P., Tinley, S., Lerman, C., and Carter, C.
(1997) Cancer Epidemiol. Biomarkers Prev., 6,
987-991.
85.Lynch, H. T., Fusaro, R. M., and Lynch, J. F.
(1997) Ann. N. Y. Acad. Sci., 833, 1-28.
86.Angioli, R., Estape, R., Mason, M., and Penalver,
M. (1998) Int. J. Oncol., 12, 1029-1034.
87.Kolodner, R. (1996) Genes Dev., 10,
1433-1442.
88.Blackwood, M. A., and Weber, B. L. (1998) J.
Clin. Oncol., 16, 1969-1977.
89.Bertwistle, D., and Ashworth, A. (1998) Curr.
Opin. Genet. Dev., 8, 14-20.
90.Marmorstein, L. Y., Ouchi, T., and Aaronson, S.
A. (1998) Proc. Natl. Acad. Sci. USA, 95,
13869-13874.
91.Zhang, H., Tombline, G., and Weber, B. L. (1998)
Cell, 92, 433-436.
92.Wang, Q., Zhang, H., Kajino, K., and Greene, M.
I. (1998) Oncogene, 17, 1939-1948.
93.Murray, A. W. (1995) Curr. Opin. Genet.
Dev., 5, 5-11.
94.Elledge, S. J. (1996) Science, 274,
1664-1672.
95.Murakami, H., and Okayama, H. (1997) Exp. Mol.
Med., 29, 1-11.
96.Paulovich, A. G., Toczyski, D. P., and Hartwell,
L. H. (1997) Cell, 88, 315-322.
97.Lanni, J. S., and Jacks, T. (1998) Mol. Cell.
Biol., 18, 1055-1064.
98.Sablina, A., Ilyinskaya, G., Rubtsova, S.,
Agapova, L., Chumakov, P., and Kopnin, B. (1998) J. Cell Sci.,
111, 977-984.
99.Khan, S. H., and Wahl, G. M. (1998) Cancer
Res., 58, 396-401.
100.Di Leonardo, A., Linke, S. P., Clarkin, K., and
Wahl, G. M. (1994) Genes Dev., 8, 2540-2551.
101.Linke, S. P., Clarkin, K., Di Leonardo, A.,
Tsou, A., and Wahl, G. M. (1996) Genes Dev., 10,
934-947.
102.Agarwal, M. L., Agarwal, A., Taylor, W. R.,
Chernova, O., Sharma, Y., and Stark, G. R. (1998) Proc. Natl. Acad.
Sci. USA, 95, 14775-14780.
103.Taylor, W. R., Agarwal, M., Agarwal, A.,
Stacey, D. W., and Stark, G. R. (1999) Oncogene, 18,
283-295.
104.Rudner, A. D., and Murray, A. W.(1996) Curr.
Opin. Cell. Biol., 8, 773-80.
105.Cahill, D. P., Lengauer, C., Yu, J., Riggins,
G. J., Willson, J. K. V., Markowitz, S. D., Kinzler, K. W., and
Vogelstein, B. (1998) Nature, 392, 300-303.
106.Orr-Weaver, T. L., and Weinberg, R. A. (1998)
Nature, 392, 223-224.
107.Linke, S. P., Harris, M. P., Neugebauer, S.,
Clarkin, K., Shepard, H. M., Maneval, D. C., and Wahl, G. M. (1997)
Oncogene, 15, 337-345.
108.Levine, A. J. (1997) Cell, 88,
323-331.
109.Fukasawa, K., Choi, T., Kuriyama, R., Rulong,
S., and Vande Woude, G. F. (1996) Science, 271,
1744-1747.
110.Harvey, M., Sands, A. T., Weiss, R. S., Heig,
M. E., Wiseman, R. W., Pantazis, P., Clovanello, B. O., Tainsky, M. A.,
Bradley, A., and Donehower, L. A. (1993) Oncogene, 8,
2456-2467.
