REVIEW: Transmembrane Ion Transport by Polyphosphate/Poly-(R)-3-hydroxybutyrate Complexes
R. N. Reusch
Department of Microbiology, Giltner Hall, Michigan State University, East Lansing, MI 48824, USA, E-mail: rnreusch@pilot.msu.edu
Received October 29, 1999
Transmembrane ion transport, a critical process in providing energy for cell functions, is carried out by pore-forming macromolecules capable of discriminating among very similar ions and responding to changes in membrane potential. It is widely regarded that ion channels are exclusively proteins, relatively late arrivals in cell evolution. Here we discuss the formation of ion-selective, voltage-activated channels by complexes of two simple homopolymers, namely, inorganic polyphosphates (polyPs) and poly-(R)-3-hydroxybutyrates (PHBs), derived from phosphate and acetate, respectively. Each has unique molecular characteristics that facilitate ion selection, solvation, and transport. Complexes of the two polymers, isolated from bacterial plasma membranes or prepared from the synthetic polymers, form voltage-dependent, Ca2+-selective channels in planar lipid bilayers that are selective for divalent over monovalent cations, permeant to Ca2+, Sr2+, and Ba2+, and blocked by transition metal cations in a concentration-dependent manner. Recently, both polyP and PHB have been found to be components of ion-conducting proteins: namely, the human erythrocyte Ca2+-ATPase pump and the Streptomyces lividans potassium channel. The contribution of polyP and PHB to ion selection and/or transport in these proteins is yet unknown, but their presence gives rise to the hypothesis that these and other ion transporters are supramolecular structures in which proteins, polyP, and PHB cooperate in forming well-regulated and specific cation transfer systems.
KEY WORDS: polyphosphate, poly-(R)-3-hydroxybutyrate, ion channel
Inorganic polyphosphates (polyPs) and poly-(R)-3-hydroxybutyrates (PHB) are simple linear polymers that are found in a wide variety of organisms, ranging from the most primitive to the most highly evolved [1-17]. Both homopolymers have unique molecular characteristics that make them valuable physiological agents, but perhaps one of their most serviceable functions is to assist in the regulation of internal ion concentrations by serving as vehicles for selective transport of ions across membranes.
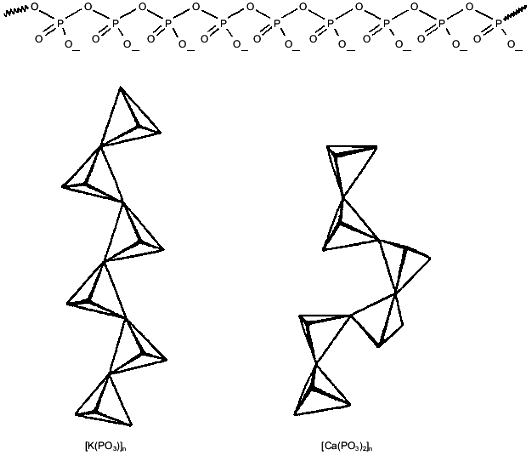
Polyphosphates attract and select for cations by charge. PolyPs are linear chains of tetrahedral phosphate residues linked through common oxygen atoms by phosphoanhydride bonds (Fig. 1). At physiological pH, each unit carries a monovalent negative charge. The resulting high density of charge gives polyPs a large capacity for cation exchange [18]. Moreover, polyPs have an inherent capacity to distinguish between monovalent and multivalent cations. This faculty is attributable to the large degree of rotational flexibility about the P-O-P backbone bond that allows conformational adjustment to the charge and coordination requirements of the binding cation (Fig. 1). As a result, unitary negative charges on adjacent phosphoryl tetrahedra can approach within 4 Å, a distance that leads to a preference for binding divalent cations [19]. The stronger electrostatic interaction of divalent cations leads to their preferential selection by polyPs over monovalent cations. Binding energy rather than ion size or coordination geometry is the rationale for the ability of polyPs to sequester Ca2+ and Mg2+, which make them effective agents in water softening [18, 20, 21]. However, when no divalent cations are available or when the charge density is reduced as, e.g., at low pH, polyPs will efficiently bind monovalent cations.
