REVIEW: Kinases of the Src Family: Structure and Functions
A. G. Tatosyan* and O. A. Mizenina
Institute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, Moscow, 115478 Russia; fax: (095) 324-1205; E-mail: tatosyan@space.ru* To whom correspondence should be addressed.
Received September 17, 1999
Tyrosine kinases of the Src family are involved in different signal transduction pathways in cells. The corresponding genes participate in such vital processes as growth, differentiation, adhesion, transcription, etc. Specific structural changes confer oncogenic properties to the Src protein. In this review, we summarize the available data on the structure, substrates, regulation mechanisms, and role of nonreceptor tyrosine kinases by the example of the src gene product (as the prototype member of this family) and a number of related proteins.
KEY WORDS: oncogenes, Src, tyrosine kinases, phosphorylation, signal transduction mechanism
In terms of contemporary biology, the harmonic functioning of a multicellular organism is determined by the ability of its cells to interact as well as their adequate response to environmental changes. Surface receptors are those "sense organs" which react to extracellular factors and "inform" the cell as appropriate. Specific interaction between the external signals, called "impulses", generated on the cellular membrane is amplified and transmitted inside the cell through certain signal chains. Some of the signals are sent to the nucleus, where they initiate the expression of particular genes. The cells' response to the signal stimulus may be different, such as cell division (or, on the contrary, repression of this process), differentiation, hormone secretion, etc.
In its simplified form the signal transduction mechanism in the cell is a direct interaction between specific proteins in strictly determined order. A number of key proteins may be used in several signal transduction pathways. Different types of kinases and adaptor molecules without any enzymatic activity are the main participants of signal transduction chains in cells. Actually, the major molecular principles underlying the signal transduction mechanism are represented by the specific protein association and their phosphorylation (or dephosphorylation). Phosphorylation of the protein targets leads to immediate changes in their configuration and properties. The balance between phosphorylation and dephosphorylation normally determines the intracellular signal transduction. During malignant transformation, the reverse picture is observed--the signal system undergoes specific changes to make the transformed cell independent of environmental conditions and surrounding tissues.
Cellular signal transduction mechanisms are very complex and despite considerable research in this field, the available data are incomplete. At the moment, among the proteins involved in phosphorylation and regulation of the cell growth and differentiation the tyrosine-specific protein kinases have been studied in the most detail.
Tyrosine kinases catalyze the transfer of phosphate from ATP to a tyrosine residue of specific cell protein targets. The major categories of tyrosine kinases are divided into two groups, receptor and nonreceptor.
All receptor tyrosine kinases share a similar structure: they contain a ligand-binding extracellular region, a hydrophobic transmembrane domain, and an intracellular (cytoplasmic) region. The latter region and the catalytic domain include regulatory sites. At the present time there are nineteen classes of receptor tyrosine kinases classified according to their primary structure homology, structural characteristics, and amino acid motifs in the cytoplasmic domain [1, 2].
Nonreceptor protein tyrosine kinases, PTK, are intermediate conductors of diverse intracellular signal pathways. Many of them are associated with transmembrane receptors, such as hormone receptors, cytokines, growth factor receptors, etc. Nonreceptor kinases are activated by means of association of receptors with extracellular ligands or cell adhesion components at particular phases of the cell cycle. Now eight classes of nonreceptor protein tyrosine kinases grouped according to their structural and functional properties, which differ in size from 50 to 150 kD, have been reported [3, 4]. The Src protein is a typical representative of the nonreceptor tyrosine kinases.
The protooncogene src was the first gene to be identified that is potentially capable of inducing cell transformation [5].Mutations in the src gene cause functional activation of the protein product; in this way, in particular, the v-src gene--the Rous sarcoma virus (RSV) oncogene--has been formed. Although many oncogenes of the nonreceptor tyrosine kinases group have been identified after the discovery of v-src, the studies are still focused on Src as a prototype oncoprotein. The most important data on properties of this family genes has been obtained during the investigation ofthe src gene and its encoded protein.
This review deals with the analysis of structural and functional properties of the Src family kinases, their regulation mechanisms, and their role in cells.
STRUCTURAL ORGANIZATION OF Src PROTEINS
The members of the Src family are divided into two classes: tyrosine kinases with a broad expression range (Fyn, Yes) and those with limited expression (Fgr, Lyn, Hck, Lck, Blk, Yrc). In vertebrates the proteins of the Src family have similar structure [6].
The proteins of this group, ranging in molecular mass from 52 to 62 kD, comprise six distinct functional domains (Fig. 1): Src homology domain 4 (SH4), a unique domain, SH3 domain, SH2 domain, a catalytic domain (SH1), and a C-terminal regulatory region.
The SH4 domain is a region containing from 15 to 17 amino acid residues which comprises signals for modification with fatty acids [7]. The glycine at position 2 is myristylated, thus binding PTK to the cell membrane. Nonmyristylated Src molecules do not bind to membranes. On the other hand, some Src molecules carrying this modification can be found unlinked in the cytosol. Myristylation probably does not guarantee association of the protein with the membrane. In addition to myristylation signals, the Src SH4 domain contains basic amino acid residues which are substrates for posttranslational palmitylation. Only myristylated molecules are palmitylated and consequently this process probably occurs on the membrane. Palmitylation is a reversible process. Regulated depalmitylation and repalmitylation may be a mechanism for the changing of the Src family kinases localization in response to corresponding stimulation [8, 9].Fig. 1. Domain structure of Src protein.
