REVIEW: Cellular Mechanisms of Multidrug Resistance of Tumor Cells
A. A. Stavrovskaya
Institute of Carcinogenesis, Blokhin Russian Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, Moscow, 115478 Russia; fax: (095) 324-1205; E-mail: Alla@Astavrovskaya.home.bio.msu.ru
Received September 17, 1999
Multidrug resistance (MDR) is the protection of a tumor cell population against numerous drugs differing in chemical structure and mechanisms of influence on the cells. MDR is one of the major causes of failures of chemotherapy of human malignancies. Recent studies show that the molecular mechanisms of MDR are numerous. Cellular drug resistance is mediated by different mechanisms operating at different steps of the cytotoxic action of the drug from a decrease of drug accumulation in the cell to the abrogation of apoptosis induced by the chemical substance. Often several different mechanisms are switched on in the cells, but usually one major mechanism is operating. The most investigated mechanisms with known clinical significance are: a) activation of transmembrane proteins effluxing different chemical substances from the cells (P-glycoprotein is the most known efflux pump); b) activation of the enzymes of the glutathione detoxification system; c) alterations of the genes and the proteins involved into the control of apoptosis (especially p53 and Bcl-2).
KEY WORDS: multidrug resistance, mechanisms of cell defense, P-glycoprotein, glutathione S-transferases, apoptosis, p53, Bcl-2
Abbreviations: MDR) multidrug resistance; Pgp) P-glycoprotein; GSH) glutathione; GST) glutathione S-transferases.
Studiesofthe resistance of tumor cells to cytotoxic drugs are necessary
for understanding the mechanisms of the cells' defense against injury.
The investigation of the insensitivity of malignant cells to
chemotherapy is needed also for oncology practice because drug
resistance is often considered to be a cause of tumor therapy failures.
Ineffectiveness of the therapy may be provoked by many other causes
besides tumor cell alterations; it may be connected with peculiarities
of the pharmacokinetics of a drug, i.e., the inability of a drug to
reach the cell in adequate amounts and active form. However, in this
paper only the mechanisms of drug resistance of cells will be
described; the problems of drug pharmacokinetics will not be
considered.
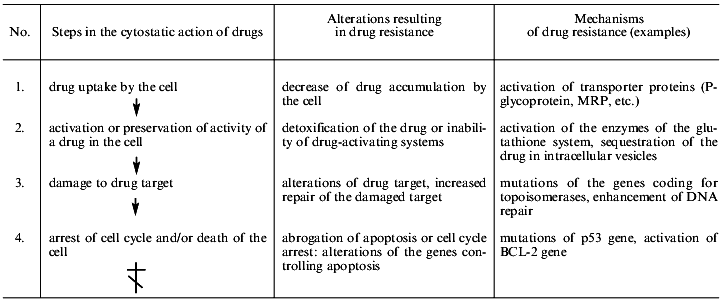
Table 1 shows the main steps of cell injury by a drug from the drug uptake by the cell to programmed death (apoptosis). Most chemotherapeutic drugs induce apoptosis [1]. The simplified list of cell alterations resulting in cell death is indicative of the ability of cells to disrupt the pathway of injury at any step. This list demonstrates the diversity of the mechanisms of cellular resistance to drugs.
Table 1. Main mechanisms of drug resistance
of tumor cells
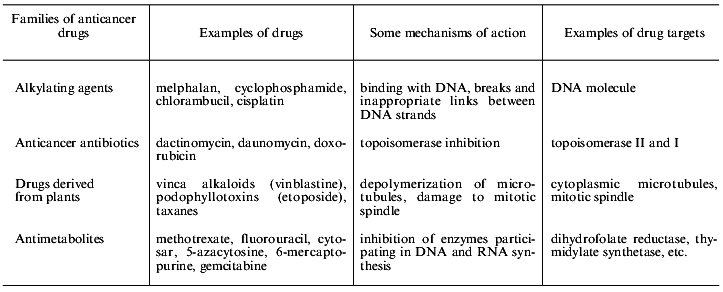
Contemporary chemotherapy uses combinations of various drugs hitting different targets [2]. Examples of some drugs and their targets are shown in Table 2. It is evident that very different drugs are damaging various targets in the cell. Thus cellular multidrug resistance (MDR) is a problem of the great importance for contemporary combined chemotherapy because of the usage of drug combinations and multiplicity of drug targets in the cell. MDR is cell resistance not to single substance but to numerous substances characterized by different mechanisms of action and by different chemical structure. MDR was first found in cell culture experiments [3]. Treatment of cultured cells by a single drug resulted in the establishment of a cell population simultaneously resistant to many other compounds (cross-resistance).
Table 2. Main groups of anticancer drugs
Naturally, MDR is not necessarily the result only of the treatment of the cells by a drug. Often it is connected with the type of cell differentiation or the localization of cells in an organism. This is intrinsic MDR (cells are drug resistant before the drug treatment). For example, brain tumors are resistant to chemotherapy due to the blood--brain barrier. Intrinsic MDR may be connected with a genetic change that initiated the tumor. For example, chromosome translocation 9;22 results in the occurrence of fused BCR/ABL protein and causes chronic myeloid leukemia; this translocation can also abolish apoptosis and so cause MDR [4]. Other examples of this type of MDR are given in the discussion of the role of p53 in MDR (section 4 of this paper). All cells of a tumor with intrinsic MDR are drug resistant.
Acquired drug resistance can arise from chemotherapy. Rare genetic variants of drug resistant cells can occur in a tumor cell population under the influence of cytostatic drug, and these cells can multiply if they have selective advantage. Selective advantage may be the result both of drug resistance and of other cell traits: acceleration of the proliferation rate, alterations of cell sensitivity to growth factors, etc. These factors can influence cell selection by the drugs. Adaptive changes are very interesting; they lead to the drug resistance of the majority of the cell population because of temporary activation of cell defense mechanisms. These alterations and their connections with subsequent drug resistance are not studied well. In the next few paragraphs we will describe the main mechanisms of drug resistance.
1. MAIN MECHANISMS OF DECREASE OF DRUG ACCUMULATION BY
CELLS
A priori decrease of a drug accumulation by cells may result both from decrease of drug influx and increase of drug efflux from the cells. Since most chemotherapeutic drugs enter cells by passive diffusion through the plasma membrane, cell changes in drug influx can be connected with changes in the cell membrane structure. Indeed, both electron microscopy and analysis of the lipids of the membranes of MDR cells revealed differences between some drug sensitive and drug resistant cells [5]. The modifications found could cause either changes in drug traffic through the cell membrane or an influence on signaling pathways controlling apoptosis. However, data concerning alterations of drug uptake by cells are scarce, so another mechanism (drug efflux from the resistant cell) is considered to be the main mechanism of the decreased drug accumulation in cells.