111.Agapova, L. S., Ilyinskaya, G. V., Turovets, N.
A., Ivanov, A. V., Chumakov, P. M., and Kopnin, B. P. (1996) Mut.
Res., 354, 129-138.
112.Fukasawa, K., Wiener, F., Woud, G. F. V., and
Mai, S. (1997) Oncogene, 15, 1295-1302.
113.Lee, J. M., Abrahamson, L. A., Kandel, R.,
Donehower, L. A., and Bernstein, A. (1994) Oncogene, 9,
3731-3736.
114.Livingstone, L. R., White, A., Sprouse, J.,
Livanos, E., Jacks, T., and Tlsty, T. D. (1992) Cell, 70,
923-935.
115.Yin, Y., Tainsky, M. A., Bishoff, F. Z.,
Strong, L. C., and Wahl, G. M. (1992) Cell, 70,
937-948.
116.Ilinskaya, G. V., Pugacheva, E. N., Sokova, O.
I., Chumakov, P. M., and Kopnin, B. P. (1995) Genetika,
31, 622-627.
117.Agapova, L. S., Ivanov, A. V., Sablina, A. A.,
Kopnin, P. B., Sokova, O. I., Chumakov, P. M., and Kopnin, B. P. (1999)
Oncogene, 18, 3135-3142.
118.Rovensky, Yu. A. (1998) Biochemistry
(Moscow), 63, 1029-1043.
119.Dietrich, C., Wallenfang, K., Oesch, F., and
Wieser, R. (1997) Oncogene, 15, 2743-2747.
120.Wieser, R. J., Faust, D., Dietrich, C., and
Oesch, F. (1999) Oncogene, 18, 277-281.
121.St Croix, B., Sheehan, C., Rak, J. W.,
Florenes, V. A., Slingerland, J. M., and Kerbel, R. S. (1998) J.
Cell. Biol., 142, 557-571.
122.Guilford, P., Hopkins, J., Harraway, J.,
McLeod, M., McLeod, N., Harawira, P., Taite, H., Scoular, R., Miller,
A., and Reeve, A. E. (1998) Nature, 392, 402-405.
123.Tlsty, T. D. (1998) Curr. Opin. Cell.
Biol., 10, 647-653.
124.Rapp, L., and Chen, J. J. (1998) Biochim.
Biophys. Acta, 1378, F1-F19.
125.Fang, F., Orend, G., Watanabe, N., Hunter, T.,
and Ruoslahti, E. (1996) Science, 271, 499-502.
126.Kumar, C. C. (1998) Oncogene, 17,
1365-1374.
127.Wu, R. C., and Schonthal, A. H. (1997) J.
Biol. Chem., 14, 29091-29098.
128.Kawada, M., Yamagoe, S., Murakami, Y., Suzuki,
K., Mizuno, S., and Uehara, Y. (1997) Oncogene, 15,
629-637.
129.Tanimura, S., Chatani, Y., Hoshino, R., Sato,
M., Watanabe, S., Kataoka, T., Nakamura, T., and Kohno, M. (1998)
Oncogene, 17, 57-65.
130.Khwaja, A., Lehmann, K., Marte, B. M., and
Downward, J. (1998) J. Biol. Chem., 273, 18793-18801.
131.Herrera, R. (1998) J. Cell Sci.,
111, 1039-1049.
132.Potempa, S., and Ridley, A. J. (1998) Mol.
Biol. Cell., 9, 2185-2200.
133.Taylor, G. A., Jeffers, M., Webb, C. P., Koo,
H. M., Anver, M., Sekiguchi, K., and Vande Woude, G. F. (1998)
Oncogene, 17, 1179-1183.
134.Jeffers, M., Fiscella, M., Webb, C. P., Anver,
M., Koochekpour, S., and Vande Woude, G. F. (1998) Proc. Natl. Acad.
Sci. USA, 95, 14417-14422.
135.Anand-Apte, B., and Zetter, B. (1997) Stem
Cells, 15, 259-267.