Poly-(R)-3-hydroxybutyrates (PHBs) solvate cations. PolyP chains can form a framework of sufficient length to cross the bilayer, attract cations, select for divalent over monovalent cations and, in response to voltage or concentration gradients, move cations along its backbone. However, their high charge density creates a strong electrostatic barrier to penetrating a lipid bilayer. Also, polyPs do not distinguish among cations of the same charge by differences in size or coordination geometry. In order to form effective and selective cation transporters, polyPs must associate with amphiphilic macromolecules that enable them to span the membrane and assist them in cation discrimination. Poly-(R)-3-hydroxybutyrates (PHBs) (Fig. 2) are perhaps the simplest and most primitive macromolecules to associate with polyP to form ion channels.Fig. 1. Molecular structure of inorganic polyphosphates (polyPs)--phosphate residues with monovalent charge joined by flexible phosphoanhydride bonds. Rotational flexibility of P-O-P bonds joining the tetrahedral allows the chains to twist into a variety of conformations, depending on the coordination preferences of binding cations.
PHBs are linear homopolymers of (R)-3-hydroxybutyrate, formed from acetyl-CoA in three steps: condensation of two acetyl-CoAs to form acetoacetyl-CoA, reduction by NADPH to form (R)-3-hydroxybutyryl-CoA, and polymerization with release of CoA to yield PHB [8-10]. PHB was first identified by Lemoigne in Bacillus megaterium [7] as high-molecular-weight polymer (from 100 to >1000 kD) deposited within cytoplasmic granules. This form of PHB has since been found in a wide variety of archaebacteria and eubacteria, in which it is presumed to serve as a carbon store. PHBs and their homologs, poly-(R)-3-hydroxyalkanoates (PHAs) have recently become of commercial interest as biodegradable plastics [22]. More recently, it was discovered that low-molecular-weight (<14 kD) PHBs are found not only in prokaryotes, but also in cells, organelles, and intracellular fluids of eukaryotes [11, 13-17]. Low-molecular-weight PHBs, like polyPs, appear to be universal constituent of biological cells.Fig. 2. Poly-(R)-3-hydroxybutyrate (PHB)--polyester chain in which hydrophobic methyl groups alternate with hydrophilic ester carbonyl oxygens.
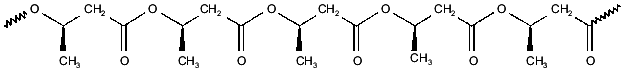
The properties of PHB that are most fundamental to its role in ion transport is its amphiphilic nature and its ability to solvate salts. The repeating unit of PHB contains both hydrophobic methyl groups and hydrophilic ester carbonyl oxygens, enabling the polyester to act as an intermediary between polyP and the bilayer (Fig. 2) [14]. This amphiphilic nature allows PHB to penetrate hydrophobic regions, such as membranes and hydrophobic pockets of proteins that are relatively inaccessible to water. Perhaps of even greater importance is the remarkable capacity of PHB to “dissolve” salts. Indeed PHB may be unique among biological polymers in having the structural features common to a small group of polymers, most notably polyethylene oxides, that form ion-conducting salt complexes known as polymer electrolytes [23-26]. Important characteristics of this class are flexible backbones, heteroatoms (in this case, ester carbonyl oxygens) that form coordinate bonds with cations, and a suitable distance between heteroatoms along the backbone to permit multiple coordinate bonds between the polymer backbone and cations.
The capacity of polymers (>50 units, PHBs) and oligomers (<50 units, OHBs) of R-3-hydroxybutyrate to “dissolve” salts has been well established experimentally. Seebach et al. [27] have shown that the triolide of R-3-hydroxybutyrate forms crown ester complexes with alkali metals, Bürger and Seebach [28] have shown that OHBs transport alkali and alkaline earth salts across methylene chloride layers in U-tubes, Reusch and Reusch [29] have prepared conducting complexes from PHB and its homolog, poly-(R)-3-hydroxyvalerate, with lithium perchlorate, and Fritz and Seebach have demonstrated concentration-gradient-driven transport of Ca2+ into liposomes by OHBs [30, 31]. Finally, as discussed further below, PHBs and OHBs make membranes permeant to ions.