Drug efflux from cells is mediated by the activity of transporter proteins, i.e., P-glycoprotein (Pgp), transporters of the MRP family, and some other proteins (Table 3). Pgp has been studied for comparatively long time (25 years) and many facts are gathered in this field. Thus it is possible to consider Pgp mediated MDR (Pgp-MDR) as a tool for the analysis of problems which are not resolved in other types of MDR. So we will discuss Pgp in more detail.
Table 3. Examples of multidrug transporters
(proteins and genes coding the transporters)

1.1. Multidrug Resistance Mediated by P-Glycoprotein (Pgp)
Pgp: general characteristics and functional activity. Pgp functional activity mediates cellular drug resistance to diverse antitumor drugs (to anthracycline antibiotics, plant alkaloids, especially to vinca alkaloids, to podophyllotoxins, taxanes, etc.) and to numerous other substances--fluorescent dyes, ethidium bromide, puromycin, gramicidin D, etc. There is a saying that Pgp and MRP proteins (see further) cause cellular drug resistance to the half of the substances in the Sigma catalogue.
Pgp is a large transmembrane glycoprotein with molecular weight approximately 170 kD consisting of two similar halves each including six hydrophobic transmembrane segments (Fig. 1). This suggests that the Pgp molecule spans the membrane twelve times. The Pgp molecule has two ATP-binding domains (Fig. 1) showing that Pgp function is energy-dependent. One of most popular hypotheses proposes that the drug molecule binds to a specific site of Pgp within the lipid bilayer of the cell plasma membrane and by means of the energy of ATP hydrolysis is transported out of the cell. Indeed, chemotherapeutic drugs were shown to be bound by the membranes of drug resistant cells better than by the membranes of sensitive cells; experiments showed also that the drugs in a gel are bound by a protein with the molecular weight of 170 kD [6]. Labeled drugs, for example vinblastine, and fluorescent dyes (they are also Pgp substrates) were excluded with the greater speed from Pgp overexpressing drug resistant cells [6]. Data obtained recently suggest that Pgp may function as flipase which moves bound substances to the external membrane layer or outside the cell [7].
There are various techniques for determination of Pgp function as an efflux pump. Pgp activity may be evaluated by the rate of uptake by cells of a labeled drug (for example, [3H]vinblastine) or fluorescent drugs (daunorubicine) and also by the extent of uptake and the rate of efflux of florescent dyes, Pgp substrates (rhodamine-123, etc.) by cells [8, 9]. In these experiments modulators of Pgp activity are used (preferentially the most specific ones--PSC833 or verapamil). The accumulation of every drug that is a Pgp substrate is decreased in the cells characterized by increased Pgp activity. This substance effluxes with greater speed from the cells with Pgp-MDR, a modulator of Pgp activity inhibiting drug efflux [10].Fig. 1. Proposed structure ofthe multidrug resistance protein P-glycoprotein (Pgp): 1) transmembrane domains (indicated by rectangles); 2) ATP-binding domains (framed by oval) (after [11]).
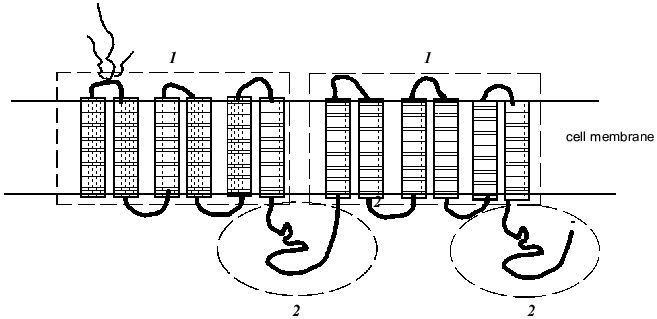
Pgp belongs to the superfamily of ABC (ATP-Binding Cassette) transporters. More than a hundred transport proteins are included in the ABC family; they have been found in different species ranging from E. coli to humans [11]. The proteins of this family transport very different substrates ranging from inorganic ions to polysaccharides and proteins. These diverse transporters are united by a common domain organization--the presence of transmembrane and ATP-binding domains, which also are named ABC domains (Fig. 1) [11]. ABC domains of prokaryotic and eukaryotic proteins of the family share 30-40% sequence homology (irrespective of their substrate specificity) [11]. Experiments with the transfection of a gene of bacterial transporter LmrA into human cells showed the high conservatism of the proteins of this family [12]. The LmrA protein determines resistance of the bacterium Lactococcus lactis to antibiotics. In human lung cells transformed by the LmrA gene a polypeptide of molecular weight 66 kD was found in the cell membrane. Transfected cells had resistance to the drugs of the Pgp-MDR group: anthracyclines, vinca alkaloids, ethidium bromide, rhodamine. Inhibitors of Pgp activity removed this drug resistance [12]. Thus these experiments have shown that bacterial transporter can carry out a role inherent in Pgp in human cells.
There is an opinion that the binding sites of the different substances on a molecule of the protein differ. Mutational analysis of the Pgp coding gene suggested that Pgp directly interacts with its substrates. The binding sites of the protein substrates are obviously located in transmembrane domains or directly under the membrane [13]. Until recently the chemical nature of wide substrate specificity of Pgp and a number of other ABC transporters remained mysterious. Their ability to transport substances of a different chemical nature and mechanisms of action greatly differs from the strict specificity of the majority of known ligand-binding proteins. A. A. Neyfakh and co-authors have shown by means of crystallography that the unfolding of an alpha-helix (the ligand-binding domain) is needed for substrate binding by the bacterial protein BmrR. Then an internal drug-binding pocket with a negatively charged glutamate become exposed and binds positively charged ligands with high affinity [14]. It is possible that the functioning of Pgp is carried out on same way. BmrR, a transcription regulator in Bacillus subtilis, activates the Bmr transporter in response to the binding with BmrR of various inducers including some Pgp substrates [14].
Gene MDR1 coding P-glycoprotein, genetic mechanisms of Pgp-MDR. Human Pgp is coded by gene MDR1 that is a member of the MDR family. The gene is located on chromosome 7 (7q21.1). The MDR family includes two genes in man (MDR1 and MDR2) and three in rodents (mdr1, 2, 3). By means of gene transfer it was shown that only one human gene (MDR1) and two rodent genes cause MDR. Mutations in some sites of the MDR1 genecan result in the changes in the cross-resistance pattern, i.e., in the binding of certain substrates by the protein. The introduction into cells of the MDR2 gene did not result in drug resistance [15]. The product of the MDR2 gene (mouse mdr2) is present at high amount on the surface of cells of bile canaliculi, functions as a flipase, and transports phosphatidylcholine into the bile [16].
MDR can occur both due to alteration of MDR1 gene expression and to increase in the dose of a gene--amplification of a fragment of the genome containing the MDR1 gene and five or six genes linked to it [17]. Amplification is found usually in cultured cell lines with high levels of Pgp-MDR, but not in a samples from patients. MDR can also result from stabilization of MDR1 mRNA, regulation at the level of synthesis, and alterations of protein processing [13]. There is a possibility also that Pgp activity may be influenced directly by its substrates [13].