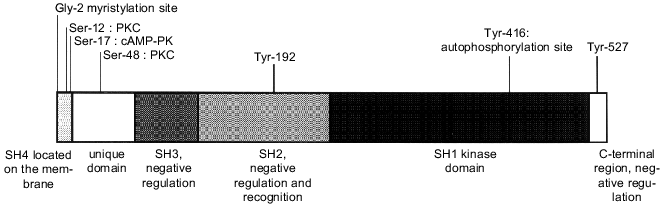
The unique domain (amino acid residues 18-84) is specific for each Src family protein. This region is suggested to be responsible for specific interaction of the PTK with particular receptors and protein targets [10]. At the same time, Src is a single member of the Src kinase group which is phosphorylated at Thr-34, Thr-46, and Ser-72 by a cyclin-dependent kinase (Cdc2) and cyclin B complex in M phase [11]. Theeffect of this phosphorylation is unclear. Moreover, the unique domain contains the protein kinase C (PKC)binding sites--Ser-12 and Ser-48--which are phosphorylated in the cells during PKC activation.
The SH3 domain (amino acid residues 85-140) is necessary for interactions with protein substrates and it also ensures the intramolecular bindings controlling catalytic activity, protein localization in the cell, and association with protein targets [12]. The SH3 domain of Src has a globular structure, one side of which is slightly hydrophobic and contains a cluster of acidic residues. This domain binds to the proline-rich regions of PTK substrates. All known SH3 ligands carry a consensus sequence PXXP. Amino acid residues adjacentto proline determine specificity of SH3 domains [13]. The SH3 ligands may bind to this domain both in NH2--COOH and COOH--NH2 orientation [14].
The SH3 sequences as well as the SH2 and the catalytic domains shown below, have been found in cellular proteins of different classes.
The SH2 domain is a second modulating region (amino acid residues 141-260), which controls the rangeof proteins interacting with the Src family kinases. The SH2 domains of different PTK recognize a short amino acid sequence carrying phosphotyrosine. From three to five amino acid residues following tyrosine determine the specificity of individual SH2 domains [15]. In kinases of the Src family this region is more conserved than the SH3 domain and can be tightly bound with specific proteins phosphorylated bytyrosine. The SH2 domains of Src and Lck kinases carry deep hydrophobic pockets for interactions of amino acid Ile at position pY + 3. Some proteins interacting with Src contain an optimum binding structure, pYEEI. However, not all proteins binding to the SH2 domain of Src possess such phosphorylated sequence.
A mutation in the SH2 domain in the region of amino acid residues from 142 to 169 leads to cell transformation [16]. This mutation probably affects Src binding to substrates, cytoskeletal proteins in particular (see below).
The kinase domain (amino acid residues 265-516) is found in all proteins of the Src family as well as in other PTK. It is responsible for tyrosine kinase activity and plays a crucial rolein substrate specificity [17]. Certain amino acid residues within this domain are identical in all kinases and involved in ATP binding and the phosphotransferase reaction [18].
The amino acid sequence preferably phosphorylated by the Src kinase is as follows: EEEIYG/EEFD. The kinases of the Src family may bind some substrates after phosphorylation, thus promoting phosphorylation of other sequences of one or several neighboring substrate molecules [19]. A strict specificity towards Tyr but not towards Ser or Thr is due to the close proximity of aconserved loop present in all tyrosine kinases (FP425IKWTA in Src) to the main chain of the substrate. Proline facilitatesbinding to the phenylalanine ring of tyrosine, but is ineffective in binding substrates carrying serine or threonine.
Phosphorylation of Tyr-416 stimulates complete activation of Src and provides a binding site for SH2 domains of other cellular proteins. The elimination of Leu-516, highly conserved in all protein tyrosine kinases and located in the catalytic domain, interferes with the transforming activity of p60 v-Src [20].
The C-terminal region (amino acid residues from 517 to ~536) plays a significant role in regulation of Src kinase activity. All kinases of the Src family have a C-terminal region of 15-19 amino acid residues with tyrosine at the constant position surrounded by conserved amino acids (Tyr-527 in Src). It has been shown that elimination of phosphotyrosine from the normal Src increases its kinase activity [21]. Phosphorylation of the C-terminal Tyr inhibits kinase activity by more than 98% and suppresses all stimulating effects caused by phosphorylation of Tyr-416 in the catalytic domain [22].
Structural differences between v-Src and c-Src proteins. The main difference between v-Src and c-Src is found in the structure of their C-terminal regions. The last "tail" 19 amino acids of c-Src contain Tyr-527, which plays a regulatory role controlling kinase activity [20, 23]. In v-Src these 19 amino acids are replaced by 12 amino acids present in all known RSV strains. The only exception is v-SrcLM whose atypical structure is associated with a decreased metastatic potential of transformed cells [24]. It has been recently demonstrated that in metastases of human colon cancer, the c-Src protein has mutations just in this region. It should be mentioned that now this is the most reliable example of the role of mutations in the src gene in human carcinogenesis [25].
REGULATION OF Src KINASES
The SH2 and SH3 domains play a key role in regulation of catalytic activity of the Src family kinases. X-Ray analysis has demonstrated how intramolecular interactions between SH2 and SH3 domains stabilize inactive conformational structure of Src kinases. Both domains are adjacent to the kinase domain from the side opposite to the catalytic cleft. The SH3 domain interacts with the catalytic domain and linker sequences located between SH2 and catalytic domains (Fig. 2) [26]. The SH2 domain interacts with phosphotyrosine at position 527 localized in the C-terminal region of the protein. Tyr-527 in c-Src, as well as the corresponding tyrosine residues in other PTK are the primary phosphorylation sites in vivo. This base is phosphorylated by the cytoplasmic kinase Csk [27]. The loss of Tyr-527 or its dephosphorylation leads to stimulation of Src catalytic activity. This conclusion is based on a number of experimental data: the substitution of Tyr-527 by another amino acid residue constitutively activates c-Src [28]; this region is absent from the v-Src protein [29]; the inhibition of the csk gene activity stimulates activity of PTK of the Src family [30]. Therefore, it has been suggested that phosphorylation of the C-terminal tyrosine by Csk kinase provides intramolecular interaction of this region with the SH2 domain thus preserves the Src protein in a closed inactive form.