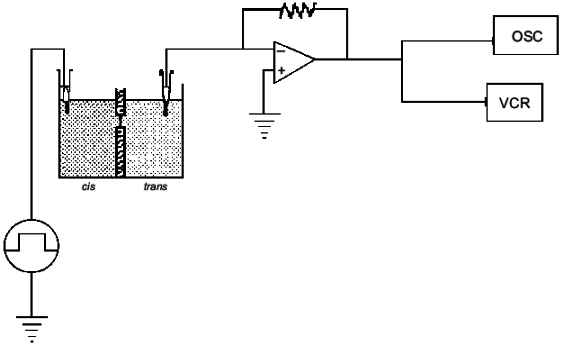
PHBs and OHBs form non-selective ion channels. The ability of PHBs and synthetic OHBs to conduct salts across bilayers was examined in a planar bilayer voltage-clamp setup (Fig. 3) [32, 33]. PHBs of 130-150 units (isolated from E. coli) [34] and unidisperse OHBs (8, 16, 32, 64, and 96 units), prepared synthetically by an exponential fragment-coupling strategy [35, 36], were individually incorporated into planar bilayers of synthetic 1-palmitoyl-2-oleoyl-phosphatidylcholine (16:0, 18:1 PC) [37]. When concentrations of the polymers or oligomers were 0.1-5% of the phospholipid concentration, discrete current fluctuations were observed at positive and negative potentials for chain lengths of 16 monomer units or multiples of 16 units. The conductance at a given potential was reasonably constant for a given preparation, but varied for different preparations of the same oligomer, so that current-voltage relationship could not be established. As expected, the PHBs and OHBs did not discriminate well among ions. The current records were similar to those observed with ionic and non-ionic detergents such as Triton X-100, octyl glucoside, or sodium dodecyl sulfate [38, 39], and for pure phosphatidylcholine bilayers at temperatures of the gel to liquid-crystalline phase transition [40, 41].
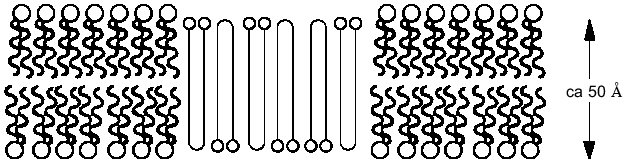
If it is assumed that the oligomers in the bilayer preserves the 2 1 helicity, 6 Å pitch determined from fiber X-ray scattering for the crystalline state of the polymer [42], then the length of 16 units corresponds closely to the ~50 Å lamellar thickness of synthetic crystalline OHBs (>16 units), as determined from fiber X-ray scattering, transmission electron microscopy, and atomic force microscopy [43, 44], and to the 48 Å width of 16:0, 18:1 PC bilayers, estimated from electrical capacitance measurements [45]. This suggests that the PHB molecules fold in the 16:0, 18:1 PC bilayer in the same lamellar morphology, 16 residues per turn. It is assumed that each OHB molecule crosses the hydrophobic region of the 16:0, 18:1 PC bilayer in 16 units per turn, and is stabilized at each interface by the formation of hydrogen bonds fromthe terminal hydroxyl and carboxyl groups to the ester groups of the phospholipids (Fig. 4). Ion permeability presumably results from areas of mismatch at the interfacial regions between islands of lamellar crystallites of PHBs or OHBs in the bilayer and phospholipids [46].Fig. 3. Planar bilayer system. The system consists of two aqueous solutions (labeled cis and trans) separated by a planar lipid bilayer. External voltage commands are applied to the cis side with the trans side maintained at ground (defined as zero voltage); OSC, oscilloscope; VCR, recording tape system [52].