The MDR1 gene has two promoter regions, the lower and upper promoters. In tissues and cultured cells the expression of the lower promoter is usually found. Thus this promoter is considered as a basic element regulating the activity of this gene [18]. Transcription of the MDR1 gene is increased due to very different influences, for example, effects of chemotherapeutics including those which are not Pgp substrates [19]. It is obvious, however, that inducibility of a gene MDR1 depends on a cell's context: diverse inducers in different ways influence the activity of this gene in different cells. Rather differing inducers can cause Pgp-MDR. For example, irradiation of the cultured Chinese hamster cells (the line CHO) resulted in cell resistance to vincristine, probably connected with translational or posttranslational Pgp modifications [20]. Chemical carcinogens induce the activity of a mdr1 gene in the cells of rat liver; mdr1 activity increases after partial hepatectomy in a regenerating rat liver [21]. Some cytokines can induce Pgp-MDR in some types of the cultured cells. Heat shock and inducers of cell differentiation were found to influence the activity of MDR1/Pgp [13]. It is noteworthy, however, that most of these effects were found only in particular cell lines; they were strictly dependent on species and tissue origin of the cells. Thus, it is evident that the increaseof MDR1/Pgp activity resulting from the action of different inducers is dependent on the particular conditions (evidently on the activation of certain signaling pathways in the cell). It is evident also that this system is easily activated in different situations (not only under the influence of stress).
Studies of genes and signaling pathways involved in the regulation of Pgp activity showed that these pathways (genes) are numerous (or at least there are several pathways). For example, experiments using transient transfection of the MDR1 promoter region (linked to a reporter gene) into the cells as well as stable transfection of some other genes showed that genes p53, ras, raf, RARalpha and RARbeta (receptors of retinoic acid) can influence the activity of introduced MDR1 promoter or the expression of the endogenous cellular MDR1 [22-24]. Genes c-fos and c-jun also were shown to influence MDR1 activity (the responsive elements for these transfactors were found in the MDR1 promoter) [13, 25]. Protein kinase C, protein kinase A, and, probably, some other protein kinases were shown to take part in the regulation of Pgp activity: the results of experiments using inhibitors of protein kinases support this statement [13, 26]. The evidence for multiple regulators of MDR1/Pgp activity supports the notion of the great importance of this defense system for the life of the cell (the regulation is duplicated many times for cell safety).
Expression of Pgp in normal tissues and cells. Pgp is expressed in different tissues differently--from a very low, indefinable level of expression (for example, in epidermal cells or pneumocytes) up to high levels (for example, in cells of the cortex of adrenal gland, mucosal cells of the colon) [27]. The degree of MDR1 expression in these tissues greatly differs (Table 4). Immunochemical technique revealed intensive and comparatively homogeneous staining of Pgp expressing tissues. This is true for the adrenal cortex, epithelial cells lining cavities (colon mucosal cells, cells of kidney tubular epithelium, placental trophoblasts) and also for endothelial cells of the brain and testicular capillaries. The different degree of the Pgp expression was also revealed in the blood cells. Stem cells and early progenitors in normal bone marrow (cells expressing CD34 antigen) also express functionally active Pgp [28]. Then both Pgp expression and its functional activity are gradually reduced in the process of myeloid cell differentiation [29]. It is clear that malignancies occurring from the Pgp-expressing cells will have an intrinsic MDR, while the tumors arising from Pgp non-expressing cells will not have this type of drug resistance. It is clear also, that normal lymphocytes can sometimes determine the activity of Pgp in the samples of peripheral blood of a patient.
Table 4. Expression of the MDR1 gene
in normal human tissues (after [27])

The tissue specificity of Pgp expression raises a question of its physiological function in the organism. A study of mice with MDR1 gene knockout show that the main function of this protein is the defense of an organism from a toxic injury [30]. Pgp carries out “barrier” functions, protecting cells from harmful substances contained, for example, in the cavity of the colon. There is a supposition that Pgp supports the necessary level of steroid hormones in the adrenals. The pattern of Pgp and MRP expression in the choroid plexus of the brain suggests a role of these proteins in the blood--brain barrier [31]. The high inducibility of MDR1/Pgp is probably connected with physiological functions of Pgp in the organism.
Significance of Pgp-mediated MDR (Pgp-MDR) for the therapy of malignancies. In some malignant diseases expression of active Pgp (or overexpression of the MDR1 gene) is often found at diagnosis of the malignancy. Usually it is connected with the type of the cells from which the tumor arises. Thus this is an intrinsic MDR connected with the tissue origin of tumor cells. Untreated carcinomas of the colon, kidney, hepatomas, adrenal tumors, pheochromocytomas, and some other malignancies demonstrate high levels of MDR1 gene expression [32]. Some hematological malignancies--acute myeloid leukemia, chronic lymphatic leukemia, adult T-cell lymphomas--are also usually characterized by the MDR connected with the type of the cells from which a malignancy arose [32]. The investigation of the cell differentiation pattern in the material from the patients with some leukemias (for example, the cells from patients in blast crisis of chronic myeloid leukemia) shows that some cases of MDR can be connected with the proliferation of the cells of certain type of differentiation (with intrinsic Pgp expression), although this type of tumor usually originates from the Pgp-non-expressing cells [33]. Thus, in some cases Pgp may be a marker of the certain type of cell differentiation.
The increase in the number of cases with Pgp overexpression after tumor therapy is often used as a reason in favor of the role of acquired MDR in tumor therapy failure. The correlation between MDR1/Pgp overexpression and ineffective treatment is also employed as this reason. The results suggesting the importance of Pgp-MDR for outcome of tumor treatment were obtained in experiments of this kind for lung and ovarian adenocarcinomas, breast cancers, and sarcomas (including osteosarcomas) [32]. The significance of Pgp-MDR has been most thoroughly documented in hematological malignancies. Pgp-positivity was revealed in 30-50% of acute myeloid leukemia cases (data of different studies), and this protein was more often found after courses of chemotherapy in the patients resistant to the treatment [34]. Multiple myeloma is a good example of a disease where Pgp-MDR results from the treatment. While at diagnosis of the disease approximately 6% of the patients are found to be Pgp-positive, after therapy (especially treatment using the VAD scheme--vincristine, doxorubicin, dexamethasone) up to 85% resistant to treatment patients become Pgp-expressing [34].
