Natural Selection and Early Changes of Phenotype of Tumor Cells in vivo: Acquisition of New Defense Mechanisms
G. I. Deichman
Institute of Carcinogenesis, Blokhin Russian Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, Moscow, 115478 Russia; fax: (095) 324-1205; E-mail: antitum@space.ru
Received October 5, 1999
This review summarizes results obtained in the author's and collaborating laboratories within the last decade and is designed to attract the attention of researchers to discrete biochemical mechanisms of protection acquired in vivo by cells of malignant tumors against effectors of innate antitumor immunity. Tumor progression in vivo is associated with the appearance and selection of tumor cells with new specific characteristics: a high level of H2O2-catabolizing (antioxidant) activity (H2O2CA) and the ability for immediate release of E2-type prostaglandin (PGES) on contact with natural killers, macrophages, and neutrophils; the expression of the [H2O2CA + PGES] phenotype provides the tumor cells with two mechanisms of local protection against effectors of innate and acquired antitumor immunity. This results in a 10-100-fold less effective rejection of tumor cells in immune and normal animals and corresponding increase of tumorigenicity. The in vitro transformation of normal fibroblasts, spontaneous or induced by oncogenes LTSV40, E1a,b, Ha-ras, myc, and also by p53175 and bcl-2 does not result in the [H2O2CA + PGES] phenotype expression, but during subsequent in vivo growth of the above-mentioned transformants the selection of tumor cells of the [H2O2CA + PGES] phenotype is correlated with a 30-200-fold increase in their tumorigenicity (accompanied or not accompanied by spontaneous metastatic activity). Unlike the transformation induced by the above-mentioned oncogenes, the transformation of normal cells by the v-src gene results in the [H2O2CA + PGES] phenotype expression. The data presented confirm the determining role of the v-src gene in the expression of the [H2O2CA + PGES] phenotype. In various primary viral carcinogenesis (SV40, SA7(C8)) the natural selection of cells expressing the [H2O2CA + PGES] phenotype begins even within the latent period and can be completed by the appearance of primary tumors.
KEY WORDS: transformation, carcinogenesis, in vivo tumor progression, antioxidant and PGE2-releasing activity of tumor cells, protection mechanisms of tumor cells, innate and specific antitumor immunity
The problem of interrelations between neoplastic cells and in vivo systems of the innate and specific immunity includes three main aspects: 1) the host's protection, i.e., mechanisms of recognition and elimination of mutant, transformed, and tumor cells by effectors of the system of the innate antitumor immunity (and of the specific antitumor immunity in the case of antigenic tumors); 2) the defense mechanisms of the transformed and tumor cells against effectors of the innate antitumor immunity system; 3) the effect of selective pressure of the host's innate and specific antitumor immunity on the tumor progression.
The first aspect of the problem has been studied for more than 30 years mainly in experiments on tumor transplantation in laboratory animals including those with certain genetic deficiencies of the immune system and on in vitro cytotoxic activity (CTA) of resident and activated macrophages (MPh), dendrite cells (DC), natural killers (NK), neutrophils (NPh), and also of T-lymphocytes directed against transformed and tumor cells of various origin. The nonspecific recognition of tumor cells by effectors of the innate antitumor immunity and the inhibition of their growth (or, on the contrary, the stimulation of their proliferation) are associated with a complex of intercellular interactions and the induction of various cytokine networks which are produced in cooperation with DC, MPh, NK, NPh, and the tumor cells and with the involvement (or without it during the early stages of carcinogenesis) of activated T-cells. This aspect of the innate antitumor immunity has been long and intensively studied and is considered in various reviews [1-8].
Tumor cells during their in vivo appearance, growth, and dissemination suffer permanent stress caused by various growth-inhibiting and cell-damaging signals, including the CTA of DC, MPh, NK, and NPh. Apoptosis is the mechanism of death for the great majority of tumor cells. Tumor cells that survive in the primary focus of tumor growth (and also in remote deposits) develop under conditions of continuous stress and selective pressure of effectors of the innate antitumor immunity. Highly malignant tumor cells selected in vivo usually show 10-100-fold increased tumorigenic activity (TGA) compared to their in vitro transformed precursors, whereas the spontaneous and experimental metastatic activities (SMA, EMA) and also some other new characteristics arise less regularly. These new characteristics include the increased resistance of tumor cells to the CTA of MPh, NK, and NPh. Obviously, the appearance of a monoclonal tumor is associated with selection of extremely rare variants of tumor cells which have different (sometimes very specific) mechanisms to overcome the host's control.
Growth-regulating positive and negative signals of normal surrounding cells are another type of selective pressure of the host on the heterogenous population of tumor cells [9]. But unlike the effectors of the innate antitumor immunity, such as DC, MPh, NK, and NPh, the normal surrounding cells seem to be not activated during the contact interaction with the tumor cells (at least, this is not shown) and their effect on the tumor cells is, as a rule, either cytostatic or growth-stimulating, but not cytolytic.
The great advantage of the innate antitumor immunity system versus the specific antitumor immunity is its permanent readiness to recognize in vivo the appearance of a very small number of transformed and tumor cells (even single cells), no matter whether they express the specific tumor antigens or not. It is obvious a priori that primary tumors can be most efficiently controlled by the innate antitumor immunity within the initial latent period when the number of tumor cells is not great and their evolutionary genetic changes are, as a rule, minimal [4, 5]. During this period, only a few of the in vivo transformed cells can escape the recognition and elimination by the innate antitumor immunity system and start the proliferation. Therefore the latent period in the tumor development seems to be crucial in carcinogenesis because it is just the time when the collision is solved in favor of either the proliferative activity of the in vivo transformed cells or the ability of the host's immunity system to detect their appearance and to reject them.
Until recently, the role of natural (innate) immunity in the protection of the host organism, intensively studied in bacterial and viral infections, was considered separately from mechanisms of the specific T-cell immunity. The current experimental and theoretical analysis of innate immunity reactions during infectious and autoimmune processes suggests the innate immunity not only involves the mechanisms of inherent nonclonal recognition and elimination of dangerous exogenous agents but plays instructive and controlling roles in the induction of specific (adoptive) immunity [10-13].
It seems that these new concepts concerning the interrelations between innate and acquired immunity may be also applied to the problem of antitumor immunity; this especially concerns the nonclonal mechanisms of recognition of tumor cells by effectors of the innate immunity. But unlike bacterial and viral infections which usually have short latent periods, acute course, and the effective development of humoral and/or cell immune reactions, in the case of spontaneous tumors the system of innate antitumor immunity deals with the absent or insignificant antigenic differences between the normal and autologous tumor cells and, as a rule, with the long-term carcinogenesis developing by a number of oncogenetic stages in the tumor evolution which significantly change biological properties of the tumor cells.
The present review considers phenotypic changes in tumor cells which are associated with their in vivo acquisition of discrete resistance mechanisms to the CTA of effectors of the innate antitumor immunity system and also the role of these mechanisms in the in vivo survival of tumor cells under conditions of normal and immune organism, natural selection, and progression of tumors. This aspect of carcinogenesis has been the area of main interest and experimental study in our laboratory and in the laboratories of our collaborators for the last decade.
In these studies our main approach included the following of tumor progression beginning from in vitro (or in vivo) transformation of normal cells by various oncogenes or spontaneously, with subsequent natural selection in vivo of their descendants and their acquisition of malignant phenotype. This approach allows us to study the in vivo dynamics of the progression of individual tumors and to identify discrete changes in the tumor cells which regularly appear during their malignant development, i.e., the secondary phenotypic changes of tumor cells specific for the tumor progression. It was found that during in vivo growth of cells transformed in vitro, natural selection resulted in tumor cell variants with high antioxidant activity that was displayed by H2O2-catabolizing activity (H2O2CA) and newly acquired ability to immediately release E2-type prostaglandin (PGES) during contact interaction with NK, MPh, and NPh. The remarkable selective in vivo advantages of tumor cells due to acquisition of the [H2O2CA + PGES] phenotype and, respectively, the local protection against effectors of the innate and adopted T-cell immunity, the indispensable acquisition of these properties during the tumor progression, and the relation with tumorigenicity are unique for the above-described phenotype compared to some other secondary changes in tumor cells which more or less regularly appear during tumor progression in vivo.
I. EXPRESSION OF [H2O2CA +
PGES] PHENOTYPE DURING in vivo PROGRESSION OF CELLS
SPONTANEOUSLY TRANSFORMED in vitro
We first observed [H2O2CA + PGES] phenotype expression more than 10 years ago during natural selection in vivo of cell variants of Syrian hamster tumors which were different in the levels of TGA and SMA. These cells were originated from spontaneously transformed in vitro hamster embryo cells of the STHE strain obtained in our laboratory. The cells of this strain have low levels of TGA and EMA, fail to spontaneously metastasize, lack immunogenicity, and are highly sensitive to the cytotoxic activity of MPh and NK. They had not been selected in vivo before being used in that work. It seemed important to determine the discrete differences between the parental cells of the STHE strain and their in vivo selected malignant descendants.