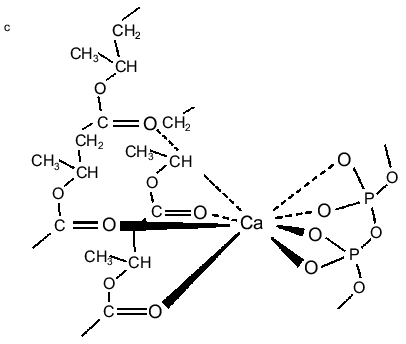
PolyP and PHB form selective ion channels. PHBs solvate polyPs salt by encircling them and replacing the water of hydration about the cations with coordinate bonds to its ester carbonyl oxygens. The relatively weak solvating ability of carbonyl ester oxygens (as compared to the oxygens of water) and the absence of hydrogen bond donors for solvation of anions means that PHB will preferentially solvate salts composed of cations with high solvation energies and anions with diffused charge. As stated above, critical factors in achieving this solvation are the flexible backbone of PHB and the optimal distance between carbonyl oxygens along its backbone [11, 23-25]. The result is a flexible structure of two discrete polymers bridged together by lanes of cations [11, 34, 47] (Fig. 5, a and b). Since polyP is fully charged at physiological pH, it will preferentially bind divalent cations. The major physiological divalent cations are Mg2+ and Ca2+. PolyP does not distinguish between these two cations, but the geometry of the ligands in the irregular binding cavities formed by the phosphoryl oxygens of polyP with the ester carbonyl oxygens of PHB strongly favor Ca2+ (Fig. 5c) [48]. Sr2+ and Ba2+ are not physiological cations but they have the same coordination geometry as Ca2+: consequently, they are also permeant [34, 48].Fig. 4. Schematic representation of (R)-3-hydroxybutyrate oligomers (OHBs) of 32 units incorporated into planar phospholipid bilayer [37].
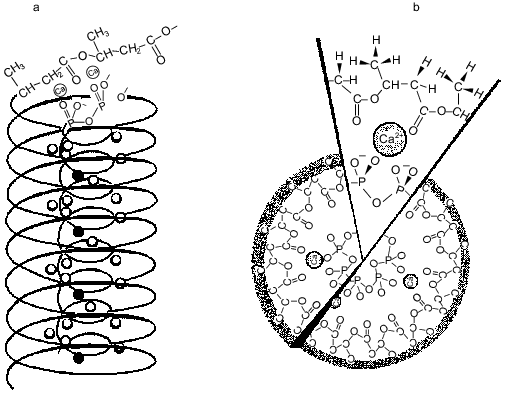
Natural polyP/PHB ion channels. PolyP/PHB complexes were first discovered in the plasma membranes of bacteria [47, 49-52]. The complexes are at low concentrations in log-phase cells, substantially higher concentrations in stationary-phase cells, and highest concentrations in genetically transformable cells. The channel activity of the complexes was examined in planar bilayers of 16:0, 18:1 PC between symmetric solutions of 200 mM CaCl2, 5 mM MgCl2, 10 mM Tris-Hepes, pH 7.4 [34]. The current records at positive and negative potentials of the complexes in plasma membrane vesicles of competent E. coli vesicles were essentially the same as those for complexes extracted from competent E. coli cells into chloroform (crude or HPLC-purified) (Fig. 6).Fig. 5. a) Drawing depicting putative relationship between polyP, PHB, and Ca2+ in the complexes. The PHB forms a helix with a lipophilic shell of methyl groups and polar lining of ester carbonyl oxygens surrounding a core helix of polyP with Ca2+ bridging the two polymers [11]. b) A view looking down the channel structure [11]. c) Coordination of Ca2+ by polyP and PHB [47]. Ca2+ forms ionic bonds with four phosphoryl oxygens of polyP and coordinate bonds with four ester carbonyl oxygens of PHB to form a neutral complex with irregular cubic geometry.
The physiological role of polyP/PHB complexes has not been established, but bacterial cells maintain low internal Ca2+ [53-55], and there is increasing evidence pointing to calcium involvement in a number of important cellular functions, such as chemotaxis, cell division, heat shock, pathogenicity, and differentiation [56-59]. Systems for calcium export have been identified [60, 61], but mechanisms for calcium entry are less well known, although an L-type channel was reported in Bacillus subtilis [62, 63], and Jones et al. [55] demonstrated the presence of two putative Ca2+ influx channels, one inhibited by La3+ and the other not. It was suggested that the properties of the former were consistent with the polyP/PHB calcium channel complexes.Fig. 6. Single channel records of polyP/PHB complexes from genetically competent cells of E. coli DH5alpha, in planar lipid bilayers.Channels were incorporated into planar lipid bilayers, composed of 1-palmitoyl-2-oleoyl-phosphatidylcholine and cholesterol (5:1w/w), between symmetric bathing solutions of 200 mM CaCl2, 5 mM MgCl 2 , 10 mM Tris-Hepes, pH 7.4 at 22°C. Channel activities of complexes in plasma membrane vesicles and extracts are essentially similar. Clamping potential was +80 mV with respect to ground (trans side). The solid triangles at the side of each profile indicate the fully closed state of the channel. The data was filtered at 1 kHz. a) Plasma membranes; b) chloroform extract; c) HPLC purification.