In acute myeloid leukemia MDR1 overexpression is often associated with the phenotype of immature cells--expression of CD34 antigen [34]. A positive correlation between Pgp and CD34 expression was found also in myelodysplasias. Thus, in the most cases of acute myeloblastic leukemia and myelodysplastic syndrome Pgp-MDR is connected with the proliferation of the early hematopoietic progenitors. The same is true for another myeloproliferative disorder: in blast crisis of chronic myeloid leukemia we have found the correlation of the expression of functionally active Pgp and the expression of the antigens of early myeloid differentiation--CD34 and CD13 [33]. Pgp detection in patients with acute myeloid leukemia (especially in association with CD34) means poor prognosis for treatment outcome. However, it is not clear whether all the cases of the disease progression are due to Pgp activity. It is necessary to do repeated examinations during the evolution of the disease in order to reveal the significance of Pgp for the therapy failure. These data are scarce. Our studies of chronic myeloid leukemia show that among the patients Pgp expressing cells disappear under therapy using drugs that are Pgp substrates (instead of multiplying under these conditions) [33]. Thus, in these cases expression of the active Pgp most likely does not produce cell viability under therapy pressure. However, we have found that detection of Pgp on blast cells in peripheral blood of a patient at the blast crisis diagnosis seems to predict more rapid progression of the disease [33]. All these data show that connections between the expression of MDR proteins and the outcome of a tumor therapy are not very simple. Our results may be explained by a combination of different MDR mechanisms in the cells, while Pgp serves as a marker of the activation of several systems defending cells against cytostatic drugs.
Although caution is needed for the comparison of the results of various studies, it is possible to state that in a part of the cases of malignant diseases Pgp plays a role in the reaction of the patients to therapy: sometimes Pgp expression can serve as a sign of a poor prognosis of the disease; however, even in these cases Pgp not always determines by itself the resistance of the patients to the treatment.
Reversion of Pgp-MDR. The clinical significance of Pgp called the attention of many researchers and pharmaceutical companies to the examination of ways to inhibit Pgp activity. Many types of compounds were found to overcome Pgp-MDR in experiments: calcium channel blockers (e.g., verapamil, nifeldipin), hypotensive drugs (reserpine), antibiotics (cephalosporins, gramicidin, puromycin) immunosuppressors (cyclosporin A and its derivatives), and many other lipophilic compounds [35]. All of these diverse compounds are hydrophobic and have a low-molecular-weight aromatic ring in the molecule. Some of these compounds (for example, verapamil) bind to the Pgp molecule and inhibit in a competitive manner the binding or transport of drugs that are Pgp substrates. These numerous Pgp inhibitors were found in experiments with cultured cells selected for Pgp-MDR. The experiments show that the problem of Pgp reversal is complex: for example, in cells with 200-fold resistance to vinblastine, verapamil induced twofold decrease of vinblastine resistance, but cross resistance to doxorubicin remained unchanged [36]. Clinical testing showed that it is difficult to obtain the necessary concentration in the patient's blood for most of the Pgp-MDR modifiers because of their toxicity and grave side effects [35]. Less toxic and more effective Pgp inhibitors were developed, among them cyclosporin derivative PSC-833 and verapamil R, a variant of verapamil. However, these substances also have no clinical success, probably due to the fact that MDR is caused by the activity of several proteins (not only by Pgp activation). In recent years anti-Pgp monoclonal antibodies were suggested as modifiers of Pgp-MDR; however, they are still under clinical testing.
1.2. Multidrug Resistance Mediated by MRP Protein
MRP (Multidrug Resistance-associated Protein) was discovered in 1992, thus the results elucidating its role in the evolution of malignant tumors are not so numerous as the data obtained in the studies of Pgp. This protein, with molecular weight ~190 kD contributes resistance to tumor cells to almost the same antitumor drugs as Pgp. It also functions as an energy-dependent (ATP-dependent) pump excluding toxic substances from the cell. MRP (like Pgp) belongs to the family of ABC transporters [37]. The role of MRP in drug resistance is shown by means of the introduction of the MRP gene into cells [38]. Functional activities of MRP and Pgp transporters are different: for its function as a drug effluxing pump, MRP needs cellular glutathione. The protein is one of the transporters of glutathione conjugates (so-called GS-X pumps) [39, 40]. Recently several genes and proteins of the MRP family were found in very different species--from man to bacteria [40]. One of them is the cMOAT protein (MRP2); it is also a GS-X pump specifically located on the apical surface of hepatocytes lining the cavities of the bile canaliculi [40]. Rats lacking this protein are unable to secrete bilirubin glucuronides into the bile [40]. The role of cMOAT in MDR needs to be determined. It has been shown, however, that this protein can transport vinblastine, which is a classical Pgp and MRP substrate.
MRP determines the resistance of cells almost to the same substances as Pgp; however, the patterns of cross-resistance have some differences. MRP-expressing cells usually have lower cross-resistance to taxol than Pgp-MDR cells [39]. In normal (drug sensitive) cells MRP proteins are apparently transporting glutathione conjugates, which are important both for the normal life of the cell and play a role in inflammation. MRP expression is increased in the bronchial epithelium, its overexpression possibly being involved in the pathogenesis of bronchial asthma [37].
MRP has been found to be expressed at a low basal level in many tissues including all the peripheral blood cells (regardless of cell lineage) [37]. Blast cells of peripheral blood from patients with acute myeloid leukemia also demonstrated a similar low level of expression [41]. Some new results show that in drug resistant cases of acute myeloid leukemia MRP expression is higher than in the cases where complete remission is obtained. More cases in which cells expressed higher MRP levels were found in chronic lymphoid leukemia. There are few studies of the determination of MRP functional activity, but the necessary assay is already established. The application of this assay together with the techniques evaluating protein expression by the cells will give in the future more definite results.
Although the clinical significance of MRP is not yet clear, the search for inhibitors of the activity of this MDR protein is active. It was already found that effective inhibitors for MRP and Pgp are different, though some Pgp modifiers can in part inhibit MRP functional activity. Genistein was shown to inhibit daunorubicine uptake in cells with MRP overexpression but not in Pgp-expressing cells. Genistein is very toxic and cannot be used for the therapy of patients; however, it is used in assays determining MRP functional activity. Inhibitors of glutathione synthesis and protein kinase C inhibitors are also used [42].
1.3. Multidrug Resistance Mediated by LRP Protein
The MDR protein LRP (Lung-Resistance-related Protein, molecular weight 110 kD, discovered in 1993) is found not on the cell membrane (like Pgp and MRP), but in the cytoplasm. It is expressed by the cells of normal epithelium and cells of tissues which are exposed to toxic substances. LRP is a major protein of ribonucleoprotein cell particles named "vaults" [43]. The function of these organelles is not yet known, but it was suggested that they can participate in the transport of a substrates from the nucleus to the cytoplasm. In the cell LRP is often associated with vesicles and lysosomes; this fact connects its function with drug transport and drug sequestration into vesicles. Probably, after this a drug can be excluded from the cell by exocytosis [44]. There are data suggesting that LRP may cause MDR in patients with ovarian cancer and acute myeloid leukemia [45, 46].