In preliminary experiments in vivo selected variants of these cells of high tumorigenicity, metastasizing or not metastasizing (SMA+ and SMA-), were found by quantitative tests on TGA (determined as the logarithm of 50% transplantation dose (log TrD50)), EMA, and SMA to have a significantly higher resistance to the CTA of MPh and NK than the parental cells, and thus, the previous reports of other authors [14-19] were confirmed. In studies [20] and [21] the resistance of various cell lines of mouse tumors of different histogenesis to the CTA of MPh was found to correlate with their antioxidant activity determined by the total activity of catalase, CuZn-superoxide dismutase [CuZn-SOD], and glutathione peroxidase. In the same period, the resistance of some tumor cell lines to the CTA of NK was independently reported to be associated with their ability to secrete E2-type prostaglandins which neutralized the CTA of NK [22-25].
In connection with these data, twenty-six individual cell lines of the same origin (strain STHE) selected in vivo were compared by the following characteristics: the sensitivity (or resistance) of the cells to damage by H2O2 (H2O2R); the ability of extracts (standardized against the protein) from the cells to catabolize H2O2 (H2O2CA); the activities of catalase, CuZn-SOD, glutathione, and glutathione oxidase and peroxidase (in cell extracts); the ability of tumor cells to secrete PGE2 during contact interaction with NK and inactivate the CTA of the NK; the possible relation of PGE2-secreting activity (PGE2S) with the cell sensitivity to the CTA of NK and also to the TGA, EMA, and SMA [5, 26-28].
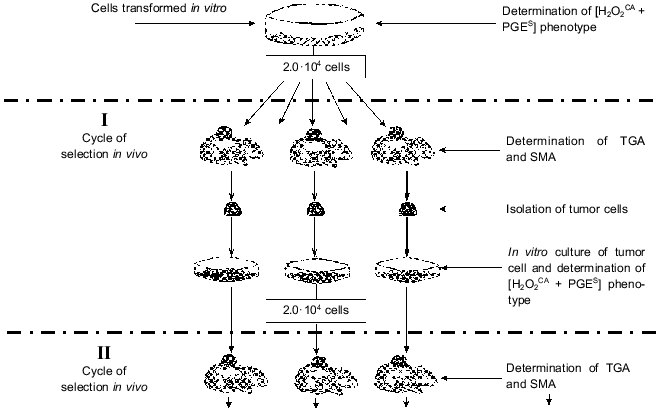
A procedure was elaborated for in vivo selection of variants of these cells during their subcutaneous growth in normal animals. Each cycle of the selection was started with a subcutaneous transplantation of 2.0·104 tumor cells grown in the in vitro culture; their growth in vivo was continued for 55-60 days (Fig. 1). Then the animals were sacrificed, cells from the tumor nodes from individual animals were again put into tissue culture, and their characteristics were studied after 3-5 in vitro passages (to remove the host's stroma cells).
The sensitivity of tumor cells to damage by H2O2 (H2O2R) was tested by incorporation of [3H]TdR into intact cells (control) and into cells treated with five twofold different doses of H2O2 (from 1.65 to 27.0 mM). The H2O2CA was determined using luminol-dependent chemiluminescence which was recorded at the time required for inactivation of 95% of H2O2 in the presence of an extract from the cells under study (versus the time required for inactivation of H2O2 in solution without the cell extract). The H2O2CA was considered positive when the inactivation of 95% of H2O2 added to the tumor extract required 20 sec - 3 min. These two tests were convenient for the evaluation of total antioxidant activity of the cells, and their results were essentially the same. The standard methods used for determination of the H2O2R and H2O2CA and also of some enzymatic antioxidants are described elsewhere [26-29].Fig. 1. Natural selection of tumor cells under conditions of subcutaneous growth of the tumor. Abbreviations: [H2O2CA + PGES] phenotype: H2O2CA) H2O2-catabolizing activity, PGES) PGE2-secreting activity; TGA) tumorigenic activity; SMA) spontaneous metastatic activity.
The PGE2S activity of tumor cells was determined with a standard radioimmunoassay (RIA) [24] and a very sensitive and reproducible biological test based on the ability of PGE2 to inactivate the CTA of NK was especially elaborated for this work. This method makes it possible to determine the biological activity of native PGE2 secreted by tumor cells and has some important benefits versus the RIA [30].
The quantitative test of the TGA includes the subcutaneous transplantation into five or six normal Syrian hamsters with four tenfold different doses of the tumor cells with subsequent recording of final (55-60 days later) results of the transplantation and determination of 50% transplantation dose (log TrD50). This approach appears to be very informative, and its high reproducibility enables the correct evaluation of the TGA level of tumor cell lines under study. Standard approaches were also used for quantitative determination of the EMA, SMA, and also of the cell sensitivity to the CTA of MPh, NPh, and NK.
Using these methods the in vivo selected high-tumorigenic variants of STHE cells were shown to have more than the tenfold increased level of H2O2R compared to the low-tumorigenic variants and the parental STHE cells and also significantly accelerated H2O2CA (from 15-20 min characteristic for the extract from the parent STHE cells to 20-180 sec for the high-tumorigenic variants). The high-tumorigenic cell variants also had constantly increased activity of catalase by 20-60% and decreased activity of CuZn-SOD and of lipid peroxidation. Changes in other antioxidant enzymes were less regular [26-28].
The in vivo selected high-tumorigenic variants of STHE cells were significantly more resistant to the CTA of NK, and, moreover, direct contact resulted in the rapid loss of the CTA by the NK. Pretreatment of the tumor cells with indomethacin canceled these effects and thus confirmed the earlier reports that PGE2 secreted by tumor cells was involved in the inactivation of the CTA of NK [22-24, 30-33]. The inhibition of the CTA of NK was not observed during the interaction of NK with the parental STHE cells and their in vivo selected low-tumorigenic variants which did not secrete PGE2.
The tumor cells began to release PGE2 into the medium immediately after the contact signal of NK and the release continued for 3-4 h with a peak at 30-60 min [34]. The transfer of PGE2 produced by the high-tumorigenic cell variants onto the intact NK completely inactivated the CTA of the latter against standard targets. Pretreatment for 2 h of the high-tumorigenic tumor cells with indomethacin before their contact with NK or MPh prevented the release of PGE2 [33, 35].
It was unexpectedly found that two biochemically different characteristics of tumor cells, their antioxidant activity (H2O2CA or H2O2R) and the PGE2S, seemed to be simultaneously acquired as a cluster of characters during the in vivo selection of high-tumorigenic variants of the STHE strain. No intermediate variants were found among the great number of STHE cell lines studied: the two features were either expressed or not expressed in all lines selected in vivo [27]. A positive correlation was also found between the [H2O2CA + PGES] phenotype expression and the high TGA level of the tumor cells, but until recently its relation to local mechanisms of tumor cell defense was unclear (see section V). However, the [H2O2CA + PGES] phenotype expression specific for all high-tumorigenic lines of the STHE strain could be accompanied or unaccompanied by the SMA. Thus, the [H2O2CA + PGES] phenotype expression was likely to be necessary but insufficient for manifestation of the SMA by the tumor cells.
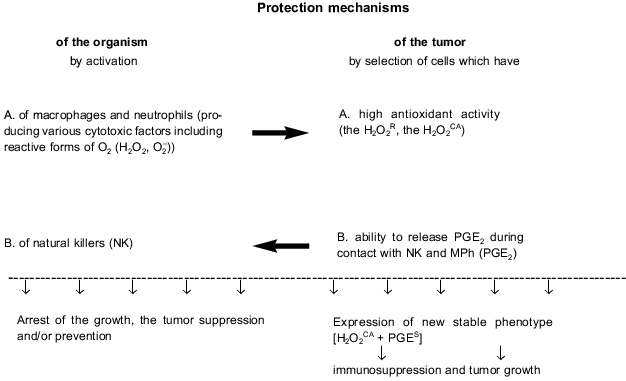
The biochemically different properties of the tumor cells which are expressed in the [H2O2CA + PGES] phenotype (i.e., H2O2CA and PGES) seem to be responsible for two different protection mechanisms of these cells against the CTA of MPh and NPh on one hand, and against the CTA of NK on the other (Fig. 2). Thus, in most of the STHE variant lines expressing and non-expressing the [H2O2CA + PGES] phenotype the [H2O2CA + PGES] phenotype expression clearly correlated with the two-to-fivefold increased resistance to the CTA of resident and activated MPh compared to the parental STHE cells and variants non-expressing the [H2O2CA + PGES] phenotype [36, 37]. The increased antioxidant activity of malignant variants of the tumor cells correlated, as a rule, with the increased activity of catalase, and this probably protected the tumor cells against damage by reactive oxygen forms and H2O2 produced by MPh and NPh.