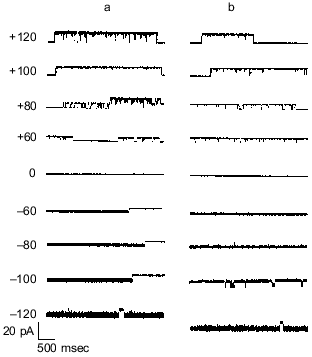
Synthetic polyP/PHB ion channels. To resolve remaining doubts as to whether the channel activity was actually effected by trace protein contaminants, Das et al. [64] performed a total synthesis of the channel complex from (R)-3-hydroxybutanoic acid, sodium polyphosphate and calcium chloride. An exponential fragment-coupling strategy was employed to prepare the 128-mer of R-3-hydroxybutyrate (PHB 128 ) [65]. This synthetic polymer has a molecular weight of ~11.4 kD, which is close to that of E. coli PHB (~12 kD). Calcium polyphosphate was prepared from sodium polyphosphate glass (average residue number 65) and calcium chloride. The size of complexed polyP was determined by acrylamide gel electrophoresis to be in the same range (55-65 residues) as that in the E. coli complexes [66]. The current records of channels formed in planar bilayers by the synthetic complexes were indistinguishable from those of polyP/PHB complexes extracted from E. coli (Fig. 7), and the conductances of the synthetic and E. coli channels were equivalent, 101 ± 6 and 104 ± 12 pS, respectively (Fig. 8).
Fig. 7. Representative single-channel current fluctuations of synthetic polyP/PHB128 (a) and E. coli-derived polyP/PHB complexes (b) at various clamping potentials [64]. Complexes were incorporated into planar lipid bilayers composed of 1-palmitoyl-2-oleoyl-phosphatidylcholine and cholesterol (5:1 w/w) between symmetric aqueous solutions of 200 mM CaCl2, 5 mM MgCl2, 10 mM Tris-Hepes, pH 7.4 at 22°C. Bars at side of each profile indicate fully closed state of channel. Clamping potentials with respect to ground are indicated at left (in mV).
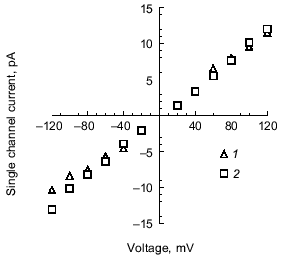
Selectivity of polyP/PHB channels. The channels formed by E. coli or synthetic polyP/PHB, show strong selectivity for divalent over monovalent cations [36, 64]. The selectivity was examined by determining the reversal potential (zero-current potential) when the channels were incorporated into bilayers formed between aqueous solutions of unequal ion composition. The reversal potential was estimated graphically from the single-channel current-voltage relationships, and this value was compared with the equilibrium potential (calculated from concentrations) for each cation in a perfectly selective channel, calculated from the Nernst equation [67].Fig. 8. Current-voltage relations for E. coli polyP/PHB (1) and synthetic polyP/PHB128 (2)channels [64]. The conductance of the channel for Ca2+ in symmetric solutions under the same experimental conditions as in Fig. 7 is 104 ± 12 pS for the E. coli channels and 101 ± 12 pS for the synthetic channels. The data points represent mean values of ten observations.
The selectivity for divalent cations of E. coli polyP/PHB complexes was demonstrated in 16:0, 18:1 PC bilayers between unequal solutions of Sr2+ over Na+. The reversal potential is -38 mV, close to the equilibrium potential for Sr2+ of -40 mV, whereas the equilibrium potential for Na + is nominally plus infinity and the Cl- equilibrium potential is +7 mV. Correspondingly, selectivity for divalent cations of synthetic polyP/PHB complexes was established in 16:0, 18:1 PC bilayers between unequal solutions of Ca2+ and Na+ (Fig. 9). The reversal potential is -67 mV, close to the equilibrium potential for Ca2+ of -82 mV, whereas the equilibrium potential for Na+ is +76 mV and the Cl- equilibrium potential is +9 mV.