2. DRUG RESISTANCE MEDIATED BY DETOXIFICATION OF THE DRUG IN THE
CELL
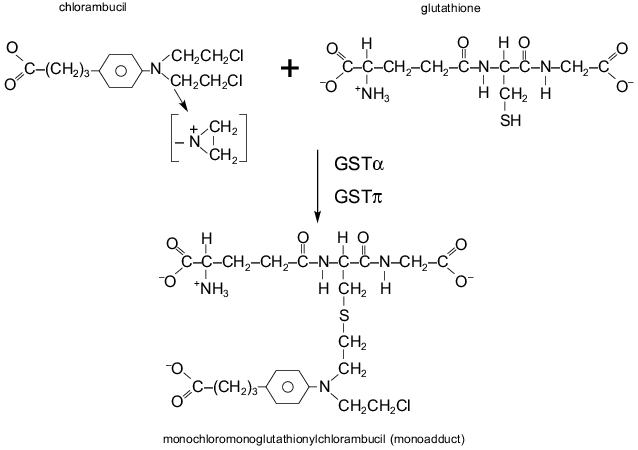
The cellular glutathione (GSH) system is a critical component of detoxification of cytostatics in the cell. Glutathione, a non-protein thiol, can interact via its thiol with the reactive site of a drug, resulting in conjugation of the drug with glutathione (Fig. 2). The conjugate is less active and more water-soluble, and it is excluded from the cell with the participation of transporter proteins named GS-X (including MRP). Increased levels of glutathione were found in cell lines resistant to alkylating agents (e.g., nitrogen mustard, chlorambucil, melphalan, cyclophosphamide, BCNU) [47]. The enzymes glutathione S-transferases (GST) catalyze the interactions between glutathione and alkylating drugs, increasing the rate of a drug detoxification (Fig. 2). So activation of these enzymes can cause cellular drug resistance [47]. Numerous studies have been dedicated to these enzymes. Enzymes which catalyze glutathione synthesis also could mediate drug resistance; however, their role in this phenomenon is not yet clear. Resistance of tumor cells to drugs of Pgp-MDR group (anthracyclines, vincristine) can also be connected with alterations of the GSH system [47, 48]. In cells with Pgp-MDR, increased levels of a GST isoenzyme, GSTpi, is frequently found [47]. It is probable that genes of several defense systems are coordinately regulated in these cells.
Introduction into mammalian cells of the genes of various GSTs has produced some lines with modest levels of resistance to alkylating agents [47]. Although two-three-fold resistance to alkylating agents may not have clinical significance, these data are important: it is probable that several defense system are activated simultaneously in the cell and this can give ground for further selection of MDR cells. Transfection of human GSTs genes into the cells of the yeast Saccharomyces cerevisiae conferred significant levels of resistance to adriamycin and chlorambucil [49]. These experiments, like the experiments with Pgp, show that this cell defense system is very old.Fig. 2. Reaction of the binding of alkylating drug (chlorambucil) with the glutathione molecule and conjugate (monoadduct) formation. The SH group of glutathione binds the reactive group of chlorambucil. GSTalpha and GSTpi are some glutathione S-transferases catalyzing the reaction.
Inhibitors of the activity of glutathione S-transferases are considered as most potent modifiers of this type of drug resistance. Thus ethacrynic acid (usually used as diuretic) is now under clinical trial as a modulator of GSH-mediated drug resistance [50]. BSO (buthioninesulfoximine) decreases intracellular levels of glutathione and thereby overcomes resistance to alkylating agents [47]. BSO is used as a modifier of MRP-mediated MDR in experiments with cell cultures [38]. GSH is implicated in the results of the treatment of lymphomas. Increased GST activity has been observed in patients with chlorambucil-resistant chronic lymphocytic leukemia [51]. There was a supposition that GST expression may have prognostic value at early cases of breast cancer [52, 53]. It must be noted, however, that like in the Pgp studies, drug resistance connected with the alterations of the GSH system cannot completely explain drug resistance of cancer patients to a treatment. Moreover, there is much less reason for the benefit of the clinical importance of this type of MDR than for the Pgp-MDR, although the number of studies is large.
3. DRUG RESISTANCE MEDIATED BY ALTERATIONS OF DRUG TARGETS OR BY
ENHANCEMENT OF TARGET REPAIR
Some anticancer drugs are inhibitors of topoisomerases (topoisomerase I, more often topoisomerase II). These drugs stabilize the DNA--topoisomerase complex which in normal circumstances is easily decomposed. In cell lines selected for the resistance to topoisomerase II-inhibiting drugs the activity or the quantity of this enzyme are reduced [54]. Mutations in topoisomerase II gene were found; presumably they are the cause of some cases of this drug resistance [54]. This type of drug resistance arises to adriamycin, daunorubicine, mitoxantrone, etoposide, etc. Thus, the majority of the substances interacting with topoisomerase II are also substrates of Pgp and MRP. The clinical significance of this type of MDR is not yet clear. Experimental studies showed that the combination of a drug and Pgp modifier can produce MDR mediated by topoisomerase II alterations. It is evident that this type of drug resistance sometimes occurs in cells in association with other drug resistance mechanisms.
Another probable way of drug target alteration is an increase in the quantity of target protein in the cell. For example, in a number of cultured cell lines resistant to methotrexate increased levels of dihydrofolate reductase (DHFR, target protein of methotrexate) was found. This increase was due to the amplification of the DHFR gene. However, this type of drug resistance is not multiple drug resistance.
Enhanced DNA repair is probably implicated in drug resistance to the drugs interacting with DNA, for example, to nitrosomethylurea or platinum derivatives. Resistance to BCNU (carmustine) is correlated with the expression of chloro-O-6-alkylguanine-DNA-alkyltransferase. Changes in the quantities of proteins recognizing and repairing DNA injury (ERCC1, ERCC2, and ERCC3/XPB) were found in cultured cells with altered sensitivity to platinum complexes [55]. Some of these cells are resistant to several drugs; however, the number of drugs to which cells are cross-resistant is not as large as in cells with Pgp-MDR.
4. THE ROLE OF KEY GENES CONTROLLING APOPTOSIS IN THE DRUG
RESISTANCE OF TUMOR CELLS
4.1. p53 and Multidrug Resistance of Tumor Cells
The gene p53 and the genes regulated by p53 are critical elements of the cell response to stress, including cell reactions upon the influence of antitumor drugs (see reviews of B. P. Kopnin and P. M. Chumakov in this issue). Normal p53 proteinis activated in response to different cell injuries and its alterations result in cell cycle arrest or apoptosis. Thus, injured cells either are eliminated from the cell population or they repair damaged DNA. Alterations of p53 are very frequent in tumors, and these alterations result in impaired p53 function: the cells become unable to die by apoptosis or to stop in cell cycle checkpoints after damage. Thus, alterations of p53 function can a priori result in changes of tumor cell sensitivity to the drugs, in particular, in multidrug resistance.