In contrast, the release of PGE2 by the same tumor cells during contact interaction with NK and MPh inhibited the CTA of NK and thus, an active and essentially aggressive protection of the tumor cells against the CTA of NK cells was displayed. Moreover, pretreatment of such tumor cells with indomethacin recovers their sensitivity to the CTA of NK [38]. Thus, the discrete mechanism of resistance of the STHE high-tumorigenic variants to the CTA of NK is determined by their ability to inactivate the CTA of NK by release of PGE2 in response to contact interaction with these effectors. Paradoxically, during this interaction, the tumor cells and NK interchange their roles: the first play as effectors, whereas NK become their unprotected targets. The CTA of MPh was significantly (by two orders of magnitude) less sensitive to the suppression by PGE2; however, the PGE2-induced decrease in the production by macrophages of H2O2 which is toxic for tumor cells and may be an additional protection mechanism of tumor cells against these effectors. Such an autoregulation mechanism of H2O2 release and production of PGE2 was described for activated MPh. Its biological significance seems to be in protecting MPh against excess damage with H2O2 [21]. Certain mechanisms of tumor cell protection against MPh and NK are schematically shown in Fig. 2.Fig. 2. Innate immunity of the host and tumor progression.
The two described protection mechanisms of malignant variants of tumor cells against MPh, NPh, and NK which are based on the [H2O2CA + PGES] phenotype expression do not exhaust all possibilities of tumor cell protection; in particular, tumor cells can produce unidentified factors which suppress the innate antitumor immunity and thus allow the tumor to "sneak through" the host's protection barriers [39]. Apoptosis of NK and T cells induced during the FAS--FASL-interaction of these effectors with tumor cells should be noted among recently found new mechanisms of tumor cell protection. This mechanism of tumor cell protection has already been described elsewhere [40-43].
The mechanism of genetic regulation of characters responsible for the [H2O2CA + PGES] phenotype is still unknown. While the H2O2R (and H2O2CA) of tumor cells presents the total activity of various genes responsible for catalase, CuZn-SOD, and glutathione peroxidase, the releasing of PGE2 (which is one of ultimate metabolites of arachidonic acid) is determined by the activity of one enzyme, cyclooxygenase. Until recently, these characters (the antioxidant activity and production of PGE2) were usually considered separately, disregarding their interrelations. However, the co-expression of the characters which determine the antioxidant and PGE2-secreting activities suggest a common regulation of these two biochemically different properties of tumor cells. In this case, the in vivo selection of H2O2R variants of tumor cells (e.g., by activated MPh or NPh) would result in the acquisition of PGE2S without involvement of NK, and, on the contrary, NK would be responsible for selection of cells expressing both these characteristics.
It is still unclear whether cell variants expressing the [H2O2CA + PGES] phenotype preexist in the population of the parental STHE strain. However, it is obvious that the in vitro cultivation fails to reveal their selective advantages. We were unsuccessful in our attempts to select in vitro cells of the STHE strain expressing [H2O2CA + PGES] phenotype by a long-term treatment with low cytotoxic doses of H2O2 (unpublished data) or by a long-term co-culture of the tumor cells with the resident or activated MPh. In these experiments the acquisition by the STHE cells of a certain resistance to the H2O2-induced damage was not accompanied by the expression of PGES [44]. It seems to indicate that cell variants expressing the [H2O2CA + PGES] phenotype do not preexist in the parental population of cells which were spontaneously transformed in vitro and that the expression of this phenotype more likely is a new characteristic of the tumor cells which is acquired under conditions of their in vivo growth and natural selection (including the selection determined by the CTA of MPh, NK, and NPh) and provides them selective advantages in vivo during the interaction with these effectors. The [H2O2CA + PGES] phenotype expression correlated with a 30-200-fold increase in the TGA (accompanied or unaccompanied with SMA) of variant lines of the STHE strain, and this prompted us to study other models of in vitro cell transformation especially those specified by the high TGA level induced during transformation.
II. EXPRESSION OF [H2O2CA +
PGES] PHENOTYPE BY CELLS TRANSFORMED in vitro BY ROUS
SARCOMA VIRUS; POSSIBLE ROLE OF THE v-src GENE
It was shown for the first time in the 1960s and then confirmed that cells of mice, rats, and hamsters transformed in vitro by the Rous Sarcoma Virus (Schmidt--Ruppin strain) (RSV-SR) had an extremely high level of TGA and often of the SMA [45-47]. Much later, the v-src-induced carcinogenesis was characterized as an exclusive one-step type of neoplasm different from other types of carcinogenesis. The mechanism of one-step carcinogenesis still remains unclear [48].
Therefore, in the light of the data presented in section I, it was interesting to study characteristics of cells transformed in vitro by RSV-SR, including the possible expression of the [H2O2CA + PGES] phenotype, TGA, SMA, and the sensitivity to the CTA of MPh and NK. For this purpose we subjected normal embryonic cells of Syrian hamster to in vitro transformation with RSV-SR and thus acquired four independent strains of v-src-transformants.
It was shown that during the in vitro transformation these four strains acquired the expression of the [H2O2CA + PGES] phenotype, extremely high levels of catalase, TGA, and SMA (the latter was found in three of the four lines), high resistance to the CTA of MPh and NK. The [H2O2CA + PGES] phenotype of these cells was expressed on the level of both, i.e., cell populations and clones [28, 29, 49, 50]. Some characteristics of two lines of the v-src-transformants are presented in Table 1.
Table 1. Characteristics of Syrian hamster
cell lines transformed in vitro by the Rous sarcoma virus
(RSV-SR) compared to normal cells and spontaneously transformed cells
of the STHE strain
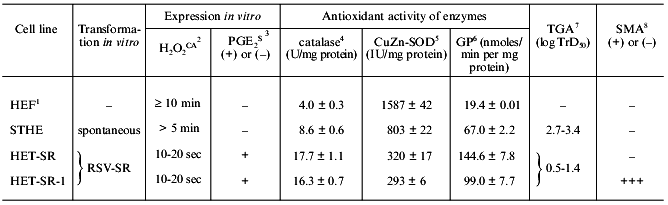
1 Normal fibroblasts of Syrian hamster.
2 H2O2-catabolizing activity of
cell extracts (min, sec); details in [28].
3 PGE2-secreting activity, (+) or (-), during
contact interaction of target cells with NK [28].
4 Spectrophotometric determination, units per mg protein
(after Aebi, 1984; details in [28]).
5 Spectrophotometric determination, international units
per mg protein (after Beauchamp and Fridovich, 1971; details in [28]).
6 Glutathione peroxidase (GP) was determined in nanomoles
per mg protein per min (after Tietze, 1969; details in [28]).
7 Tumorigenic activity determined as log of 50%
transplantation dose of tumor cells; details in [49].
8 Spontaneous metastatic activity determined with
standard tests (+) or (-); details in [49].
A nonrandom correlation between the [H2O2CA + PGES] phenotype expression and a high level of TGA of cells of RSV-SR-transformants was confirmed by experiments when the antioxidant activity (H2O2R) of these cells and PGES were preliminary suppressed in vitro, separately or combined, by nontoxic doses of BCNU and indomethacin and the TGA level of the treated and intact cells was subsequently determined in vivo. The treatment with both agents or only with BCNU more than 150-fold suppressed the TGA of the cells; a single treatment with indomethacin alone insignificantly suppressed the TGA [49].
The unusually high antioxidant activity of the cell strains RSV-SR-transformed in vitro is mainly due to the high activity of catalase which in these cells is 1.8-2.5-fold higher than in spontaneously transformed cells of the STHE strain and fivefold higher than in normal fibroblasts (Table 1).
The [H2O2CA + PGES] phenotype expression by cells of the RSV-SR-transformants (similarly to the earlier shown pattern for the STHE variants selected in vivo) was directly responsible for the cell resistance to the CTA of MPh and NK. Pretreatment of the RSV-SR-transformants with indomethacin made them highly sensitive to the CTA of NK [38]. The initially high resistance of v-src-transformants to the CTA of MPh and NK was especially surprising because these cells had not been selected in vivo and had no preliminary experience of interaction with these effectors of the innate antitumor immunity.
Evidently, in the RSV-SR-transformants the [H2O2CA + PGES] phenotype expression, high level of the TGA, and the acquisition of resistance to the CTA of MPh and NK are determined by the transforming activity of the v-src gene and do not depend on their selection in vivo by MPh or NK, as it was earlier supposed for the in vivo selection of STHE variants (see section I).