136.Graf, K., Xi, X. P., Yang, D., Fleck, E.,
Hsueh, W. A., and Law, R. E. (1997) Hypertension, 29,
334-339.
137.Adam, L., Vadlamudi, R., Kondapaka, S. B.,
Chernoff, J., Mendelsohn, J., and Kumar, R. (1998) J. Biol.
Chem., 273, 28238-28246.
138.Choudhury, G. G., Karamitsos, C., Hernandez,
J., Gentilini, A., Bardgette, J., and Abboud, H. E. (1997) Am. J.
Physiol., 273, 931-938.
139.Xie, H., Pallero, M. A., Gupta, K., Chang, P.,
Ware, M. F., Witke, W., Kwiatkowski, D. J., Lauffenburger, D. A.,
Murphy-Ullrich, J. E., and Wells, A. (1998) J. Cell Sci.,
111, 615-624.
140.Gloushankova, N. A., Alieva, N. A., Krendel, M.
F., Bonder, E. M., Feder, H. H., Vasiliev, J. M., and Gelfand, I. M.
(1997) Proc. Natl. Acad. Sci. USA, 91, 8597-8601.
141.Gloushankova, N., Ossovskaya, V., Vasiliev, J.,
Chumakov, P., and Kopnin, B. (1997) Oncogene, 16,
536-539.
142.Serrano, M., Lin, A. W., McCurrach, M. E.,
Beach, D., and Lowe, S. W. (1997) Cell, 88, 593-602.
143.Zohn, I., Campbell, S., Khosravi-Far, R.,
Rossman, K. L., and Der, C. J. (1998) Oncogene, 17,
1415-1438.
144.Rodriguez-Viciana, P., Warne, P. H., Khwaja,
A., Marte, B. M., Pappin, D., Das, P., Waterfield, M. D., Ridley, A.,
and Downward, J. (1997) Cell, 89, 457-467.
145.He, H., Watanabe, T., Zhan, X., Huang, C.,
Schuuring, E., Fukami, K., Takenawa, T., Kumar, C. C., Simpson, R. J.,
and Maruta, H. (1998) Mol. Cell. Biol., 18,
3829-3837.
146.Khosravi-Far, R., Solski, P. A., Clark, G. J.,
Kinch, M. S., and Der, C. J. (1995) Mol. Cell. Biol., 15,
6443-6453.
147.Khosravi-Far, R., White, M. A., Westwick, J.
K., Solski, P. A., Chrzanowska-Wodnicka, M., van Aelst, L., Wigler, M.
H., and Der, C. J. (1996) Mol. Cell. Biol., 16,
3923-3933.
148.Okazaki, K., and Sagata, N. (1995)
Oncogene, 10, 1149-1157.
149.Oldham, S. M., Clark, G. J., Gangarosa, L. M.,
Coffey, R. J., Jr., and Der, C. J. (1996) Proc. Natl. Acad. Sci.
USA, 93, 6924-6928.
150.Greulich, H., and Erikson, R. L. (1998) J.
Biol. Chem., 273, 13280-13288.
151.Topol, L. Z., Marx, M., Calothy, G., and Blair,
D. G. (1995) Cell Growth Differ., 6, 27-38.
152.Van Aelst, L., and D'Souza-Schorey, C. (1997)
Genes Dev., 11, 2295-2322.
153.Cobellis, G., Missero, C., and di Lauro, R.
(1998) Oncogene, 17, 2047-2057.
154.Okuno, H., Suzuki, T., Yoshida, T., Hashimoto,
Y., Curran, T., and Iba, H. (1991) Oncogene, 6,
1491-1497.
155.Ljungdahl, S., Linder, S., Franzen, B.,
Binetruy, B., Auer, G., and Shoshan, M. C. (1998) Cell Growth
Differ., 9, 565-573.
156.Dong, Z., Lavrovsky, V., and Colburn, N. H.
(1995) Carcinogenesis, 16, 749-756.