Different influences activate p53: various DNA damaging agents, altered ribonucleotide pools, changes in redox potential, disruption of mitotic spindle, etc. [56]. Many anticancer drugs kill the cell using some of these mechanisms, thus it is evident that most cytostatics have to be p53 activators. Indeed, a large study done in the National Cancer Institute of the USA (60 cell lines and more than 100 anticancer drugs were examined) revealed a positive correlation between p53 status and cell sensitivity to cytotoxic drugs [57]. Cells with mutant p53 more often were more resistant to the drugs with different mechanisms of action (for example, to cisplatin and 5-fluorouracil) than cells with wild-type p53. These data stress the importance of p53 in the tumor sensitivity to chemotherapy.
It is evident, however, that this problem is not very simple. Studies of p53 regulation show that the type of modifications activating this protein is stress-, tissue-, and species-dependent and also depends on the type of amino acid mutation in p53 [56]. Different injuries activate p53 in different ways, this activation being realized through different signaling pathways [56]. These and perhaps some other peculiarities of p53 regulation determine significant differences of the reactions of the different tumor cells to the same injury as well as the different effects of various agents on p53 in the cell. Examples of these different effects are: p53 activation by DNA damage resulting in death (apoptosis) of human fibroblasts; however, the proliferation of a rodent fibroblasts was only temporary delayed [58]. One of the studies showed that p53 hyperexpression in human lung cells H1299 increased cisplatin cytotoxicity, but protected the cells from the toxic action of vepeside [59]. In other cells (mouse thymocytes) p53 induced apoptosis under the influence of the same drug (vepeside) [58]. Antiapoptotic effects of p53 (instead of the expected proapoptotic action) have been described in several papers.
Thus, it is evident that p53 function is very important in the tumor cell sensitivity to cytostatics; however, the type of the p53 influence on the drug resistance depends on many parameters: on mechanisms of the action of a drug, species and tissue lineage of the cells, and, evidently, on the genetic changes arising during carcinogenesis and tumor progression (including the differences of the genetic changes of p53 itself).
The proteins whose syntheses are controlled by the transcriptional regulator p53 (p21Waf-1, MDM) canalso influence cell sensitivity to the drugs. The results in favor of their significance for the drug resistance of tumor cells are like the data for p53. Sometimes the effects of p53 and of the genes controlled by this protein may differ.
4.2. Antioncogene PTEN and Drug Resistance of Tumor Cells
Another system influencing drug resistance is the signaling pathway which involves the PTEN gene. PTEN is the tumor suppressor with sequence homology to tyrosine phosphatases and tensin, a protein associated with the actin cytoskeleton in focal adhesions (see review of B. P. Kopnin in this issue). PTEN protein is capable of dephosphorylating both phosphotyrosine and phosphothreonine-containing substrates. The function of PTEN as a tumor suppressor is evidently connected with its ability to be a negative regulator of phosphatidylinositol 3'-kinase (PI3'K) PKB/Akt signaling pathway [60]. Protein kinase B/Akt is a crucial regulator of cell survival. Thus, negative influence of PTEN on cell survival in harmful surrounding can increase cell sensitivity to chemotherapy. Inactivation of PTEN can result in drug resistance.
Indeed, PTEN-deficient cells demonstrated resistance to a number of injuring (apoptosis inducing) agents: UV irradiation, TNFalpha, cycloheximide, anisomycin, sorbitol, heat shock [60]. In these resistant cells the activity and phosphorylation of kinase PKB/Akt were increased.
4.3. Influence of the BCL-2 Oncogene and Other Genes of the BCL-2 Family on the Drug Resistance of Tumor Cells
BCL-2 is an oncogene which contributes to tumor occurrence due to inhibition of apoptosis (see review of B. P. Kopnin in this issue). BCL-2 is a member of a large family of genes coding both anti-apoptotic proteins (for example, Bcl-2, Bcl-XL ) and pro-apoptotic proteins (Bax, Bad Bic, etc.) [61]. Polypeptides coded by these genes can heterodimerize and the combination of the members of these heterodimers can determine their influence on apoptosis [61]. Bcl-2 protein is able to inhibit the apoptosis induced by p53 in response to genotoxic stress [62]. There are data showing that Bcl-2 overexpression results in the resistance of a cells to different drugs [63]. It was shown also that BCL-2 gene expression may have a negative prognostic value for some tumors, for example for bladder cancer and hematological malignancies (both lymphoid and myeloid leukemia) [64].
4.4. CD95-L/CD95 (FasL/Fas) System and Drug Resistance
The ligand/receptor system CD95-L/CD95 (FasL/Fas or APO1) is very important for the apoptosis of some cells. Ligand CD95-L (ligand, the substance binding with receptor) is an integral membrane protein which can be released from the cells and can act in the same way as soluble growth factor or cytokine [65]. There are another cytokines which, like CD95-L, induce apoptosis--tumor necrosis factor (TNFalpha), TRAIL, etc. When CD95-L (or other cytokine with the same mode of action) binds to its receptor, the apoptotic signaling pathway is switched on.
There are data demonstrating the involvement of the CD95-L/CD95 system in drug-induced death of cancer cells and suggesting a contributory role of the alterations of the function of this system in the occurrence of drug resistance [66]. It was shown that lymphocytes of mice with the mutation in the gene coding CD95 receptor and cell lines resistant to CD95-L are resistant to a number of chemotherapeutic drugs [67-69]. The inhibitors of this signaling pathway inhibited drug induced apoptosis. However, some studies (using several cell lines) showed that the CD95-L/CD95 system is not involved into the drug induced cell death [70, 71]. Thus, the influence of this system on the cellular drug sensitivity is not straightforward and depends on a number of parameters (as was described here for other mechanisms of drug resistance).
5. CONCLUSIONS
The results of studies of the mechanisms of multidrug resistance of tumor cells show that these mechanisms are many. So far we have described only some of these mechanisms. For example, the influence of changes in the rate of proliferation on cell drug sensitivity was not analyzed in this paper. New discoveries of genes and proteins mediating cell defense from injury are on the way. The multiplicity and diversity of MDR mechanisms hamper precise diagnosis of the causes of the a patient's resistance to therapy as well the creation of reasonable protocols of MDR reversion.
The analysisof MDR mechanisms shows that the sensitivity of tumor cells to the therapy is dependent on cell context--on the combination of peculiarities of the regulatory pathways connected with the species and tissue lineage of the cell and also with genetic alterations arising in the cells during carcinogenesis and tumor progression. These phenomena also make difficult the diagnosis of the causes of MDR.