The specific role of the v-src gene and its transforming activity in the induction of the [H2O2CA + PGES] phenotype and also of high levels of the TGA, SMA, and resistance to the CTA of MPh and NK became clearer due to following studies. It occurred that two of our RSV-SR-transformants (lowly metastatic HET-SR and highly metastatic HET-SR-1) were transfected with plasmid DNA which contained the activated gene N-ras (neo). It was shown earlier that p21ras and pp60v-src used the same signal pathway--via phosphatidylinositol-3-kinase [51, 52]. Expression of the v-src gene (mRNA and enolase activity of pp60) was significantly (nearly completely) inhibited in the HET-SR cells transfected with N-ras, whereas the expression of p21N-ras increased tenfold and more [53]. Simultaneously with the inhibition of activity of the v-src gene, the N-ras-transfected HET-SR cells completely lost the expression of the [H2O2CA + PGES] phenotype [54]. In the N-ras-transfectants of the HET-SR-1 strain the expression of pp60v-src was significantly less suppressed and the expression of the gene N-ras (p21ras) increased only 2.0-2.5-fold. In this case cells of most of the transfected clones under study retained the H2O2CA but lost the PGES. It should be noted that the N-ras-transfection of the RSV-SR-transformed cells was found to be the only condition that resulted either in the complete inhibition of activity of the v-src-gene and the complete loss of the [H2O2CA + PGES] phenotype or, in the case of partial inhibition of the v-src-gene and low activity of the transfected N-ras, in the dissociation of characters which contribute to the [H2O2CA + PGES] phenotype (that always occurred only due to the loss of PGES).
The interaction of pp60v-src and p21N-ras in various recipient cells can occur in more than one site of the signal transduction pathway initiating from the G-protein and moving through PI-3 [55]; the level of genetic instability of transfectant cells is suggested to influence this interaction. The complete inhibition of the [H2O2CA + PGES] phenotype during the N-ras-transfection of the HET-SR cells is likely to be associated with the initial part of the signal pathway above the activation of PLC (phospholipase C), while the inhibition only of PGE2 with the retained H2O2CA in the N-ras-transfected HET-SR-1 cells seems to be associated with the interaction of pp60v-src and p21N-ras downstream of the signal pathway [53, 54]. In any case, the complete or partial inhibition of the v-src-gene activity in the RSV-SR-transformants in most N-ras-transfectant clones is accompanied by complete or partial suppression of the [H2O2CA + PGES] phenotype, respectively, and this indicates that the expression of this phenotype in the RSV-SR-transformants depends on v-src-gene activity. Later we showed that the transfection of cells of both the HET-SR and STHE strains with the genes N-ras and Ha-ras suppressed their antioxidant activity due to significant (virtually complete) inhibition of catalase which was gradually recovered during the subsequent two-three cycles of in vivo selection [28]. Thus, it was shown for the first time that the significantly increased activity of catalase along with the PGE2S are specific biochemical manifestations of cell transformation in vitro by the v-src-gene, whereas genes N-ras and Ha-ras inhibited these two v-src-induced cell characteristics.
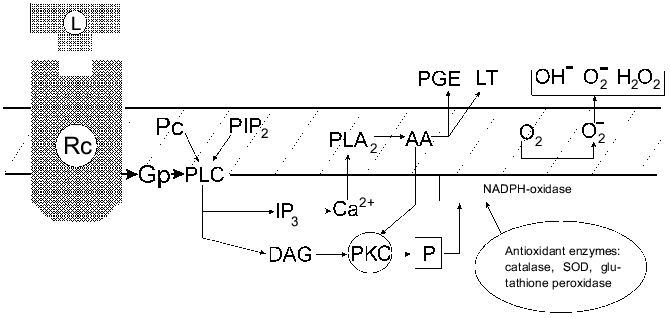
It seems that only one type of normal cells can express a similar [H2O2CA + PGES] phenotype, namely, activated MPh which have H2O2R and the ability to release PGE2 in response to certain signals [56]. The authors' scheme suggests that the signal transmission in the activated MPh after the receptor contact with the ligand involves a G-protein, hydrolysis of PIP2, activation of PLC, and then, possibly, divides into two interrelated pathways which provides the secretion of PGE2 (or LT) and results in the oxygen burst (and its subsequent suppression with PGE2). Figure 3 presents a simplified scheme of these events.
Thus, the [H2O2CA + PGES] phenotype expression during the activation of MPh and its stable expression in the cells of v-src-transformants and in the in vivo selected malignant variants of STHE cells suggest the existence of a common regulation mechanism of the [H2O2CA + PGES] phenotype expression which seems to be associated with the activation of the c-src gene. Expression of the activity of tyrosine kinase of the src family genes in the activated MPh and also in normal fibroblasts under the influence of growth factors such as PDGF, CSF-1, etc., was shown in several studies. The list of exogenous agents activating transmembrane receptors of src-tyrosine kinase is rapidly increasing [57]. Oxidative and radiation stresses activate genes of the src family. The encoding SH2- and SH3-domains of c-src are involved in the control of signal proteins of cytoplasm; their site-directed mutagenesis can induce the oncogenic activation of pp60c-src [58-61]. The mutation-induced activation of pp60 [c-src] of tyrosine kinase accompanied by an increase in TGA was found during the in vivo selection of preneoplastic cells of Syrian hamster initially transformed in vitro spontaneously or by various agents [62]. Recently many observations are accumulating on activation of the c-src-gene in cells of various human tumors, particularly in cases of colon cancer, especially during late stages of the tumor development [63, 64].Fig. 3. Modified scheme of signal transduction during two metabolic outbursts in activated MPh: releasing of PGE (or LT) and of oxygen free radicals and the inactivation of H2O2 by antioxidant enzymes. Abbreviations: GP) G-protein; PC) phosphatidylcholine; PLC) phospholipase C; DAG) 1,2-diacylglycerol; PLA2) phospholipase A2; PKC) protein kinase C; PGE) E-type prostaglandin; LT) leukotrienes; AA) arachidonic acid [56].
It remained unclear whether the expression of the [H2O2CA + PGES] phenotype is specific only for v-src-transformation of cells or whether other oncogenes or suppressor genes can induce the expression of this phenotype in in vitro transformed cells. This question is considered in the next section of this paper.
III. TRANSFORMING GENES AND TUMOR PROGRESSION in vivo
In connection with data presented in sections I and II, it was interesting to study the possible expression of the [H2O2CA + PGES] phenotype by cells of same origin (Syrian hamster) which were transformed (or transduced) in vitro with oncogenes other than the v-src gene and different in their transforming mechanisms. For this purpose, we studied the possible induction in vitro of the [H2O2CA + PGES] phenotype expression by transforming genes LTSV40, E1a and b, Ha-ras, myc, p53175, and bcl-2 and also the TGA and SMA levels of the transformed cells. Similar studies were also carried out on a variety of their variant cell lines selected in vivo.
None of the parent cell lines transformed (or transduced) in vitro with the genes LTSV40, E1a and b, myc, Ha-ras, p53175, and bcl-2, similarly to the spontaneously transformed in vitro cells of the STHE strain which were studied earlier, expressed the [H2O2CA + PGES] phenotype, were lowly tumorigenic, and did not spontaneously metastasize. Results of studies on all the above-listed parent cell lines including the v-src-transformants and the STHE strain cell are presented in Table 2 and Fig. 4. All parent cell lines of the transformants and transducents which initially failed to express the [H2O2CA + PGES] phenotype were subcutaneously transplanted into normal animals in order to subject them to the standard (Fig. 1) cycles of natural selection in vivo. After each cycle of the in vivo selection (for 48-60 days) variant lines of tumor cells isolated each from the individual animals (in total more than 100 lines) were tested again on the [H2O2CA + PGES] phenotype expression, TGA, and SMA.
Table 2. Transforming genes, expression of
the [H2O2CA + PGES]
phenotype, and tumorigenicity of tumor cells before and after in
vivo selection

Note: Notations same as in Table 1.
One can see from the data presented that all variant cell lines have acquired the expression of the [H2O2CA + PGES] phenotype and a significant (30-200-fold) increase in TGA which was sometimes accompanied by SMA within the second and third cycles of the in vivo selection (i.e., in total, during 120 or some more days of subcutaneous growth in vivo). The SMA expression by the cells of some clones transformed with LTSV40 and p53175 was observed earlier, already after two cycles of the in vivo selection.Fig. 4. Transforming genes and in vivo progression of tumor: expression of the [H2O2CA + PGES] phenotype (dark rectangles); TGA (x50-150) (light rectangles); *, SMA.