157.Malliri, A., Symons, M., Hennigan, R. F.,
Hurlstone, A. F., Lamb, R. F., Wheeler, T., and Ozanne, B. W. (1998)
J. Cell Biol., 143, 1087-1099.
158.Li, J., Hu, S. X., Perng, G. S., Zhou, Y., Xu,
K., Zhang, C., Seigne, J., Benedict, W. F., and Xu, H. J. (1996)
Oncogene, 13, 2379-2386.
159.Folkman, J. (1995) in The Molecular Basis of
Cancer (Mendelson, J., Howley, P. M., Israel, M. A., and Liotta, L.
A., eds.) W. B. Saunders Press, N. Y., pp. 206-232.
160.Dameron, K. M., Volpert, O. V., Tainsky, M. A.,
and Bouck, N. (1994) Science, 265, 1582-1584.
161.Adolf, K. W., Liska, D. J., and Bornstein, P.
(1997) Gene, 193, 5-11.
162.Sugihara, T., Kaul, S. C., Mitsui, Y., and
Wadhwa, R. (1994) Biochim. Biophys. Acta, 1224,
365-370.
163.Mukhopadhyay, D., Tsiokas, L., and Sukhatme, V.
P. (1995) Cancer Res., 55, 6161-6165.
164.Graeber, T. G., Osmanian, C., Jacks, T.,
Housman, D. E., Koch, C. J., Lowe, S., and Giaccia, A. J. (1996)
Nature, 379, 88-91.
165.Volpert, O. V., Dameron, K. M., and Bouck, N.
(1997) Oncogene, 14, 1495-1502.
166.Grugel, S., Finkenzeller, G., Weindel, K.,
Barleon, B., and Marme, D. (1995) J. Biol. Chem., 270,
25915-25919.
167.Rak, J., Mitsuhashi, Y., Bayko, L., Filmus, J.,
Shirasawa, S., Sasazuki, T., and Kerbel, R. S. (1995) Cancer
Res., 55, 4575-4580.
168.Saez, E., Rutberg, S. E., Mueller, E.,
Oppenheim, H., Smoluk, J., Yuspa, S. H., and Spiegelman, B. M. (1995)
Cell, 82, 721-732.
169.Arbiser, J. L., Moses, M. A., Fernandez, C. A.,
Ghiso, N., Cao, Y., Klauber, N., Frank, D., Brownlee, M., Flynn, E.,
Parangi, S., Byers, H. R., and Folkman, J. (1997) Proc. Natl. Acad.
Sci. USA, 94, 861-866.
170.Meade-Tollin, L. C., Boukamp, P., Fusenig, N.
E., Bowen, C. P., Tsang, T. C., and Bowden, G. T. (1998) Br. J.
Cancer, 77, 724-730.
171.Giambernardi, T. A., Grant, G. M., Taylor, G.
P., Hay, R. J., Maher, V. M., McCormick, J. J., and Klebe, R. J. (1998)
Matrix Biol., 16, 483-496.
172.Himelstein, B. P., Lee, E. J., Sato, H., Seiki,
M., and Muschel, R. J. (1997) Oncogene, 14,
1995-1998.
173.Newberry, E. P., Willis, D., Latifi, T.,
Boudreaux, J. M., and Towler, D. A. (1997) Mol. Endocrinol.,
11, 1129-1144.
174.Christofori, G., and Hanahan, D. (1994)
Semin. Cancer Biol., 5, 3-12.
175.Richards, F. M., Webster, A. R., McMahon, R.,
Woodward, E. R., Rose, S., and Maher, E. R. (1998) J. Int. Med.,
243, 527-533.
176.Wizigmann-Voos, S., Breier, G., Risau, W., and
Plate, K. H. (1995) Cancer Res., 55, 1358-1364.
177.Seimeister, G., Weindel, K., Mohrs, K.,
Barleon, B., Martiny-Baron, G., and Marme, D. (1996) Cancer
Res., 56, 2299-2301.