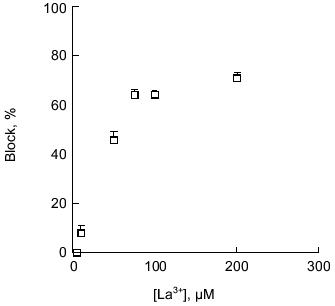
Block of polyP/PHB channels. PolyP/PHB channel complexes are blocked by transition metal cations in a concentration-dependent manner. The E. coli-derived complexes were completely blocked by 0.6 mM La 3+ , 1.5 mM Co2+, or 8 mM Cd2+ [34]; nearly complete blocking of single-channel currents was observed in the synthetic complexes at concentrations >0.1 mM La3+ (0.1% of Ca2+) [64] (Fig. 10). Mg2+ also has an affect on the currents. At low concentrations (1% of Ca2+ concentration), it appears to increase the stability of the channel complexes and acts as a mild blocker, thus making it easier to observe channel openings and closures. For this reason, low concentrations of Mg2+ are customarily included in the buffers. Raising Mg2+ concentrations to higher levels significantly diminished single-current magnitudes, and nearly complete blocking of Ca2+ currents was observed in the E. coli channels when Mg2+ was present in amounts in excess of 10% of Ca2+.Fig. 9. Cation selectivity of synthetic polyP/PHB channels. Selectivity of synthetic polyP/PHB128 channel complexes for Ca2+ over Na+ [64]. Single-channel current-voltage relations in planar lipid bilayers composed of 1-palmitoyl-2-oleoyl-phosphatidylcholine and cholesterol (5:1 w/w) between asymmetric aqueous solutions--cis (65 mM CaCl2, 10 mM NaCl, 5 mM MgCl2, 10 mM Tris-Hepes, pH 7.4) and trans (0.1 mM CaCl2, 200 mM NaCl, 5 mM MgCl2, 10 mM Tris-Hepes, pH 7.4) at 22°C. The equilibrium potentials calculated from concentrations were ECa = -82 mV, ECl = +9 mV, and ENa = +76 mV. Error bars indicate standard deviation from the mean.
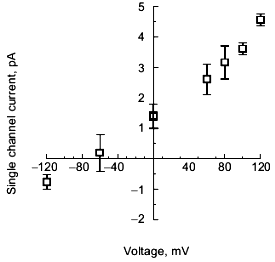
Gating of polyP/PHB channels. PolyP/PHB complexes isolated from E. coli and synthetic polyP/PHB complexes exhibit two distinct gating modes (modes 1 and 2) in planar lipid bilayers. Although either mode can be observed at both positive and negative potentials, mode 1 is illustrated at positive potentials and mode 2 at negative potentials in Fig. 7 [64]. In mode 1, the channels display long openings of the order of several seconds with infrequent and brief closures to the fully closed state [68]. In mode 2, the channel activity is characterized by long bursts with flickering between the fully open state of 87 ± 3 pS and a major subconductance state of 56 ± 2 pS, interrupted occasionally by closures to the fully closed state (<0.5% of total time). The channel exhibits a number of other subconductance states but with much lower frequency. In both modes, the channel occasionally enters a long closed state of several seconds duration. Switching between modes 1 and 2 is observed infrequently. The switching can take place in either direction--from mode 1 to mode 2 or the reverse. The factors that determine preference for a specific mode or effect switching between modes are presently unknown, but the complex gating kinetics of this channel may be attributable to the multitude of conductive conformations that can be adopted by these multi-lane, multi-binding site amorphous complexes in response to potential differences.Fig. 10. Block of synthetic polyP/PHB128 channels by transition metal cation La3+ [64]. The bilayer was composed of 1-palmitoyl-2-oleoyl-phosphatidylcholine and cholesterol (5:1 w/w) between symmetric aqueous solutions of 200 mM CaCl2, 5 mM MgCl2, 10 mM Tris-Hepes, pH 7.4 at 22°C. After incorporation of the channel, activities were recorded for 5 min. Then La3+ was added to the trans side at the indicated final concentrations. The bath was stirred and activities were recorded after 1 min. Data points represent mean values of the amplitude histograms at -80 mV clamping potential. Error bars show the standard deviation from the mean.