Although the mechanisms of drug resistance are multiple, some of them seem to be more significant--those which are found more often and whose clinical significance has been determined. These mechanisms are: alterations of the genes and proteins controlling apoptosis and survival of tumor cells (especially alterations of p53 and Bcl-2, sections 4.1 and 4.2); increase in the activity of the transporter protein Pgp; activity of the glutathione system (sections 1.1 and 2). Negative results of studies of the clinical significance of these key mechanisms are not rare, although most of the data show that they play a role in the determination of drug resistance. Thus, further studies are needed to prove the significance of the different resistance mechanisms for to occurrence of MDR in patients. Probably, the number of the genes and proteins studied for the determination of the causes of MDR have to be different for different tumors; the choice may depend on the pattern of protein (for example, Pgp) expression in the normal tissue and also on the alterations of the genes causing this type of malignancy.
In choosing the techniques for MDR studies it is necessary to remember that usually several different mechanisms of drug resistance are operating in the same cell, although there are mechanisms of resistance more often found after the influence of a particular drug on particular cells. For example, the resistance to cisplatin can be connected with the activation of the glutathione system, increased efflux of the drug, alterations in apoptosis regulation, and increased DNA repair [55].
Different mechanisms of drug resistance may co-exist in the same cell and they may be interconnected. For example, the inhibition of wild type p53 (inhibition of drug induced apoptosis) may result in the activation of Pgp [72]. Thus two different types of MDR proved to be connected: MDR mediated by the impaired function of p53 and MDR connected with the activation of Pgp. In section 1.2 we have already discussed the interconnections between MRP protein and the glutathione system.
The different methods for the determination of the individual sensitivity of a patient's cells to the number of the drugs were developed. The most popular now is a set of techniques using primary cultures of a patient's cells and the testing of the sensitivity of these cells to the set of different drugs. The techniques used are: the tests of drug influence on cell proliferation; evaluation of the expression of the MDR genes and proteins (Pgp, Bcl-2, enzymes of glutathione system, etc.) and some other tests [73]. The set of these tests already permitted the choice of the necessary treatment of some children with leukemia.
For studies of the defense systems of the cell, the most important are the investigation of the signaling pathways participating in the regulation of the different systems and the evaluation of the connections between different signaling pathways.
This study was supported by a grant from the Russian Foundation for Basic Research No. 99-04-48617.
REFERENCES
1.Hannun, Y. A. (1997) Blood, 89,
1845-1853.
2.Chissov, V. I. (ed.) (1989) Combined and Complex
Therapy of the Patients with Malignant Diseases [in Russian],
Meditsina, Moscow.
3.Biedler, J. L., and Reihm, H. (1970) Cancer
Res., 30, 1174-1179.
4.Jones, R. J. (1997) Curr. Opin. Oncol.,
9, 3-7.
5.Simon, S. M., and Schindler, M. (1994) Proc.
Natl. Acad. Sci. USA, 91, 3497-3504.
6.Bradley, G., Juranka, P. F., and Ling, V. (1988)
Biochim. Biophys. Acta, 948, 87-128.
7.Higgins, C. F., Callaghan, R., Linton, K. J.,
Rosenberg, M. F., and Ford, R. C. (1997) Sem. Cancer Biol.,
8, 135-142.
8.Dano, K. (1973) Biochim. Biophys. Acta,
323, 466-483.
9.Neyfakh, A. A. (1988) Exp. Cell Res.,
174, 168-176.
10.Egudina, S. V., Stromskaya, T. P., Frolova, E.
A., and Stavrovskaya, A. A. (1993) FEBS Lett., 329,
63-66.
11.Higgins, C. F. (1995) Cell, 82,
693-696.
12.Van Veen, H. W., Callaghan, R., Soceneuntu, L.,
Sardini, A., Konings, W. N., and Higgins, C. F. (1998) Nature,
391, 291-295.
13.Bosh, I., and Croop, J. (1996) Biochim.
Biophys. Acta, 1288, F37-F54.
14.Zheleznova, E. E., Markham, P. N., Neyfakh, A.
A., and Brennen, R. G. (1999) Cell, 96, 353-362.
15.Gros, P., and Buschman, E. (1993) Int. Rev.
Cytol., 137C, 169-197.
16.Smit, J. J. M., Schinkel, A. H., Oude Ellerink,
R. P. J., Groen, A. K., Wagenaar, E., van Deemeter, L., Mol, C. A. A.
M., Ottenhoff, R., van der Lugt, N. M. T., van Roon, M. A., van der
Valk, M. A., Offerhaus, G. J. A., Berns, A. J. M., and Borst, P. (1993)
Cell, 75, 451-462.
17.Borst, P. (1991) Acta Oncol., 30,
87-101.
18.Ueda, K., Pastan, I., and Gottesman, M. M. (1987)
J. Biol. Chem., 268, 17432-17436.
19.Chaudhary, P. M., and Roninson, I. B. (1993)
J. Natl. Cancer Inst., 85, 632-639.
20.Hill, B. T., Deucharts, K., Hosking, L. K., Ling,
V., and Whelan, R. D. H. (1990) J. Natl. Cancer Inst.,
82, 607-611.
21.Burt, R. K., and Thorgeirsson, S. S. (1988) J.
Natl. Cancer Inst., 80, 1383-1386.
22.Teeter, L. D., Ecksberg, T., Tsai, Y., and Kuo,
K. T. (1991) Cell Growth Differ., 2, 429-437.
23.Chin, K.-V., Ueda, K., Pastan, I., and Gottesman,
M. M. (1992) Science, 255, 459-462.
24.Stromskaya, T. P., Rybalkyna, E. Yu., Shtil, A.
A., Zabotina, T. N., Filippova, N. A., and Stavrovskaya, A. A. (1998)
Brit. J. Cancer, 77, 1718-1725.
25.Bhushan, A., Abramson, R., Chiu, J. F., and
Tritton, T. R. (1992) Mol. Pharmacol., 42, 69-74.
26.Chaudhary, P. M., and Roninsin, I. B. (1992)
Oncol. Res., 4, 281-290.
27.Roninson, I. B. (ed). (1990) Molecular and
Cellular Biology of Multidrug Resistance in Tumor Cells, Plenum
Press, N. Y.-London.
28.Chaudhary, P. M., and Roninsin, I. B. (1991)
Cell, 66, 85-94.
29.List, A. F. (1996) Leukemia, 10,
937-942.
30.Schinkel, A. H. (1997) Sem. Cancer Biol.,
8, 161-170.
31.Vallabhaneni, V., Rao, J. L., Dahlheimer, M. E.,
Bardgett, A. Z., Snyder, R. A., Finch, A. C., and Sartorelli,
D.(1999)Proc. Natl. Acad. Sci. USA, 96, 3900-3905.
32.Goldstein, L. G. (1997) Eur. J. Cancer,
32A, 1039-1050.
33.Stavrovskaya, A., Turkina, A., Sedyakhina, N.,
Stromskaya, T., Zabotina, T., Khoroshko, N., and Baryshnikov, A. (1997)
Leukemia and Lymphoma, 28, 469-482.
34.Marie, J.-P., Zhou, D.-C., Gurbuxani, S.,
Legrand, O., and Zittoun, R. (1996) Eur. J. Cancer, 32A,
1034-1038.