178.Nikiforov, M. A., Kwek, S. S., Mehta, R.,
Artwohl, J. E., Lowe, S. W., Gupta, T. D., Deichman, G. I., and Gudkov,
A. (1997) Oncogene, 15, 3007-3012.
179.Deichman, G. I., Matveeva, V. A., Kashkina, L.
M., Dyakova, N. A., Uvarova, E. N., Nikiforov, M. A., and Gudkov, A. V.
(1998) Int. J. Cancer, 75, 277-283.
180.Mashimo, T., Watabe, M., Hirota, S., Hosobe,
S., Miura, K., Tegtmeyer, P. J., Rinker-Shaeffer, C. W., and Watabe, K.
(1998) Proc. Natl. Acad. Sci. USA, 95, 11307-11311.
181.Guo, X. Z., Friess, H., di Mola, F. F.,
Heinicke, J. M., Abou-Shady, M., Graber, H. U., Baer, H. U.,
Zimmermann, A., Korc, M., and Buchler, M. W. (1998) Hepatology,
28, 1481-1488.
182.Sho, M., Adachi, M., Taki, T., Hashida, H.,
Konishi, T., Huang, C. L., Ikeda, N., Nakajima, Y., Kanehiro, H.,
Hisanaga, M., Nakano, H., and Miyake, M. (1998) Int. J. Cancer,
79, 509-516.
183.Huang, C. I., Kohno, N., Ogawa, E., Adachi, M.,
Taki, T., and Miyake, M. (1998) Am. J. Pathol., 153,
973-983.
184.Friess, H., Guo, X. Z., Berberat, P., Graber,
H. U., Zimmermann, A., Korc, M., and Buchler, M. W. (1998) Int. J.
Cancer, 79, 349-355.
185.Higashiyama, M., Kodama, K., Yokouchi, H.,
Takami, K., Adachi, M., Taki, T., Ishiguro, S., Nakamori, S., Yoshie
O., and Miyake, M. (1998) Cancer, 83, 466-474.
186.Gao, A. C., Lou, W., Dong, J. T., and Isaacs,
J. T. (1997) Cancer Res., 57, 846-849.
187.Takaoka, A., Hinoda, Y., Satoh, S., Adachi, Y.,
Itoh, F., Adachi, M., and Imai, K. (1998) Oncogene, 16,
1443-1453.
188.Takaoka, A., Hinoda, Y., Sato, S., Itoh, F.,
Adachi, M., Hareyama, M., and Imai, K. (1998) Jpn. J. Cancer
Res., 89, 397-404.
189.Sherbet, G. V., and Lakshmi, M. S. (1998)
Anticancer Res., 18, 2415-2421.
190.Ambartsumian, N. S., Grigorian, M. S., Larsen,
I. F., Karlstrom, O., Sidenius, N., Rygaard, J., Georgiev, G., and
Lukanidin, E. (1996) Oncogene, 13, 1621-1630.
191.Maelandsmo, G. M., Hovig, E., Skrede, M.,
Engebraaten, O., Florenes, V. A., Myklebost, O., Grigorian, M.,
Lukanidin, E., Scanlon, K. J., and Fodstad, O. (1996) Cancer
Res., 56, 5490-5498.
192.Lloyd, B. H., Platt-Higgins, A., Rudland, P.
S., and Barraclough, R. (1998) Oncogene, 17, 465-473.
193.Keirsebilck, A., Bonne, S., Bruyneel, E.,
Vermassen, P., Lukanidin, E., Mareel, M., and van Roy, F. (1998)
Cancer Res., 58, 4587-4591.
194.Andersen, K., Maelandsmo, G. M., Hovig, E.,
Fodstad, O., Loennechen, T., and Winberg, J. O. (1998) Anticancer
Res., 18, 3299-3303.