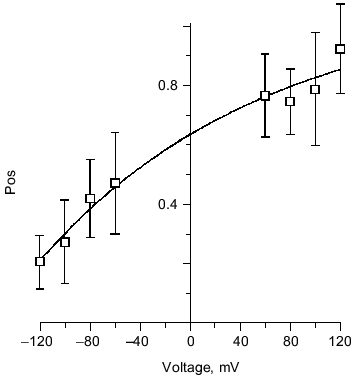
Several kinetic properties of the channel are voltage-dependent, indicating an asymmetry in the channel structure. For a given single channel, the fraction of time spent in each conductance level is strongly voltage-sensitive (Fig. 11). A given single channel displayed one of two voltage-dependent responses that were mirror images of each other. This suggests two opposite orientations of the complexes in the bilayer. PHB has a carboxyl group at the head and hydroxyl group at the tail, creating a structural asymmetry that permits the complex to assume a head to tail or tail to head alignment across the bilayer. The importance of end residue structure was demonstrated by the strong inhibition of ion transport activity by PHB, itself, when the hydroxyl or carboxyl end groups were derivatized [37]. Synthetic tris(macrocycle)-cation channels have also shown remarkable sensitivity to structural changes at the distal ends [69]. In cells, PHB synthesis takes place at the cytoplasmic side, thus it is likely that all PHB/polyP channels in the plasma membrane are oriented in the same direction and display the same pattern of voltage dependence.
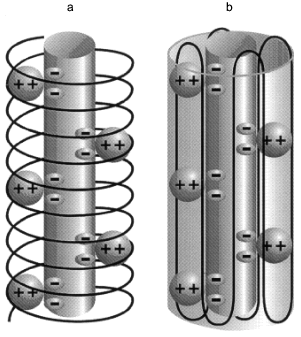
Structure of polyP/PHB ion channels. The detailed structure of polyP/PHB complexes is unknown. However, some assumptions can be made concerning the general organization of the complexes from the physical properties and sizes of the polymers and the low dielectric environment they inhabit. It is clear that the highly polar polyanionic polyP must be shielded from the hydrophobic region of the bilayer by the amphiphilic PHB. The two models proposed for the channel complexes by Reusch et al. [34, 65] and Seebach et al. [45, 64] have this general structure. The former model proposes that PHB has a coiled conformation such as it displays in solution [70] (Fig. 12a) while the latter holds that PHB maintains the folded helix form of its solid state [44] (Fig. 12b). A consequence of both arrangements is the formation of multiple parallel lanes between the two polymers, with multiple cation-binding sites lining each lane [34, 64]. Normal molecular motions of the polymers, such as twisting or stretching movements, or sliding or rotation of polyP within the homogeneous environment provided by PHB may alter channel geometry and effect changes in current amplitude and gating.Fig. 11. Voltage-dependence of polyP/PHB channels in mode 2 [68]. Complexes were extracted from E. coli DH5alpha and incorporated into planar lipid bilayers composed of 1-palmitoyl-2-oleoyl-phosphatidylcholine and cholesterol (5:1 w/w) between symmetric aqueous solutions of 200 mM CaCl2, 5 mM MgCl2, 10 mM Tris-Hepes, pH 7.4 at 22°C. The data show the probability of opening of the channel (Pos) to the fully open state at indicated clamping potentials. Fit by nonlinear regression (Boltzman).
It may not be possible to resolve the structure of the complexes with certainty. In the Reusch model, the complexes have the liquid properties of polymer electrolytes and this suggests a family of conformations rather than a single defined structure. In the Seebach model, several PHB molecules are involved in surrounding polyP. The individual PHB chains are free to adopt various positions in the phospholipid lattice; hence a well-defined structure is again unlikely. Further studies may help to decide between the two views of the arrangement of PHB molecules in the complexes and delineate the more probable conformations.Fig. 12. Diagrammatic representations of polyP/PHB channels [64]. The central cylinder represents the polyP helix, which contains pairs of closely spaced monovalent negative charges that provide a ladder of binding sites for Ca2+. The Ca(polyP) is surrounded and solvated by PHB: a) PHB forms a helix encircling Ca(polyP); b) PHB forms a beta sheet type structure surrounding Ca(polyP).