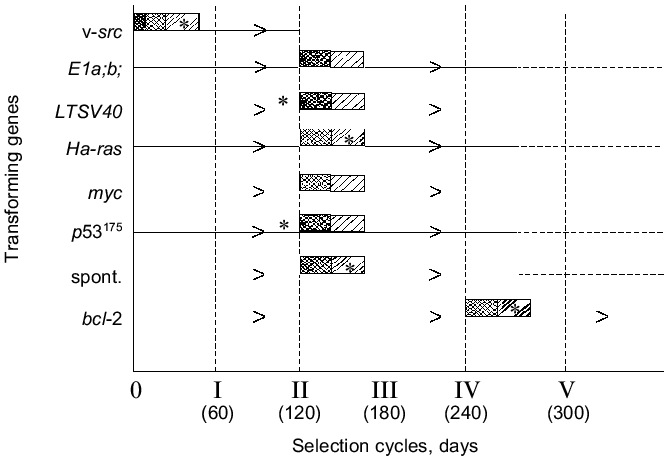
Unlike all other cell lines, the in vivo selection of cells of the STHE-bcl-2 transducent was significantly delayed, and the [H2O2CA + PGES] phenotype expression and significant increase in TGA (and in SMA) occurred only after four-to-five cycles of the selection of these cells (i.e., after 240-300 days of their subcutaneous growth). During the first and partially the second cycles of the in vivo selection of the STHE-bcl-2 variants, subcutaneous tumor nodes formed by these variants were nearly free of the central necrosis which is very specific for tumors arising after the transplantation of the parent STHE cells and all other transducents of these cells. However, necrotic zones in the tumor nodes of HETR-bcl-2 gradually increased during the subsequent growth and selection in vivo and became indistinguishable from large necrotic zones of the parent STHE cells.
As a whole, the data presented in sections I-III indicate that after the transformation (spontaneous or induced in vitro by genes v-src, LTSV40, or E1a;b) of normal hamster embryonic cells and also after the transduction of STHE cells with genes Ha-ras, myc, p53175, and bcl-2 all cell lines (except for the v-src-transformants) did not express the [H2O2CA + PGES] phenotype and remained lowly tumorigenic. Regardless of the genes which initially transformed the cells (except for the v-src-transformants), the tumor progression in vivo inevitably resulted in replacement of the in vitro transformed parent cells by the tumor cells which express the [H2O2CA + PGES] phenotype, high level of TGA, and less regularly SMA. The time required for the in vivo selection of cells expressing the [H2O2CA + PGES] phenotype in the subcutaneous tumor nodes was surprisingly the same for most of the transformants with only one exception: the transduction of the STHE cells by the gene bcl-2 significantly decreased the in vivo progression of these tumors (Table 2 and Fig. 4).
To explain such a significant delay in the in vivo progression of tumors expressing the gene bcl-2, three possibilities are suggested as follows: the bcl-2-expressing cells due to their high antiapoptotic activity have an increased (or the same) selective advantage in vivo versus the cell variants expressing the [H2O2CA + PGES] phenotype which rarely appear in the cell population (in this case the latter ones have no chances to be selected in vivo because the former ones dominate in the cell population); the selective advantages of the rarely appearing [H2O2CA + PGES] + bcl-2 cell variants are higher than those of the STHE-bcl-2 cells (in this case the [H2O2CA + PGES] + bcl-2 variants should gradually displace the parent STHE-bcl-2-cells from the subcutaneously growing tumor, and this really seems to occur); a gradual decrease in the in vivo expression of the bcl-2-gene and the correspondingly increased chances for appearance and selection of the [H2O2CA + PGES] variants also remain quite possible. In any of these possibilities, the expression of the bcl-2 gene seems to prevent the selection of cells expressing the [H2O2CA + PGES] phenotype and thus (paradoxically.) to suppress the tumor progression.
Two polar variations in the rate of the in vivo acquisition of the [H2O2CA + PGES] phenotype by the in vitro transformed cells (Fig. 4) attract attention: the v-src-transformants (unlike all other transformants studied) acquire the [H2O2CA + PGES] phenotype and extremely high level of the TGA during the cell transformation in vitro, i.e., at the apparently zero point of the tumor progression. The subsequent experience of the cells transformed in vivo by the v-src gene seems to add nothing to the characteristics under study (or adds SMA in cases of its initially absent expression), whereas the bcl-2-transducents acquire the same [H2O2CA + PGES] phenotype and the high level of TGA (and SMA) only during a very long-term selection in vivo. It is likely that the activity of the bcl-2 gene suppressing the apoptosis in vivo competes with the selection of cells which express the [H2O2CA + PGES] phenotype and, consequently, with the tumor progression [29]. The rate of natural selection and of the in vivo acquisition of the [H2O2CA + PGES] phenotype expression by the cells transformed in vitro with the genes LTSV40, E1a,b, Ha-ras, myc, and p53175 was approximately the same and did not depend on the type of transforming genes.
In general, the findings permit the [H2O2CA + PGES] phenotype expression as a marker of a definite relatively early (premetastatic) step in the in vivo progression of tumors.
IV. SELECTION OF CELLS BY THE [H2O2CA +
PGES] PHENOTYPE DURING PRIMARY VIRAL CARCINOGENESIS
Until recently the earliest steps of primary carcinogenesis in vivo and phenotypical changes in tumor cells which seem to occur during its latent period were inaccessible for investigation. However, according to Nowell [65], "the fundamental nature of this initial step, and degree to which it is specific for each neoplasm, remains a basic problem in cancer research". To study this problem, we have used a new opportunity, namely, the determination of the [H2O2CA + PGES] phenotype as a marker of the definite and, possibly, one of the earliest steps in tumor progression in vivo after transformation.
As shown in the previous sections, the acquisition of the [H2O2CA + PGES] phenotype expression along with the 30-200-fold increase in TGA by the cells which were initially transformed or transduced in vitro with various antigens occurred during the in vivo selection in the subcutaneous tumors at surprisingly similar rates (with the above-described two polar variations). This could indicate that the initial population of cells transformed in vitro with various agents did not contain variants expressing the [H2O2CA + PGES] phenotype and/or there was no conditions for their selection in vitro. This hypothesis is supported by the finding that the probability of their appearance de novo and the selection rate during the tumor growth in vivo were nearly the same for various transformants. However, the possibility and time of the acquisition of the [H2O2CA + PGES] phenotype expression by the cells during the appearance of primary tumors remained unknown.
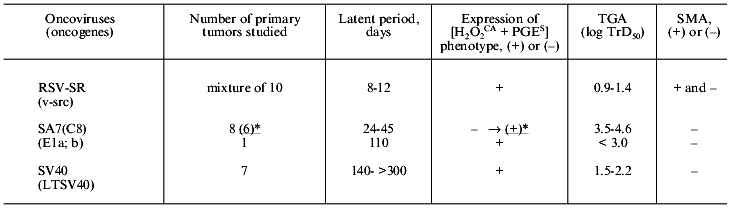
To study these problems, we used newborn Syrian hamsters for the three following models of primary viral carcinogenesis: RSV-SR-induced tumors characterized by a very short (few days) latent period and multiple (nearly uncountable) confluent tumor nodes in the site of the virus injection; tumors induced by the Simian adenovirus SA7(C8) in many cases characterized only by a somewhat longer latent period but significantly lower number of primary tumor nodules [1-6]; and tumors induced by SV40 virus characterized by a long latent period and appearance of usually no more than one or two tumor nodes.
The duration of latent periods till the appearance of primary tumors after the injection of these viruses into the newborn animals was carefully recorded; usually 15 days after the appearance of the palpable tumor and not more than after 20 days the resulting individual primary tumors were taken into in vitro culture. On the level of three-to-five in vitro passages, the cultures of the primary tumors were tested in vitro on the [H2O2CA + PGES] phenotype expression and in vivo on TGA, SMA, and sensitivity to the CTA of MPh and NK.
It was shown that the cells of the RSV-SR-induced primary tumors a few days after their appearance expressed the [H2O2CA + PGES] phenotype, a high level of TGA, and were resistant to the CTA of MPh and NK (Table 3). Thus, the in vivo development of the RSV-SR-induced primary tumors was likely to add nothing to the properties (see section II) which were acquired by these cells during the transformation in vitro. Thus, this type of primary carcinogenesis was shown to express the [H2O2CA + PGES] phenotype and other characteristics under study independently of conditions of the in vivo growth. This conclusion was also supported by finding of the extremely short latent period (8-12 days) of the RSV-SR-induced primary tumors (correlating with the transformation period of the cells in vitro) and the high multiplicity of these neoplasms.
Table 3. Acquisition of
[H2O2CA + PGES] phenotype
during primary viral carcinogenesis
Note: Notations same as in Table 1.
*In parentheses the additional cycle of in vivo selection
(the subpassage) of six primary SA7(C8) tumors; acquisition of
[H2O2CA + PGES] phenotype
expression.
Unlike them, eight of nine primary SA7(C8) tumors which appeared somewhat later (24-45 days after the virus inoculation to newborn animals) and were studied after 39-65 days of their in vivo development did not express the [H2O2CA + PGES] phenotype and were two-to-three orders of magnitude less tumorigenic (Table 3). However, six of these eight tumors transplanted into animals and thus subjected to an additional cycle of in vivo selection (during 62 days of subcutaneous growth, i.e., in total, 100-120 days of their development in vivo) demonstrated the [H2O2CA + PGES] phenotype expression and about tenfold increase in TGA. It is suggested that by the end of latent periods of the early SA7(C8) primary tumors or shortly after their appearance, i.e., within 39-65 days of the tumor development in vivo some tumor cells in the primary tumor nodes (it seems to be a minor undetectable fraction) already expressed the [H2O2CA + PGES] phenotype, but the period of 30-65 days seemed to be too short for these cells to replace their parent cells in the tumor node. This hypothesis was confirmed by findings on the ninth primary SA7(C8) tumor which appeared after 110 days of the latent period: the cells of this tumor expressed the [H2O2CA + PGES] phenotype immediately after their appearance (and also an increased level of TGA) (Table 3).