35.Sandor, V., Fojo, T., and Bates, S. E. (1998)
Drug Res. Updates, 1, 190-200.
36.Beck, W. T., Cirtain, M. C., Look, A. T., and
Ashmun, R. A. (1986) Cancer Res., 46, 778-784.
37.Loe, D. W., Deeley, R. G., and Cole, S. P. C.
(1996) Eur. J. Cancer, 32A, 945-957.
38.Grant, C. E., Valdimarsson, G., Hipfner, D. R.,
Almquist, K. C., Cole, S. P. C., and Deeley, R. G. (1994) Cancer
Res., 54, 357-361.
39.Deeley, R., and Cole, S. P. C. (1997) Sem.
Cancer Biol., 8, 193-204.
40.Borst, P., Kool, M., and Evers, R. (1997) Sem.
Cancer Biol., 8, 205-213.
41.Broxterman, H. J., and Schuurhuis, G. J. (1997)
J. Int. Med., 242, Suppl. 740, 147-151.
42.Sarkadi, B., and Muller, M. (1997) Sem. Cancer
Biol., 8, 171-182.
43.Scheffer, G. L., Wijngaard, P. L. G., Flens, M.
J., Izquierdo, M. A., Slovak, M. L., Pinedo, H. M., Meijer, C. J. L.
M., Clevers, Y. C., and Scheper, R. J. (1995) Nature Med.,
1, 578-582.
44.Isquierdo, M. A., Scheffer, G. L., Flens, M. J.,
Schrorijers, A. B., van der Valk, P., and Scheper, R. J. (1996) Eur.
J. Cancer, 32A, 979-984.
45.Isquierdo, M. A., van der Zee, A. G. J.,
Vermorken, J. B., van der Valk, P., Belien, J. A. M., Giaccone, G.,
Scheffer, G. L., Flens, M. J., Pinedo, H. M., Kenemans, P., Meijer, C.
J. L. M., de Vrie, E. G. E., and Scheper, R. J. (1995) J. Natl.
Cancer Inst., 87, 1230-1237.
46.List, A. F., Spier, C. G., Grogan, T. M.,
Johnson, C., Roe, D. J., Greer, J. P., Wolff, S. N., Broxterman, H. J.,
Scheper, R. J., Scheffer, G. L., and Dalton, W. S. (1996)
Blood, 87, 2464-2469.
47.Tew, K. D. (1994) Cancer Res., 54,
4313-4320.
48.Sinha, B. K., Mimnaugh, E. G., Rajagopalan, S.,
and Myers, C. E. (1989) Cancer Res., 49, 3844-3848.
49.Black, S. M., Beggs, G. D., Hayes, J. D.,
Bartozek, A., Muramatsu, M., Sakai, M., and Wolf, C. R. (1990)
Biochem. J., 268, 309-315.
50.Shen, H., Kauvar, L., and Tew, K. D. (1997)
Oncol. Res., 9, 295-302.
51.Schisselbauer, J., Silber, R., Papadopoulous, E.,
La Creta, F. P., and Tew, K. D. (1990) Cancer Res., 50,
3569-3573.
52.Gilbert, L., Etwood, L., Merino, M., Masood, S.,
Barnes, R., Steinberg, S., Lazarus, D., Pierce, L., D'Angelo, T.,
Moscow, J. A., Townsend, A. J., and Cowan, K. H. (1993) J. Clin.
Oncol., 11, 49-58.
53.Toffoli, G., Frustaci, S., Tumiotto, L.,
Gherlizoni, F., Picci, P., and Boicci, G. (1992) Ann. Oncol.,
3, 63-69.
54.Pommier, Y., Leteurte, F., Fesen, M. R.,
Fujimori, A., Bertrand, R., Solary, E., Kohlhagen, G., and Kohn K. W.
(1994) Cancer Invest., 12, 530-542.
55.Chu, G. (1994) J. Biol. Chem., 269,
787-790.
56.Glaccia, A. J., and Kastan, M. B. (1998) Genes
Devel., 12, 2973-2983.
57.O'Connor, P. M., Jackman, J., Bai, I., Myers, T.
G., Fan, S., Mutoh, M., Scudiero, D. A., Monks, A., Sausville, E. A.,
Weinstein, J., Friend, S., Fornace, A. J., and Kohn, K. W. (1997)
Cancer Res., 57, 4285-4300.
58.Clarke, A. R., Purdie, C. A., Harrison, D. J.,
Morris, R. G., Bird, C. C., Hooper, M. L., and Wyllie, A. H. (1993)
Nature, 362, 849-852.
59.Wang, Y., Blandino, G., Oren, M., and Givol, D.
O. (1998) Oncogene, 17, 1923-1930.
60.Stambolic, V., Suzuki, A., de la Pompa, J. L.,
Brothers, G. M., Mirtsos, C., Sasaki, T., Ruland, J., Penninger, J. M.,
Siderivski, D. P., and Mak, T. W. (1998) Cell, 95,
29-39.
61.Adams, J. M., and Cory, S. (1998) Science,
281, 1322-1326.
62.Chiou, S. K., Rao, L., and White, E. (1994)
Mol. Cell. Biol., 14, 2556-2563.
63.Dive, C. (1997) J. Int. Med., 242,
Suppl. 740, 139-145.
64.Allouche, M., Bettaieb, A., Vindis, C., Rousse,
A., Grignon, C., and Laurent, G. (1997) Oncogene, 14,
1837-1835.
65.Nagata, S. (1997) Cell, 88,
355-365.
66.Herr, I., Wilhelm, D., Bohler, T., Angel, P., and
Debatin, K.-M. (1999) Int. J. Cancer, 80,
417-424.
67.Reap, E. A., Roof, K., Maynor, K., Borrero, M.,
Booker, J., and Cohen, P. L. (1997) Proc. Natl. Acad. Sci. USA,
94, 5750-5755.
68.Friezen, C., Herr, I., Krammer, P. H., and
Debatin, K.-M. (1996) Nature Med., 2, 574-577.
69.Landowski, T. H., Gleason-Guzman, M. C., and
Dalton, W. S. (1997) Blood, 89, 1854-1861.
70.Eichen, C. M., Kottke, T. J., Martins, L. M.,
Basi, G. S., Tung, J. S., Earnshow, W. S., Leibson, P. J., and
Kaufmann, S. H. (1997) Blood, 3, 935-943.
71.Villunger, A., Egle, A., Kos, M., Hartmann, B.
L., Gleley, A., Kofler, R., and Greil, R. (1997) Cancer Res.,
57, 3331-3334.
72.Thottassery, J. V., Zambetti, G. P., Arimori, K.,
Schuetz, E. G., and Schuetz, J. D. (1997) Proc. Natl. Acad. Sci.
USA, 94, 11037-11042.
73.Fruebauf, J. P., and Bosanquet, A. G. (1993)
Principles Practice Oncol., 7, 1-16.