195.Kriajevska, M., Tarabykina, S., Bronstein, I.,
Maitland, N., Lomonosov, M., Hansen, K., Georgiev, G., and Lukanidin,
E. (1998) J. Biol. Chem., 273, 9852-9856.
196.Deichman, G. I., Kashkina, L. M., Misenina, O.
A., Gorojanskaya, E. G., Nikiforov, M. A., Gudkov, A. V., Dyakova, N.
A., Komelkov, A. V., Prilutskaya, M. O., Kushlinsky, N. E., and
Tatosyan, A. G. (1996) Int. J. Cancer, 66, 747-752.
197.Deichman, G. I., Kashkina, L. M., Kluchareva,
T. E., Matveeva, V. A., Uvarova, E. N., and Burdelya, L. G. (1997)
Adv. Exp. Med. Biol., 400A, 473-477.
198.Hayflick, L. (1965) Exp. Cell. Res.,
37, 614-636.
199.Duncan, E. L., and Reddel, R. R. (1997)
Biochemistry (Moscow), 62, 1263-1274.
200.Vaziri, H. (1997) Biochemistry (Moscow),
62, 1306-1310.
201.Shay, J. W. (1997) J. Cell. Physiol.,
173, 266-270.
202.Garkavtsev, I., Hull, C., and Riabowol, K.
(1998) Exp. Gerontol., 33, 81-94.
203.Olovnikov, A. M. (1973) J. Theor. Biol.,
41, 181-190.
204.Harley, C. B., Futcher, A. B., and Greider, C.
B. (1990) Nature, 345, 458-460.
205.Hastie, N. D., Dempster, M., Dunlop, M. G.,
Thompson, A. M., Green, D. K., and Allshire, R. C. (1991)
Nature, 346, 866-868.
206.Holt, S. E., Wright, W. E., and Shay, J. W.
(1997) Eur. J. Cancer, 33, 761-766.
207.Reddel, R. R., Bryan, T. M., and Murnane, J. P.
(1997) Biochemistry (Moscow), 62, 1254-1262.
208.Wynford-Thomas, D. (1997) Eur. J.
Cancer, 33, 716-726.
209.Greider, C. W. (1998) Proc. Natl. Acad. Sci.
USA, 95, 90-92.
210.Counter, C. M., Avilion, A. A., LeFeuvre, C.
E., Stewart, N. G., Greider, C. W., Harley, C. B., and Bacchetti, S.
(1992) EMBO J., 11, 1921-1929.
211.Bodnar, A. G., Ouellette, M., Frolkis, M.,
Holt, S. E., Chiu, C. P., Morin, G. B., Harley, C. B., Shay, J. W.,
Lichtsteiner, S., and Wright, W. E. (1998) Science, 279,
349-352.
212.Wang, J., Xie, L. Y., Allan, S., Beach, D., and
Hannon, G. J. (1998) Genes Dev., 12, 1769-1774.
213.Klingelhutz, A. J., Foster, S. A., and
McDougall, J. K. (1996) Nature, 380, 79-82.
214.Kiyono, T., Foster, S. A., Koop, J. I.,
McDougall, J. K., Galloway, D. A., and Klingelhutz, A. J. (1998)
Nature, 396, 84-88.
215.Chadeneau, P., Siegel, P., Harley, C. B.,
Muller, W. J., and Bacchetti, S. (1995) Oncogene, 11,
893-898.
216.Sugrue, M. M., Shin, D. Y., Lee, S. W., and
Aaronson, S. A. (1997) Proc. Natl. Acad. Sci. USA, 94,
9648-9653.
217.Xu, H. J., Zhou, Y., Ji, W., Perng, G. S.,
Kruzelock, R., Kong, C. T., Bast, R. C., Mills, G. B., Li, J., and Hu,
S. X. (1997) Oncogene, 15, 2589-2596.
218.Vogt, M., Haggblom, C., Yeargin, J.,
Christiansen-Weber, T., and Haas, M. (1998) Cell Growth Differ.,
9, 139-146.
219.De Lange T., and Jacks, T. (1999) Cell,
98, 273-275.