The duration of the latent periods of seven SV40-induced primary tumors varied from 140 to more than 300 days, and each of these tumors after its appearance expressed the [H2O2CA + PGES] phenotype (Table 3). The TGA level of primary SV40 tumors was about two orders of magnitude higher than both, i.e., the early SA7(C8) primary tumors and the cells transformed by SV40 in vitro, and varied from 1.5 to 2.2 log TrD50.
Based on the time of acquisition of the [H2O2CA + PGES] phenotype by the cells of SA7(C8)- and SV40-induced primary tumors, the minimal time could be approximately determined that was required for the replacement in vivo of the [H2O2CA + PGES] phenotype-negative cells in the subcutaneous nodes by cells expressing this phenotype. This time was more than 65 days, and periods about 100-140 days of the tumor growth in vivo appeared to be sufficient. Thus, the dynamics of the process is the function of time required for the appearance and in vivo selection of tumor cells expressing the [H2O2CA + PGES] phenotype.
Unlike the growth of the in vitro transformed cells in tissue culture, the appearance of primary monoclonal tumors in vivo is usually a multistep process which includes the transformation of cells and selection of new variants of the tumor cells possessing some selective advantages for growth in vivo. The RSV-SR-induced one-step carcinogenesis is the exception from this rule. Findings on the primary SA7(C8)- and SV40-carcinogenesis indicated that the appearance and selection of cell variants which express the [H2O2CA + PGES] phenotype and, correspondingly, can protect themselves against effectors of the innate antitumor immunity starts during the latent period and, depending on its duration, could be completed before its termination. Primary tumors appearing in vivo are usually monoclonal (and neoplasms induced by SV40 and adenoviruses are not an exclusion from this rule), and this supports the suggestion that the occasional appearance of rare cell variants expressing the [H2O2CA + PGES] phenotype is associated with their strict selection that provide the maintenance of tumor cells with selective advantages in vivo.
The RSV-SR-transformed cells are the striking exception from this scenario: the cell transformation by the v-src gene both in vitro and in vivo occurs simultaneously with their acquisition (without a stage of selection in vivo) of the [H2O2CA + PGES] phenotype, the maximal TGA, and even SMA in some cases. The strikingly short latent period (8-12 days) till the appearance of the primary palpable tumors induced with RSV-SR, their appearance in all inoculated animals, and the multiplicity of these neoplasms suggest a nonrandom association between the expression by these cells of the v-src gene, the [H2O2CA + PGES] phenotype, and the high level of their TGA. It remains important to elucidate to what degree the probability and incidence of various tumors and also the duration of their latent periods are determined by the appearance of cells expressing the [H2O2CA + PGES] phenotype.
V. EXPRESSION OF [H2O2CA +
PGES] PHENOTYPE AND EFFICIENCY OF SPECIFIC ANTITUMOR
IMMUNITY
It remained unclear whether the local protection of tumor cells based on the [H2O2CA + PGES] phenotype expression and directed against effectors of the innate antitumor immunity is related to specific antitumor immunity. In particular, two questions were interesting for us: a possible effect of the specific antitumor immunity on the rate of in vivo selection of tumor cells expressing the [H2O2CA + PGES] phenotype and the possible effect of the [H2O2CA + PGES] phenotype expression by the tumor cells on the efficiency of their immune rejection under conditions of normal and immune host organisms.
To study the first question, cells of Syrian hamster transformed in vitro with the SV40 virus (strain HE-wtSV40) which expressed TSTA (Tumor Specific Transplantation Antigen) and did not express the [H2O2CA + PGES] phenotype were subjected to standard cycles of selection in vivo in normal and SV40-immunized animals. After each cycle of the selection in vivo, cells of the tumor nodes which appeared in the normal and SV40-immunized animals were transferred to in vitro tissue culture and tested for the [H2O2CA + PGES] phenotype expression, whereas the expression of TGA, SMA, and TSTA was tested in vivo. The TSTA expression was determined by the specific immune sensitivity of the tumor cells (the difference between TGA (in log TrD50) of the immune and normal animals) [68]. The [H2O2CA + PGES] phenotype and high TGA were acquired during the selection in both normal and immune animals nearly simultaneously (between the second and third cycles of the in vivo selection of tumor cells); the rate of in vivo selection of immune resistant tumor cells (in the immune animals) was significantly decreased compared to the selection by the [H2O2CA + PGES] phenotype and in most cases required four selection cycles. Apparently, the selective pressure of effectors of the specific antitumor immunity on the TSTA-expressing cells is less efficient than the natural selection of the same tumor cells expressing the [H2O2CA + PGES] phenotype, and the combination of these two types of selection (on the immune animals) neither accelerates, nor decreases the rate of the process. Thus, the selective pressure of the effectors of the innate and specific antitumor immunity in vivo on the tumor cells was realized in parallel and independently.
Nevertheless, the probable effect of the [H2O2CA + PGES] phenotype expression on the cell sensitivity to rejection due to effectors of the specific antitumor immunity must not be ruled out. This probability was studied using two approaches as follows.
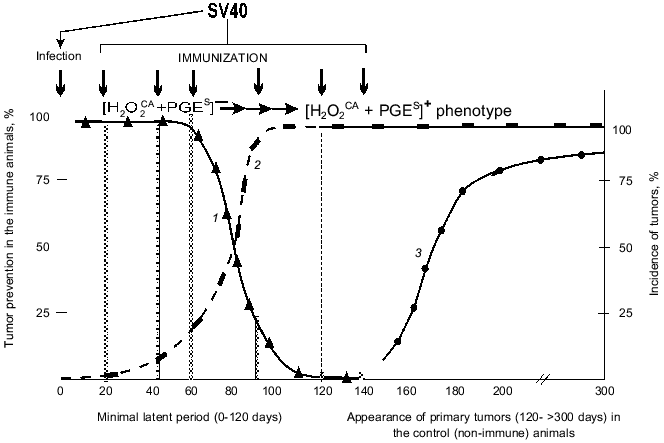
1. The possible prevention of SV40 primary tumors by a single immunization of the infected newborn animals with the SV40 virus within the latent period or after its termination. Figure 5 shows summarized results of six experiments on such prevention and includes the above-presented (Table 4) data on the approximate times of the [H2O2CA + PGES] phenotype acquisition by the cells of SV40 and SA7(C8) primary tumors during the latent period.
Table 4. Expression of the [H2O2CA + PGES] phenotype, tumorigenicity, and efficiency of immune rejectionFig. 5. Effect of selection by the [H2O2CA + PGES] phenotype on the efficiency of specific immunization against primary tumors (SV40): 1) the efficiency of immune prevention of the SV40 primary tumors depending on the immunization time; 2) the selection of tumor cells expressing the [H2O2CA + PGES] phenotype; 3) the appearance of SV40 primary tumors in the control (non-immune) animals.
Note: Notations same as in Table 1.
*Index of immune sensitivity (IS) of tumor cells was determined as the difference between values of log TrD50 for immune and normal animals.
The specific immunization of animals within the first 60-70 days was highly efficient and prevented the appearance of SV40 tumors in 90-100% of the animals (Fig. 5, grey columns). However, the efficiency of immune tumor prevention rapidly decreased on the 90-100th days of the latent period and was inefficient from the first day after the end of the minimal latent period (although it was long before the peak of appearance of the primary tumors) [66-68].
Thus, the period of maximal efficiency of the preventive specific immunization against the SV40 primary tumors (the first 60-70 days of the latent period) was strikingly the same as the early [H2O2CA + PGES] phenotype-negative stage in the development of these tumors when they could not yet protect themselves. And, on the contrary, the end of this "naive" stage (within 60-100 days of the latent period) correlated with the rapid decrease in the efficiency of the specific immunization against the SV40 tumors (Fig. 5). And the question is how the [H2O2CA + PGES] phenotype expression by the tumor cells can influence the rejection of these cells in the normal and immune organism. This question was studied using a second (and more direct) approach as follows.