Mutations in the SH3 domain also lead to activation of Src kinases, although the role of the SH3 domain in inhibition of protein enzymatic activity is obscure. The kinase domain apparently remains accessible even in "closed" conformation, namely, when the SH2 domain is associated with phosphotyrosine at position 527 (Fig. 2). The SH3 domain forms an independent intramolecular contact with the N-terminal fragment of the kinase domain. Accordingly, kinase inactivation may result from the formation of a rigid structure stabilized by double bonds between the SH2 and SH3 domains and the catalytic region of the protein. Such structure prevents any movement inside the kinase domain. Mutations in any of the interacting regions of the Src protein disrupt the rigid structure of the molecule; this, in turn, destabilizes other intramolecular interactions. Consequently, both the SH2 and SH3 domains regulate kinase activity by intramolecular contacts. Abnormal interactions may be the main activation mechanism of the Src proteins. It cannot be excluded that these domains play a similar role in regulation of kinase activity in proteins that lack phosphotyrosine in the C-terminal region (Fps, Abl) [31]. It should be noted that the above-mentioned mechanism not only precisely regulates kinase activity of the Src proteins, but controls the interactions of SH2 and SH3 domains with other molecules, thus providing different levels of Src regulation.Fig. 2. Regulation of Src activity. a) Closed autoinhibited state; b) open intermediate state induced either by interactions of the SH3 and SH2 domains with the Src protein partners or by dephosphorylation of the C-terminal Tyr-527. This makes Tyr-416 accessible for phosphorylation; c) open activated form with phosphorylated Tyr-416.
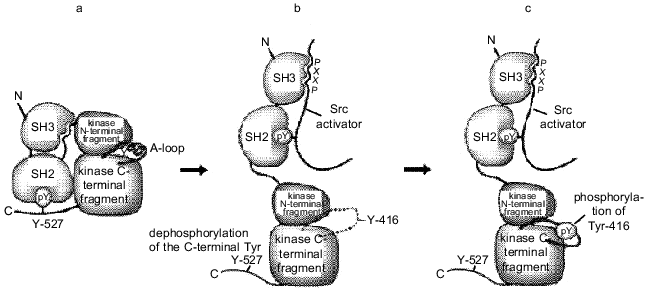
As shown above, the regulation of Src activity occurs at two sites, the modification of each of them leads to opposite results. The phosphorylation of Tyr-416 in the activating loop of the kinase domain activates the enzyme, while the phosphorylation of the C-terminal Tyr-527 causes its inactivation. Different regulatory elements controlling Src activity affect only particular regions in the kinase domain. These effectors contain amino acid residues involved in catalysis or substrate binding [32]. They may be the activating loop (amino acid residues 404-432), the catalytic loop (the region around the amino acid at position 382), and the C-helix (the region around the amino acid at position 310). The modulation of their position and conformation by phosphorylation and interaction with regulatory subunits may control catalytic activity.
In vivo the Src kinase can be phosphorylated only at one of two tyrosine residues. The model for Src tyrosine kinase activation includes three subsequent stages (Fig. 2).
The activating loop plays a central role in regulating kinase activity. Its phosphorylation at Tyr-416 in Src (or homologous amino acid residues in other tyrosine kinases) is necessary for complete activation of most kinases studied so far. In the absence of phosphorylation, the activating loop acquires different conformations, which often inhibit protein--protein interactions. A nonphosphorylated activating loop can inhibit kinase activity either directly, disturbing the region involved in activation, or indirectly, conferring a specific conformation, which prevents substrate binding. Conversely, in the phosphorylated state, the conformation of the activating loop is similar in all known kinases. In this active conformation the loop forms a part of the site recognized by the substrates [33].
Src SUBSTRATES
Presently, the list of proteins tyrosine phosphorylated as a result of src gene function is rather large. It includes both proteins directly interacting and phosphorylated by Src and those phosphorylated by tyrosine in src-transformed cells, for which direct interaction with Src has not been shown [34]. Some proteins of the second group may be true protein targets of Src; however, most of them are probably the substrates for tyrosine kinases activated by the Src protein in transformed cells (see table). The best studied protein targets of Src are described below.
Table 1. Protein targets of Src kinases
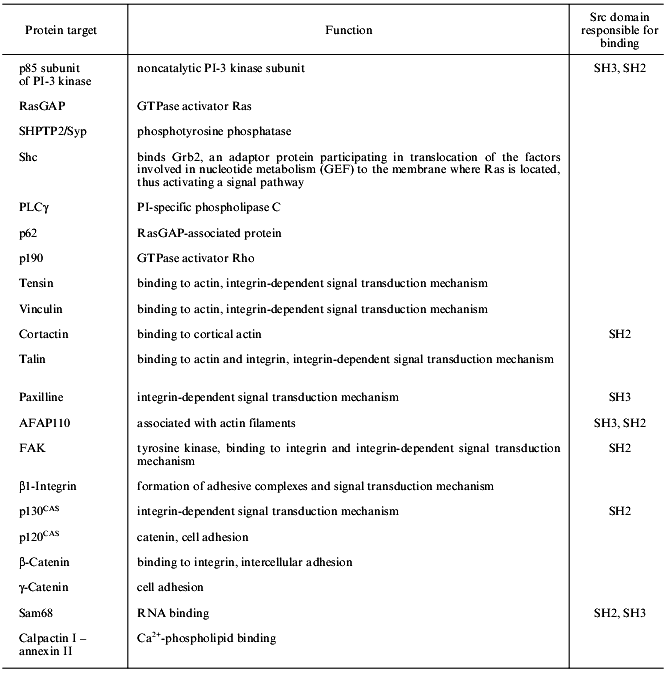
SHC. One of the main substrates phosphorylated in src-transformed cells or stimulated by growth factors is the adaptor protein SHC, which contains a proline-rich region and binds to the SH3 domain of the Src protein in vitro. This protein plays a key role in signal transduction from integrin receptors to the Ras--MAP (mitogen-activated protein kinases) signal transduction pathway [35].