220.Stunnenberg, H. G., Garcia-Jimenez, C., and
Betz J. L. (1998) Biochim. Biophys. Acta, 1423,
F15-F33.
221.Weston, K. (1998) Curr. Opin. Genet.
Dev., 8, 76-81.
222.Testa, U., Grignani, F., Hassan, H. J., Rogaia,
D., Masciulli, R., Gelmetti, V., Guerriero, R., Macioce, G.,
Liberatore, C., Barberi, T., Mariani, G., Pelicci, P. G., and Peschle,
C. (1998) Leukemia, 12, 563-570.
223.Anastasi, J., Feng, J., Dickstein, J. I., le
Beau, M. M., Rubin, C. M., Larson, R. A., Rowley, J. D., and Vardiman,
J. W. (1996) Leukemia, 10, 795-802.
224.Anastasi, J., Musvee, T., Roulston, D., Domer,
P. H., Larson, R. A., and Vardiman, J. W. (1998) Leukemia,
12, 233-237.
225.Watt, F. M. (1998) Philos. Trans. R. Soc.
Lnd. B. Biol. Sci., 353, 831-837.
226.Abelev, G. I., and Sell, S. (1999) Semin.
Cancer Biol., 9, 61-65.
227.Rotter, V., Aloni-Grinstein, R., Schwartz, D.,
Elkind, N. B., Simons, A., Wolkowicz, R., Lavigne, M., Beserman, P.,
Kapon, A., and Goldfinger, N. (1994) Semin. Cancer Biol.,
5, 229-236.
228.Raikhlin, N. T., Volodina, Yu. L., Smirnova, E.
A., Perevoshchikov, A. G., Chumakov, P. M., and Kopnin, B. P. (1995)
Arkhiv Patol., 57, 34-38.
229.Kremenetskaya, O. S., Logacheva, N. P.,
Baryshnikov, A. Y., Chumakov, P. M., and Kopnin, B. P. (1997) Oncol.
Res., 9, 155-166.
230.Shick, L., Carman, J. H., Choi, J. K.,
Somasundaram, K., Burrell, M., Hill, D. E., Zeng, Y. X., Wang, Y.,
Wiman, K. G., Salhany, K., Kadesch, T. R., Monroe, J. G., Donehower, L.
A., and El-Deiry, W. S. (1997) Cell Growth Differ., 8,
121-131.
231.Steinman, R. A., Hoffman, B., Iro, A.,
Guillouf, C., Liebermann, D. A., and El-Houseini, M. E. (1994)
Oncogene, 9, 3389-3396.
232.Tron, V. A., Tang, L., Yong, W. P., and
Trotter, M. J. (1996) Am. J. Pathol., 149, 1139-1146.
233.Gartel, A. L., Serfas, M. S., Gartel, M.,
Goufman, E., Wu, G. S., El-Deiry, W. S., and Tyner, A. L. (1996)
Exp. Cell. Res., 227, 171-181.
234.Mugita, N., Honda, Y., Nakamura, H., Fujiwara,
T., Tanaka, K., Omura, S., Shimbara, N., Ogawa, M., Saya, H., and
Nakao, M. (1999) Int. J. Mol. Med., 3, 127-137.
235.Stiegler, P., Kasten, M., and Giordano, A.
(1998) J. Cell Biochem. Suppl., 30/31, 30-36.
236.Alani, R. M., Hasskarl, J., and Munger, K.
(1998) Mol. Carcinog., 23, 226-233.
237.Maher, J., Baker, D., Dibb, N., and Roberts, I.
(1996) Leukemia, 10, 83-90.
238.Nakano, K., Mizuno, T., Sowa, Y., Orita, T.,
Yoshino, T., Okuyama, Y., Fujita, T., Ohtani-Fujita, N., Matsukawa, Y.,
Tokino, T., Yamagishi, H., Oka, T., Nomura, H., and Sakai, T. (1997)
J. Biol. Chem., 272, 22199-22206.