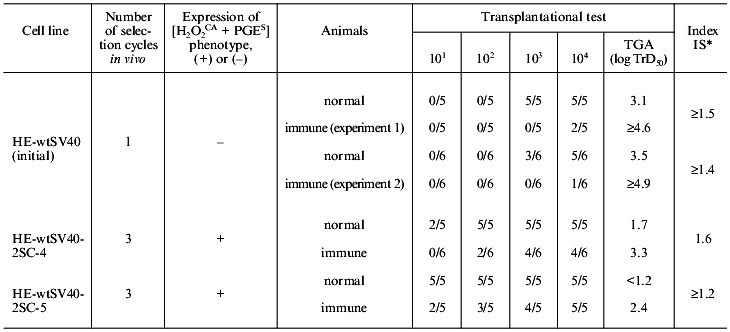
2. Determination of real number of tumor cells expressing or non-expressing the [H2O2CA + PGES] phenotype which were rejectable by the normal and immune animals. To study this problem, the parent cells (HE-wtSV40 strain) transformed in vitro with the SV40 virus (lowly tumorigenic and non-expressing the [H2O2CA + PGES] phenotype) and two cell lines of their direct descendants (highly tumorigenic and expressing the [H2O2CA + PGES] phenotype) which were subjected to three cycles of in vivo selection in normal animals were quantitatively challenged in groups of five-or-six normal and SV40-immunized animals.
In normal animals about 102 parent cells are naturally rejected, whereas the number of their variant cell lines expressing the [H2O2CA + PGES] phenotype rejected in normal animals is one-to-two orders decreased (Table 4). As a result, the TGA level of these in vivo selected lines, i.e., their ability to grow after transplantation of the decreased number of tumor cells significantly increases. The real number of parent tumor cells rejected in the immune animals is about 105, whereas the number of cells of their descendent variant line rejected due to immunity is tens or hundreds of times decreased (to 102-103); correspondingly, the TGA level of these cells is significantly increased. The efficiency of immune rejection of variant lines of the tumor cells decreases, contrary to the TSTA expression and to the remaining initial high immune sensitivity (always relative) of the lines compared [69].
Thus, cells of the in vivo selected variant lines of tumor cells differed from their in vitro transformed parental cells of the HE-wtSV40 strain in the expression of the [H2O2CA + PGES] phenotype, the significantly decreased number of cells rejected in both the immune and normal animals, and the significantly higher TGA level at the transplantation to normal and immune animals.
It was earlier shown that during the latent period of development of the SV40 primary tumors animals infected with the SV40 virus were neither tolerant to the TSTA of SV40 nor immune [67, 68]. The comparison of data presented in Fig. 5 and Table 4 suggests an explanation of the rapidly decreased efficiency of the specific immunoprevention of the SV40 primary tumors during the second half of the latent period: during this time the number of the [H2O2CA + PGES] phenotype-expressing tumor cells is gradually increasing, while their real number which is rejectable in the immune organism is simultaneously decreasing tens or hundreds of times (despite the retained immune sensitivity). Thus, the acquired with the [H2O2CA + PGES] phenotype ability of tumor cells for local defense against effectors of the innate antitumor immunity and for their suppression at the site of tumor growth is suggested to affect the interaction and interchange of instructive information between the tumor cells and T-cell effectors of the specific antitumor immunity. Direct local inhibition of T-helpers and killers by the tumor cells expressing the [H2O2CA + PGES] phenotype is also not excluded.
The data presented in this section seem to be related to one of the basic and unsolved problems in immunology of tumors: why the efficiency of immune prevention and immune therapy of tumors and metastases is, as a rule, lower than expected [6-8, 70, 71]. It seems likely that local mechanisms of tumor cell defense against effectors of the innate and specific antitumor immunity which are acquired together with the expression of the [H2O2CA + PGES] phenotype are an important and underestimated factor responsible for the low efficiency of immune treatment of tumors in vivo.
In conclusion, the results of the 10 years of studies in our laboratory presented in this review have shown that the secondary phenotypic changes in the transformed and tumor cells regularly appear during the natural selection and tumor progression in vivo. These changes are manifested by the highly increased H2O2-catabolizing (antioxidant) activity (H2O2CA) of tumor cells and the simultaneous acquisition of a new discrete character--the ability for the immediate releasing of prostaglandin E2 (PGES) on contact interaction with NK and MPh. The simultaneous acquisition in vivo of these two biochemically different characteristics of tumor cells was found during comparative studies on parent cell lines from Syrian hamster which were transformed in vitro with various agents and on more than 100 of their descendent lines naturally selected in vivo. The expression of the [H2O2CA + PGES] phenotype was shown to provide the cells of both transplantable and primary tumors of various origin with mechanisms of active local protection against the CTA of MPh, NK, NPh, and T-lymphocytes. This significantly increases the probability of their survival in vivo and, respectively, their TGA; as well it inhibits the efficiency of their specific and nonspecific rejection in vivo caused by effectors of the host immunity. Expression of the [H2O2CA + PGES] phenotype was also found in the in vitro cultures of various human tumor cell lines (data not presented).
Apparently, the in vivo selection of rare variants of tumor cells expressing the [H2O2CA + PGES] phenotype suggests its association with the direct cytotoxic influence of effectors of the innate antitumor immunity: MPh, NPh, and NK. However, experiments with cells transformed with the v-src gene in vitro (or in vivo) seem to suggest the more complicated pattern of events resulting in the appearance of the [H2O2CA + PGES] phenotype-expressing cells.
The specific role of the v-src gene in induction of the [H2O2CA + PGES] phenotype during the transformation of normal cells with the Rous Sarcoma Virus, the inability of other transforming genes studied (including the suppressor genes and bcl-2) to induce in vitro the expression of this phenotype, and the obligatory selection in vivo of cells expressing the [H2O2CA + PGES] phenotype by the same neoplasms of other oncogenesis (i.e., unrelated with the v-src gene) suggest that the tumor cells expressing the [H2O2CA + PGES] phenotype appear de novo possibly in relation with occasional damage of DNA of the transformed and tumor cells, in particular, caused by products of the CTA of MPh, NPh, and NK. Such damage can, in particular, result in the activation of the c-src gene (or other genes of the src family) and in the expression by rare variants of tumor cells of the [H2O2CA + PGES] phenotype which is specific for the v-src-transformants; the in vivo selective advantages of the tumor cells expressing the [H2O2CA + PGES] phenotype provide the conditions for survival, growth, and gradual replacement of the initial (non-expressing [H2O2CA + PGES] phenotype) cells by their descendants of this phenotype. Thus in vivo cytotoxic effectors of the host's innate immunity system seem to play the crucial role in the selection of tumor cells: under in vivo conditions, the appearance de novo and natural selection of the tumor cells expressing the [H2O2CA + PGES] phenotype seem to be preceded by activation of certain regulatory cell genes specific for the v-src-transformed cells. Such src-like changes in the tumor phenotype in vivo are likely to be one of the earliest (premetastatic) secondary transformations of tumor cells of various origin which are functionally required for their survival in vivo during natural selection and progression.
This work was supported by the State Priorities in Science and Technique program (grant Nos. 47, 46, 22), the Russian Foundation for Basic Research (grant Nos. 93-04-20708, 96-04-48054, 99-04-48358), and also by the Soros International Science Foundation (grant No. J3Z100). The author heartily thanks G. I. Abelev and A. D. Altstein for their valuable remarks and discussion of some results presented in this review and also N. A. Dyakova for her great help in preparation of the manuscript for press.
REFERENCES
1.Klein, G. (1975) in Immunology of
Tumor**--Host Relationship (Smith, R. T., and Landy, M.,
eds.) Academic Press Inc., N. Y., pp. 201-213.
2.Old, L. J. (1981) Cancer Res., 47,
261-275.
3.Alexander, P., and Eccles, S. (1984) in Cancer
Invasion and Metastasis: Biological and Therapeutic Aspects
(Nicolson, G. L., and Milas, L., eds.) Raven Press, N. Y., pp.
293-308.
4.Deichman, G. I. (1984) Advances in Science and
Technology. Oncology [in Russian], Vol. 13, VINITI, Moscow, pp.
46-97.
5.Deichman, G. I. (1988) Cancer Surveys,
7, 675-690.
6.Schrieber, H. (1993) in Fundamental
Immunology, 3rd ed. (Paul, W. E., ed.) Raven Press, N. Y., pp.
1143-1178.
7.Klein, E., and Mantovani, A. (1993) Curr. Opin.
Immunol., 5, 714-718.
8.Boon, T., Cerrottini, J.-C., van den Eynde, B., van
den Bruggen, P., and van Pel, A. (1994) Ann. Rev. Immunol.,
12, 337-366.
9.Weinberg, R. (1989) Cancer Res., 49,
3713-3721.
10.Janeway, C. A., and Travers, P. (1997)
Immunobiology, 3rd ed., Current Biology Ltd., Garland Publishing
Inc., N. Y.-London.
11.Medzhitov, R., and Janeway, C. A. (1997) Curr.
Opin. Immunol., 9, 4-9.
12.Bandelac, A., and Fearon, D. T. (1997) Curr.
Opin. Immunol., 9, 1-3.
13.Fearon, D. T., and Locksley, R. M. (1996)
Science, 272, 50-54.
14.Sniderman, R., and Pike, M. C. (1976)
Science, 192, 370-372.