p190 is a protein containing a GTPase domain and phosphorylated at tyrosine in src-transformed cells stimulated by growth factors [36]. However, direct interaction between p190 and Src protein has not been shown. Tyrosine phosphorylated p190 associates with the Ras--GAP complex via the SH2 domain of the latter.
PI-3 kinase (phosphatidylinositol-3 kinase) involved in signal transduction from the growth factor receptor (see the review of M. Krasilnikov in this issue). This protein is associated with Src proteins in transformed fibroblasts or activated B- and T-cells [37, 38]. Direct interaction between the p85 subunit of PI-3K and SH3 domains of different Src proteins has been demonstrated in several works [39, 40].
The second class of Src substrates includes proteins that are not substrates for tyrosine kinase receptors. These protein targets reflect multiple relations and functions of Src proteins and comprise proteins involved in cytoskeleton structure, cell adhesion, intercellular contacts, and RNA processing. Among them, the protein associated with the actin filament p110 (AFAP110), cortactin, FAK (focal adhesion kinase), paxilline, tensin, talin, vinculin, and p130 CAS (a cadherin-associated Src substrate) should be mentioned.
AFAP110 and cortactin. In src-transformed cells, AFAP110 and cortactin are tyrosine hyperphosphorylated [41-43]. The interaction between Src and AFAP110 is mediated by its association with SH2 and SH3 domains, while cortactin binds only to the SH2 domain. In fibroblasts with suppressed Csk kinase, cortactin is tyrosine hyperphosphorylated, this being due to the enhanced Src activity in these cells [44].
FAK and paxilline. These proteins are hyperphosphorylated in src-transformed cells. This phenomenon is difficult to explain since activation of integrins leads to phosphorylation of focal adhesion proteins and stimulates interactions between the cells and the substrate, while in src-transformed cells the number of focal contacts is decreased and the structure is changed, although the same proteins are phosphorylated. The interaction between Src and FAK is rather complicated. FAK is autophosphorylated under activation by integrins that makes tyrosine at position 397 accessible to the SH2 domain of Src [45]. Therefore, the kinase activity of Src is stimulated and reverse phosphorylation of FAK occurs at four tyrosine residues; this in turn, induces its maximum activity. Other potential Src substrates in the zone of focal contacts may be also FAK substrates. This problem still remains unclear because it is difficult to discriminate between substrate specificity of two kinases present in one complex. For example, paxilline associated with the C-terminal fragment of FAK may interact with Src through the SH3 domain [46]. There is evidence that FAK is not the only enzyme capable of phosphorylating paxilline, vinculin, talin, and tensin since it was shown that in mouse FAK-defective cells, these proteins were nevertheless phosphorylated [47].
p120 CAS. The manifestation of Src activity in the zone of intercellular contacts is increased tyrosine phosphorylation of p120 CAS in src-transformed cells [48].
Sam68 belongs to a newly recognized class of Src substrates--the RNA-binding proteins. This protein associates with Src through the proline-rich region and SH3 domain and is selectively phosphorylated at mitosis [49, 50]. The role of Sam68 phosphorylation and its association with Src are poorly understood; however, there are speculations concerning the involvement of Src in regulation of pro-mRNA processing [50].
Using a two-hybrid technique, which allows one to identify genes coding the potentially interacting proteins, a number of proteins and peptides capable of associating with the Src protein have been recently detected [51, 52].
mDab1 is found in a complex with the Scr protein in tyrosine phosphorylated form and is synthesized in large amounts during neuronal development. It has been suggested that mDab1 is an adaptor protein in a specific signal transduction chain operating during the formation of the neural network [51].
ROLE OF Src IN REGULATION OF CELLULAR PROCESSES
In recent years a considerable body of evidence has been obtained that indicates a role of the src gene product and some other tyrosine kinases in different cellular processes. The biological functions of the Src protein are summarized below.
Gene transcription. Using v-src-transformed fibroblasts as a model, both positive and negative regulation of the transcription of several genes has been shown [53-55]. The stimulating effect of v-src has been described during the analysis of a number of factors involved in regulation of transcription, including NF-kappaB, ATF/CRE, and AP1 [56-58]. The most convincing example of transcription regulation by the c-Src protein is control of Myc expression by the PDGF receptor (see below).
The constitutive kinase activity of v-Src in tumor cells triggers kinase cascades regulating transcription, such as the MAPK cascade [59-61].
There are data indicating that the members of the STAT family (signal transducers and activators of transcription), molecules transmitting signals within the cell in response to stimulation with cytokines, are transcription factors involved in oncogenesis. These proteins (STAT3 in particular) exhibit an increased constitutive tyrosine phosphorylation level and increasing of the DNA-binding activity in v-src-transformed fibroblasts. The STAT genes are activated by several cellular signaling cascades, in particular involving Jak proteins, which are also constitutively activated in src-transformed cells [62]. In tumor cells, many proteins of the STAT family are constitutively activated, for example, in mammary gland carcinoma cells, lymphomas, leukoses, etc. [63-66].