15.Gorelik, E., Fogel, M., Segal, A., and Feldman,
M. (1979) J. Natl. Cancer Inst., 63, 1397-1404.
16.Gorelik, E., Feldman, M., and Segal, S. (1982)
Cancer Immunol. Immunother., 12, 105-109.
17.Gronberg, A., Kiessling, R., Ericksson, E., and
Hansson, M. (1981) J. Immunol., 127, 1734-1739.
18.Yamashina, K., Fulton, A., and Heppner, G. (1985)
J. Natl. Cancer Inst., 75, 765-770.
19.Patek, P. O., Lin, Y., Collins, J. L., and Cohn,
M. (1986) J. Immunol., 136, 741-745.
20.Nathan, C. F., Arrick, B. A., Murray, H. W., de
Santia, N. M., and Cohn, Z. A. (1981) J. Exp. Med., 153,
766-782.
21.Adams, D. O., and Nathan, C. F. (1983)
Immunol. Today, 4, 166-169.
22.Droller, M., Schneider, M., and Perlman, P.
(1979) Cell Immunol., 39, 165-177.
23.Brunda, M. J., Herberman, R. B., and Holden, H.
T. (1980) J. Immunol., 124, 2682-2687.
24.Fulton, A. M. (1984) Int. J. Cancer,
33, 375-379.
25.Young, R., and Knies, S. (1984) J. Natl.
Cancer Inst., 72, 919-922.
26.Deichman, G. I., and Vendrov, E. L. (1986)
Int. J. Cancer, 37, 401-409.
27.Deichman, G. I., Kluchareva, T. E., Matveeva, V.
A., Kushlinsky, N. E., Bassalyk, L. S., and Vendrov, E. L. (1989)
Int. J. Cancer, 44, 904-907.
28.Deichman, G. I., Kashkina, L. M., Mizenina, O.
A., Gorojanskaya, E. G., Nikiforov, M. A., Gudkov, A. V., Dyakova, N.
A., Komelkov, A. V., Prilutskaya, M. O., Kushlinsky, N. E., and
Tatosyan, A. G. (1996) Int. J. Cancer, 66, 747-752.
29.Deichman, G. I., Matveeva, V. A., Kashkina, L.
M., Dyakova, N. A., Uvarova, E. N., Nikiforov, M. A., and Gudkov, A. V.
(1998) Int. J. Cancer, 75, 277-283.
30.Kluchareva, T. E., Matveeva, V. A., and
Kushlinsky, N. E. (1992) Immunol. Lett., 33, 239-246.
31.Kluchareva, T. E., and Matveeva, V. A. (1983)
Byull. Eksp. Biol. Med., 10, 86-89.
32.Kluchareva, T. E., and Matveeva, V. A. (1985)
Byull. Eksp. Biol. Med., 6, 732-734.
33.Kluchareva, T. E., Matveeva, V. A., Bassalyk, L.
S., and Kushlinsky, N. E. (1988) Byull. Eksp. Biol. Med.,
2, 204-206.
34.Matveeva, V. A., Kluchareva, T. E., and Uvarova,
E. N. (1993) Byull. Eksp. Biol. Med., 2, 193-194.
35.Kluchareva, T. E., Matveeva, V. A., Kashkina, L.
M., Kushlinsky, N. F., Bassalyk, L. S., and Prilutskaya, M. O. (1989)
Eksp. Onkol., 11, 53-56.
36.Lavnikova, N. A., and Burdelya, L. G. (1990)
Byull. Eksp. Biol. Med., 109, 954-956.
37.Burdelya, L. G. (1997) Neoplasma,
44, 31-35.
38.Kluchareva, T. E., Matveeva, V. A., and Uvarova,
E. N. (1990) Byull. Eksp. Biol. Med., 9, 308-310.
39.Deichman, G. I., Kluchareva, T. E., Kashkina, L.
M., and Matveeva, V. A. (1979) Int. J. Cancer, 23,
571-584.
40.Hahne, M., Rimoldi, D., Schröter, M.,
Romero, P., Schreier, M., French, L. E., Schneider, P., Bornand, T.,
Fontana, A., Lienard, D., Cerottini, J.-C., and Tschopp, J. (1996)
Science, 274, 1363-1366.
41.Walker, P. R., Saas, P., and Dietrich, P.-Y.
(1997) J. Immun., 158, 4521-4524.
42.Von Reyher, U., Strater, J., Kittstein, W.,
Gschwendt, M., Krammer, P. H., and Moller, P. (1998) Cancer
Res., 58, 526-534.
43.O'Connel, J., Bennet, M. W., O'Sullivan, G. C.,
Collins, J. K., and Shanahan, F. (1999) Nature Med., 5,
267-268.
44.Volpe, E. A. (1992) J. Exp. Clin. Cancer
Res., 11, 109-122.
45.Svoboda, J. (1964) Natl. Cancer Inst.
Monograph, 17, 277-298.
46.Alström, C. G. (1964) Natl. Cancer Inst.
Monograph, 17, 299-317.
47.Obukh, J. B., Kryukova, I. N., Biryulina, T. I.,
and Kuznetzova, N. N. (1969) Int. J. Cancer, 4,
799-808.
48.Varmus, H. (1984) Ann. Rev. Genet.,
18, 553-612.
49.Deichman, G. I., Kashleva, E. A., Kluchareva, T.
E., and Matveeva, V. A. (1989) Int. J. Cancer, 44,
908-910.
50.Kashleva, E. A., Matveeva, V. A., Uvarova, E. N.,
and Kluchareva, T. E. (1990) Eksp. Onkol., 12, 32-34.
51.Sjolander, A., Yamamoto, K., Huber, B. E., and
Larenha, E. G. (1991) Proc. Natl. Acad. Sci. USA, 88,
7908-7912.
52.Fukui, Y., and Hanafusa, H. (1991) Mol. Cell
Biol., 11, 1972-1979.
53.Topol, L. Z., Kisseljova, N. P., Gutierres, M.
L., Deichman, G. I., Musatkina, E. A., Shtutman, M. S., Zakamaldina, T.
Z., Blair, D. G., and Tatosyan, A. G. (1993) Mol.
Carcinogenesis, 8, 167-176.
54.Deichman, G. I., Topol, L. Z., Kluchareva, T. E.,
Matveeva, V. A., Zakamaldina, T. A., Uvarova, E. N., and Tatosyan, A.
G. (1992) Int. J. Cancer, 51, 903-908.
55.Yu, C.-L., Tsai, M. N., and Stacey, D. W. (1988)
Cell, 52, 63-71.
56.Hamilton, T. A., and Adams, D. O. (1987)
Immunol. Today, 3, 151-158.
57.Brown, M. T., and Cooper, J. A. (1996)
Biochim. Biophys. Acta, 1287, 121-149.
58.Hirai, H., and Varmus, H. E. (1990) Mol. Cell.
Biol., 10, 1307-1318.
59.Koch, C. A., Anderson, D., Moran, M. F., Ellis,
C., and Powson, T. (1991) Science, 252, 668-674.
60.Ralston, R., and Bishop, J. M. (1985) Proc.
Natl. Acad. Sci. USA, 82, 7845-7849.
61.Mukhopadhyay, D., Tsiokas, L., Zhou, X. M.,
Foster, D., Brugge, J. S., and Sukhatme, V. P. (1995) Nature,
375, 577-581.
62.Kanner, S. B., Parsons, S. J., Parsons, J. T.,
and Gilmer, T. M. (1988) Oncogene, 2, 327-335.
63.Littrell, D. K., Lee, A., Lansing, T. J., Crosby,
R. M., Jung, K. D., Willard, D., Luther, M., Rodrigues, M., Berman, J.,
and Gilmer, T. M. (1994) Proc. Natl. Acad. Sci. USA, 91,
83-87.
64.Irby, R. B., Weiguang Mao, Coppola, D., Kang, J.,
Laubeau, J. M., Trudeau, W., Karl, R., Fujita, D. J., Jove, R., and
Yeatman, T. J. (1999) Nature Gen., 21, 187-190.
65.Nowell, P. S. (1986) Science, 194,
23-28.
66.Deichman, G. I., and Kluchareva, T. E. (1964)
Nature (Lond.), 4937, 1126-1128.
67.Deichman, G. I., and Kluchareva, T. E. (1964)
Virology, 24, 131-137.
68.Deichman, G. I. (1969) Adv. Cancer Res.,
12, 101-136.
69.Deichman, G. I., Dyakova, N. A., Kashkina, L. M.,
Matveeva, V. A., and Uvarova, E. N. (1999) Immunol. Lett.,
70/1, 37-42.
70.Kedar, E., and Klein, E. (1992) Adv. Cancer
Res., 59, 245-294.
71.Wick, M., Dubey, P., Koeppen, H., Siegel, C. T.,
Fields, P. E., Chen, L., Bluestone, J. A., and Schreiber, H. (1997)
J. Exp. Med., 186, 229-238.