To penetrate blood vessels, an invasive cell with metastatic potential must disrupt the extracellular matrix. In v-src-transformed cells, collagenase-1 (matrix-1 metalloproteinase (MMP-1)) performs this function whose transcription is activated by signal cascade components induced by v-src via the ERK signaling cascade [67, 68]. The proteins of the STAT family act as transcription factors controlling expression, since in the MMP-1 promoter region several STAT-binding sites were found [68].
Adhesion. Fibroblasts with a repressed src gene (src-/-) show a decreased adhesion level upon growth on a fibronectin layer. The src gene is proposed to be necessary for optimum adhesion, but not obligatory for initiating this process. Fibroblasts with a "switched off" src gene are indistinguishable from those which express this protein on a collagenic matrix, i.e., Src probably acts in a fibronectin-induced signaling chain. The Src-mediated regulation of adhesion is still poorly understood. However, it is known that the catalytic activity of this protein is not obligatory, while intact structures of SH2 and SH3 domains are required for this process. Src probably functions as an adaptor protein locating specific proteins in adhesion structures [69].
Tyrosine phosphorylation occurs during focal adhesion, since PTK suppression blocks this event. Which particular protein of the Src family plays a crucial role in this process is still unclear, because several proteins of this group are known to be associated with adhesion structures (Src, Fyn, Abl, etc.) [70, 71]. Protein interactions via SH2-binding sites play an essential role in phosphorylation of focal adhesion structures.
The components of focal adhesion are involved in an integrin-induced signal transduction mechanism. The key element triggering this signaling chain is FAK. This enzyme is associated with and/or phosphorylated by a complex of different proteins including paxilline, Src proteins, adapters, the metabolic Sos factor (see below), etc. It is also evident that an important factor in signal transduction in this case is phosphorylation and dephosphorylation of target proteins. The Src protein binds and phosphorylates FAK, thus participating in regulation of FAK-specific kinase activity [72, 73].
Structures similar to focal adhesion complexes are formed by integrins in other types of cells. For example, Src, Yes, talin, vinculin, and other proteins associate with large insoluble complexes involved in thrombocyte aggregation [74].
Migration. Different signal molecules are capable of interfering with cell migration by affecting cell adhesion and cytoskeleton rearrangements. Using different experimental systems it has been shown that Src is involved in cell migration mechanisms [75]. Mouse fibroblasts with a "knocked-out" src gene (src-/-) are less mobile than the wild-type cells. This abnormality may be compensated by the addition of active c-src [76]. c-Src is involved in migration of mouse carcinoma cells induced by the epidermal growth factor [77, 78].
The Src protein may affect cell migration at different stages of a signal transduction pathway: acting on integrin-dependent adhesion, rearrangements of actin cytoskeleton, modulation of integrin gene expression and on subsequent molecules in the signal transduction mechanism. The role of the MAPK cascade in integrin-induced cell migration has been recently shown. It is known that Src activates the MAP kinase signal transduction mechanism; therefore, it cannot be excluded that the role of these proteins in cell migration is partly connected with the activation of MAP kinases [79, 80]. Other proteins involved in cell migration (such as FAK and PI-3K) are Src substrates; however, this does not exclude the effect of Src on cell mobility via kinases of other groups.
Cell cycle. Kinases of the Src family participate in induction of DNA synthesis initiated by different tyrosine growth factor receptors. Microinjection of inactivated form of src DNA or antibodies interacting with the C-terminal region of the Src protein inhibits DNA synthesis induced by epithelial growth factor (EGF), platelet derived growth factor (PDGF), and colony-stimulating factor (CSF-1). Expression of Src with inactivated kinase domain in src-/- fibroblasts blocks PDGF-induced DNA synthesis [81]. The activation of Myc protooncogene by PDGF is blocked by Src-specific antibodies; however, DNA synthesis may be restored by expression of exogenous Myc. At the same time, DNA synthesis induced by bombesin is Src-independent [82]. Recently, the role of the Src family proteins in the PDGF-induced signal transduction mechanism has been reported to be ambiguous: in mouse cells with defective src, yes, and fyn genes the signal is efficiently transmitted from the growth factor receptor [83].
Other receptors can also use Src kinase for stimulation of different cellular processes involved in induction of DNA synthesis. For example, v-Src activates the MAP kinase cascade and PI-3K, the signal molecules involved in cell proliferation. Thus, Src participates in signal transduction mechanisms induced by various receptors and stimulates DNA synthesis by different mechanisms.
The Src protein is involved in regulation of the G2/M transition. The role of c-Src in mitosis of somatic cells was proved by the analysis of cells arrested at mitosis by Src-specific antibodies [84]. The Src activity considerably increases in the cells blocked at metaphase and kinase activation correlates with Src phosphorylation at serine-threonine residues in the unique domain of the protein. This phosphorylation is performed by CDC2. Division of mouse fibroblasts is inhibited by the injection of Src-specific antibodies at G2; consequently, this protein is necessary for the transition from G2 to cell division. It is should be noted that in addition to Src other proteins of this family (Fyn and Yes) participate in cell division. Only the antibodies cross-reacting with the C-terminal peptides of all the above-mentioned proteins have an inhibitory effect on cell division. The Sam68 protein associating with Src via SH3 and SH2 domains is the Src substrate at mitosis [85]. The Src kinases may be also involved in Ras--MAP kinase signal transduction mechanism during mitosis; this is confirmed by activation of the Raf protein (Ras effector in the signal chain) during cell division [86].
Apoptosis. The v-Src expression prevents the transition of several types of cells to apoptosis induced by cytokine deficiency, irradiation, and destruction of cell contacts with the extracellular matrix [87-89]. In other words, Src activation suppresses the activity of cytokine receptors, integrins, and other receptor-mediated signal transduction pathways; this protects the cells from the realization of different apoptosis programs. The role of Src kinases in some of these processes may be indirect: they are capable of triggering the initial events, which in turn, "switch on" the components of subsequent stages of the signal transduction mechanism.
Differentiation. In mature cells, v-Src expression leads to dramatic changes in the differentiation program: in most cells, this process is completely inhibited. For example, infection of myeloblasts, retinoblasts or chondroblasts with Rous sarcoma virus maintains these cells in a proliferative state and prevents their subsequent differentiation [90, 91]. By contrast, the introduction of v-Src into PC12 cells or immature sympathetic neurons induces growth and terminal differentiation into neuron-like cells [92, 93]. It should be emphasized that the observed effects of v-Src on differentiation do not necessarily imply that cellular analogs of this gene normally produce similar effects. It cannot be ruled out that highly active v-Src "neutralizes" normal properties of cellular tyrosine kinases by phosphorylating common target proteins. This picture is observed when the effects of v-Src and neuron growth factor (NGF) are compared. In PC12 cells the changes induced by v-Src overlap the effect of NGF by initiating phosphorylation of cell proteins, gene transcription, etc. [94]. Alternatively, c-Src is known to be necessary for NGF activity, since microinjection of monoclonal antibodies against c-Src blocks NGF-induced growth. Whether activated Src affects the inhibition of differentiation in immature cells by stimulating cell proliferation or by destruction of gene transcription controlling this process remains unclear.
"Knock-out" of the src gene in mice leads to the development of osteoporosis [95]; this proves the role of Src in normal differentiation and signaling in osteoclasts. Whether abnormal functioning of these cells results from aberrant regulation of late differentiation stages or from the inability of osteoclasts to respond to extracellular signals is unknown. This process finally results in decreased bone resorption, which leads to its abnormal growth.
These data demonstrate a significant role of kinases of the Src family in regulation of vital cellular processes. Specific interactions of these proteins with substrates account for regular and consecutive distribution of the regulating signals in cells. Therefore, the transforming effect of v-Src may be mediated by irregularities in mitogenic signal cascades normally stimulated by growth factors. There is evidence that v-src transforms fibroblasts acting as a normal cellular Src protein, but in an ill-regulated manner, rather than by phosphorylation of atypical substrates. Consequently, v-src cannot transform those cells for which such changes in the cytoskeleton are normal [6].
The surprising thing is that the signals activating a limited number of Src kinases control different cellular processes. In other words, it remains unclear how functional redistribution of signals from different receptors passing the same signal transduction stages occurs. This enigma also refers to other components of signaling chains, such as the proteins of the Ras family, PI-3K, PKC, etc., which are activated by a wide range of receptors and participate in diverse intracellular processes. These solutions are of key importance of modern molecular oncology.
REFERENCES
1.Wilks, A. F. (1993) Adv. Cancer Res.,
60, 43-73.
2.Shawver, L. K, Strawn, L. M., and Ulrich, A. (1995)
Mutat. Res., 333, 23-28.
3.Taniguchi, T. (1995) Science, 268,
251-255.
4.Bolen, J. B. (1993) Oncogene, 8,
2025-2031.
5.Jove, R., and Hanafusa, H. (1987) Ann. Rev. Cell
Biol., 3, 31-56.
6.Brown, M. T., and Cooper, J. A. (1996) Biochim.
Biophys. Acta, 1287, 121-149.
7.Rech, M. D. (1993) Biochim. Biophys. Acta,
1155, 307-322.
8.Kaplan, J. M., Mardon, G., Bishop, J. M., and
Varmus, H. E. (1988) Mol. Cell. Biol., 8, 2435-2441.
9.Pellman, D., Garber, E. A., Cross, F. R., and
Hanafusa, H. (1985) Proc. Natl. Acad. Sci. USA, 82,
1623-1627.
10.Thomas, S. M., and Brugge, J. S. (1997) Annu.
Rev. Cell Dev. Biol., 1287, 121-149.
11.Superti-Furga, G., and Courtneidge, S. A. (1995)
BioEssays, 17, 321-330.
12.Pawson, T. (1995) Nature, 373,
573-580.
13.Riskles, R. J., Botfield, M. C., Zhou, X. M.,
Henry, P. A., Brugge, J. S., and Zoller, M. J. (1995) Proc. Natl.
Acad. Sci. USA, 92, 10909-10913.
14.Yu, H., Chen, J. K., Feng, S., Dalgamo, D. C.,
Brauer, A. W., and Schreiber, S. L. (1994) Cell, 76,
933-945.
15.Songyang, Z., Shoelson, S. E., Chaudhuri, M.,
Gish, G., Pawson, T., Haser, W. G., King, F., Roberts, T., Ratnofsky,
S., and Lechleider, R. J. (1993) Cell, 72, 767-778.
16.Raymond, V. M., and Parsons, J. T. (1987)
Virology, 160, 400-410.
17.Hesketh, R. (1995) The Oncogene Handbook,
Academic Press, London.
18.Hughes, A. (1996) J. Mol. Evol.,
42, 247-256.
19.Mayer, B. J., Hirai, H., and Sakai, R. (1995)
Curr. Biol., 5, 296-305.
20.Yaciuk, P., and Shalloway, D. (1986) Mol.
Cell. Biol., 6, 20807-20829.
21.Schwartzberg, P. L. (1998) Oncogene,
17, 1463-1468.
22.Van Hoek, M. L., Allen, C. S., and Parsons, C. J.
(1997) Biochem. J., 326, 271-277.
23.Dorai, T., Levy, J. B., Kang, L., Brugge, J. S.,
and Wang, L. H. (1991) Mol. Cell. Biol., 11,
4165-4176.
24.Tatosyan, A., Yatsula, B., Shtutman, M., Moinova,
E., Kaverina, I., Musatkina, E., Leskov, K., Mizenina, O., Zueva, E.,
Calothy, G., and Dezelee, P. (1996) Virology, 216,
347-356.
25.Irby, R. B., Mao, W., Coppola, D., Kang, J.,
Loubeau, J. M., Trudeau, W., Karl, R., Fujita, D. J., Jove, R., and
Yeatman T. J. (1999) Nature Genet., 21, 187-190.
26.Xu, W., Doshi, A., Lei, M., Erc, M. J., and
Harrison, S. C. (1999) Mol. Cell, 3, 629-638.
27.Nada, S., Okada, M., MacAuley, A., Cooper, J. A.,
and Nakagawa, H. (1991) Nature, 351, 69-72.
28.Kmiecik, T. E., and Shalloway, D. (1987)
Cell, 49, 65-73.
29.Reynolds, A. B., Vila, J., Lansing, T. J., Potts,
W. M., Weber, M. J., and Parsons, J. T. (1987) EMBO J.,
6, 2359-2364.
30.Imamoto, J., and Soriano, P. (1993) Cell,
73, 1117-1124.
31.Pawson, T., and Gish, G. D. (1992) Cell,
71, 359-362.
32.Sichei, F., Moarefi, I., and Kuriyan, J. (1997)
Nature, 385, 602-609.
33.Xu, W., Harrison, S. C., and Eck, M. J. (1997)
Nature, 385, 595-602.
34.Schaller, M. D., Bouton, A. H., Flynn, D. C., and
Parsons, J. T. (1993) Proc. Natl. Acad. Sci. USA, 44,
205-227.
35.Ellis, C., Moran, M., McCormick, F., and Pawson,
T. (1990) Nature, 343, 377-381.
36.Settelman, J., Narasimhan, V., Foster, L. C., and
Weinberg, R. A. (1992) Cell, 69, 539-549.
37.Cambier, J. C., Pleiman, C. M., and Clark, M. R.
(1994) Annu. Rev. Immunol., 12, 457-486.
38.Fukui, Y., and Hanafusa, H. (1991) Mol. Cell.
Biol., 11, 1972-1979.
39.Liu, X., Marengere, L. E. M., Koch, A., and
Pawson, T. (1993) Mol. Cell. Biol., 13, 5225-5232.
40.Petch, L. A., Bockholt, S. M., Bouton, A.,
Parsons, J. T., and Burridge, K. (1995) J. Cell Sci.,
108, 1371-1379.
41.Reynolds, A. B., Kanner, H. C., Wang, R., and
Parsons, J. T. (1989) Mol. Cell. Biol., 9, 3951-3958.
42.Kanner, S. B., Reynolds, A. B., Wang, H. C.,
Vines, R. R., and Parons, J. T. (1991) EMBO J., 10,
1689-1698.
43.Wu, H., and Parsons, J. T. (1993) J. Cell
Biol., 120, 1417-1426.
44.Thomas, S. M., Soriano, P., and Imamoto, A.
(1995) Nature, 376, 267-271.
45.Cobb, B. S., Schaller, M. D., Leu, T., and
Parsons, J. T. (1994) Mol. Cell. Biol., 14, 147-155.
46.Hildebrand, J., Schaller, M., and Parsons, J. T.
(1993) J. Cell. Biol., 123, 993-1005.
47.Ilic, D., Furuta, Y., Kanazava, S., Takeda, N.,
Sobue, K., Nakatsuji, N., Nomura, S., Fujimoto, J., Okada, M., and
Yamamoto, T. (1995) Nature, 377, 539-544.
48.Reynolds, A. B., Herbert, L., Cleveland, J. L.,
Berg, S. T., and Gaut, J. R. (1992) Oncogene, 7,
2439-2445.
49.Fumagalli, S., Totty, N. F., Hsuan, J. J., and
Courtneidge, S. A. (1994) Nature, 368, 871-874.
50.Neel, H., Gondran, P., Weil, D., and Dautry, F.
(1995) Curr. Biol., 5, 413-422.
51.Howell, B. W., Gertley, F. B., and Cooper, J. A.
(1997) EMBO J., 16, 121-132.
52.Mizenina, O., Yanushevitch, U., Musatkina, E.,
Rodina, A., Camonis, J., Tavitian, A., and Tatosyan, A. (1998) FEBS
Lett., 422, 79-84.
53.Bedard, P. A., Yammoni, Y., Simmons, D. L., and
Erikson, R. L. (1989) Mol. Cell. Biol., 9, 1371-1375.
54.Qureshi, S. A., Rim, M. H., Alexandropoulos, K.,
Berg, K., Sukhatme, V. P., and Foster, D. A. (1992) J. Exp.
Med., 183, 407-414.
55.Scholz, G., Martinerie, C., and Perbal, B. (1996)
Mol. Cell Biol., 16, 481-486.
56.Xie, W., Fletcher, B. S., Andersen, R. D., and
Hershman, H. R. (1994) Mol. Cell Biol., 14,
6531-6539.
57.Wasylyk, C., Imler, J. L., and Wasylyk, B. (1993)
Cell, 72, 767-778.
58.Cao, X., Tay, A., Guy, G. R., and Tan, Y. H.
(1996) Mol. Cell. Biol., 16, 1595-1603.
59.Aftab, D. T., Kwan, J., and Martin, G. S. (1997)
Proc. Natl. Acad. Sci. USA, 94, 3028-3033.
60.Meng, F., and Lowell, C. A. (1998) EMBO
J., 17, 4391-4403.
61.Gupta, S. K., Gallego, C., Johnson, G. L., and
Haesley, L. E. (1992) J. Biol. Chem., 267, 7087-7990.
62.Turkson, J., Bowman, T., Garcia, R., Caldenhoven,
E., De Groot, R. P., and Jove, R. (1998) Mol. Cell Biol.,
18, 2545-2552.
63.Rech, M. D. (1994) Cell, 76,
411-414.
64.Weber-Nordt, R. M., Egen, C., Wehinger, J.,
Ludwig, W., Gouilleux-Gruart, V., Mertelsmann, R., and Finke, J. (1996)
Blood, 88, 809-816.
65.Stankovski, H., Gonen, A., Orien, A. L.,
Schwartz, A., and Ceichanover, A. (1995) Mol. Cell. Biol.,
15, 7106-7116.
66.Garcia, R., Yu, C. L., Hudnall, A., Catlett, R.,
Nelson, K. L., Smithgall, T., Fujita, D. J., Ethier, S. P., and Jove,
R. (1997) Cell Growth Differ., 8, 1267-1276.
67.Hansell, E. J., Frisch, S. M., Tremble, P.,
Murnane, J. P., and Werb, Z. (1995) Biochem. Cell Biol.,
73, 373-389.
68.Vincenti, M. P., Schroen, D. J., Coon, C. I., and
Brinckerhoff, C. E. (1998) Mol. Carcinogeneses, 21,
194-204.
69.Kaplan, K. B., Swedlow, J. R., Morgan, D. O., and
Varmus, H. E. (1995) Genes Dev., 9, 1505-1517.
70.Romer, L. H., Burridge, K., and Turner, C. E.
(1992) Cold Spring Harbor Symp. Quant. Biol., 57,
193-202.
71.Ridley, A. J., and Hall, A. (1994) EMBO
J., 13, 2600-2610.
72.Calalb, M. B., Polte, T. R., and Hanks, S. K.
(1995) Mol. Cell. Biol., 15, 954-963.
73.Schlaepfer, D. D., and Hanter, T. (1996) Mol.
Cell. Biol., 16, 5623-5633.
74.Fox, J. E., Lipfert, L., Clark, E. A., Reynolds,
C. C., Austin, C. D., and Brugge, J. S. (1993) J. Biol. Chem.,
268, 25973-25984.
75.Altun-Gultekin, Z. F., and Wagner, J. A. (1996)
J. Neurosci. Res., 44, 308-327.
76.Hall, C. L., Lange, L. A., Prober, D. A., Zhang,
S., and Turley, E. A. (1996) Oncogene, 13, 10.
77.Rodier, J. M., Valles, A. M., Denoyelle, M.,
Thiery, J. P., and Boyer, B. (1995) J. Cell Biol., 131,
761-773.
78.Bell, G. M., Bolen, J. B., and Imboden, J. B.
(1992) Mol. Cell. Biol., 12, 5548-5554.
79.Schlaepfer, D. D., Broome, M. A., and Hunter, T.
(1997) Mol. Cell. Biol., 17, 1702-1713.
80.Klemke, R. L., Cai, S., Giannini, A. L.,
Gallagher, P. J., De Lanerolle, P., and Cheresh, D. A. (1997) Mol.
Cell. Biol., 8, 4541-4546.
81.Broome, M. A., and Hunter, T. (1997)
Oncogene, 14, 17-34.
82.Barone, M. V., and Courtneidge, S. A. (1995)
Nature, 378, 509-512.
83.Klinghofer, R. A., Sachsenmaier, C., Cooper, J.
A., and Soriano, P. (1999) EMBO J., 18, 2459-2471.
84.Tailor, S. J., and Shalloway, D. (1996)
BioEssays, 18, 9-11.
85.Weng, Z., Thomas, S. M., Rickles, R. J., Taylor,
J. A., Brauer, A. W., Seidel-Dugan, C., Michael, W. M., Dreyfuss, G.,
and Brugge, J. S. (1994) Mol. Cell Biol., 14,
4509-4521.
86.Pathan, N. I., Ashendel, C. L., Geahlen, R. L.,
and Harrison, M. L. (1996) J. Biol. Chem., 271,
30315-30317.
87.McCubrey, J. A., Smith, S. R., Algate, P. A.,
DeVente, J. E., White, M. K., and Steelman, L. S. (1993)
Oncogene, 8, 2905-2915.
88.Frisch, S. M., and Francis, H. (1994) J. Cell
Biol., 124, 619-626.
89.Basu, A., and Cline, J. S. (1995) Int. J.
Cancer, 63, 597-603.
90.Yoshimura, M., Iwasaki, Y., and Kaji, A. (1981)
J. Cell. Physiol., 109, 373-385.
91.Alema, S., and Tato, F. (1987) Adv. Cancer
Res., 49, 1-28.
92.Haltmeier, H., and Rohrer, H. (1990) J. Cell
Biol., 110, 2087-2098.
93.Hecker, G., Lewis, D. L., Rausch, D. M., and
Jelsema, C. L. (1991) Biochem. Soc. Trans., 19,
385-386.
94.Thomas, S. M., Hayes, M., D'Arcangelo, G.,
Armstrong, R. C., Meyer, B. E., Zilberstein, A., Brugge, J. S., and
Halegoua, S. (1991) Mol. Cell Biol., 11, 4739-4750.
95.Soriano, P., Muller, O., Clark, R., Conroy, L.,
Moran, M. F., Polakis, P., and McCormick, F. (1992) Cell,
69, 551-558.