Putative mechanism of ion conduction by polyP/PHB channels. The mechanism of ion conduction by polyP/PHB channel complexes can be rationalized in terms of the structures and properties of the component polymers. One view of how the channel may operate in the cell membrane or planar bilayer follows.
Ca(polyP), surrounded and solvated by PHB, forms a salt bridge extending from the cytoplasm to the periplasm. A multi-lane channel is formed between the two polymers; the outer wall is lined with solvating oxygens, the inner wall is girdled by monovalent, phosphoryl anions. At the outer interface, cations are drawn to the mouth of the channel by polyP, and divalent cations are preferentially bound. Most of the binding sites within the channel are occupied by Ca2+, and the strong bonds between Ca2+ and polyP prevent ion movement, and so the channel is “closed”.
The polyP “wire” of negative charges across the bilayer acts as a sensor of membrane potential. PolyP reacts to membrane depolarization (or a voltage step of sufficient strength) by stretching or sliding within the PHB pore, thus dislodging the resident Ca2+ and initiating an ion flow. Ca2+ at the interface then preferentially permeates into the binding cavities at the end of the channel by virtue of their well-suited coordination geometry and the relatively rapid rate at which they undergo replacement of water of hydration. Since binding sites on polyP are identical and spaced at frequent intervals, there is no net potential energy cost to cation movement within the channel. Segmental motions of the PHB backbone and librational movements of ester carbonyl oxygens carry Ca2+ from site to site in parallel single-file lanes until internal concentrations rise to an appropriate level or the membrane is again polarized. Transition metal cations, particularly trivalent cations like La3+, bind tightly to polyP at the interface but have difficulty entering because of their unsuitable coordination preferences, and consequently they block the ion flow.
PolyP/PHB complexes as calcium pumps. Bacterial cells, like those of eukaryotes, maintain low internal Ca2+ concentrations. The means by which they do this is still uncertain, although secondary systems for Ca2+ export have been identified in a number of bacteria [60, 61]. PolyP/PHB complexes have many structural features that make them attractive agents for Ca2+ extrusion. The ionic bond between Ca2+ and polyP is sufficiently strong to withstand the considerable Ca2+ gradient across the membrane, estimated as 2 mM outside and 0.1 µM inside for E. coli [54]. At the same time, Ca2+ is held to PHB only by weak ion-dipole bonds. As a result, the Ca(polyP) helix can slide within the PHB envelope in response to an increase in the concentration or voltage gradient. This organization implies that Ca2+ could be transported out of the cell by extending the polyP chain on the cytoplasmic side of the membrane. As the appended phosphate units move into the PHB channel, Ca2+ would be sequestered from the cytoplasm; at the outer face of the membrane, Ca(polyP) is exported.
The enzymes required to carry out this mechanism are known in many organisms [3, 9, 71]. PolyP occupies an intermediate position in the free energy scale of phosphorylated compounds, and can act as both a donor and acceptor of phosphate groups. It has a free energy of hydrolysis similar to that of ATP, and in bacterial cells is formed enzymatically from ATP and other high-energy phosphates. Polyphosphate kinase, discovered by Kornberg et al. [72] in E. coli, catalyzes the reversible transfer of the gamma-phosphate of ATP to the end of a polyphosphate chain. Similar kinases have since been reported in many other organisms [71]. Phosphorylated compounds with still higher free energy of hydrolysis, such as 1,3-diphosphoglycerate, can also donate high-energy phosphates to polyP [3]. At the periplasmic face, polyP may be degraded by polyphosphatases [3-6, 73]. The ultimate effect of these reactions is to draw Ca2+ from the cytoplasm, and extrude it into the periplasm.
Inasmuch as both PHB and polyP have been conserved in eukaryotes, as evidenced by their presence in a wide variety of plant and animal tissues [13-17], the question arose as to whether PHB/polyP complexes have also been conserved in eukaryotic calcium transport systems. Reusch et al. [74] examined the Ca2+-ATPase of the human erythrocyte membrane for the presence of these two polymers. This protein was the first plasma membrane enzyme demonstrated to work as a Ca2+ pump [75, 76] and it is the sole transporter of Ca2+ in the cell. Moreover, it has subsequently been determined that the Ca2+-ATPase is ubiquitous in plasma membranes of a variety of cell types in eukaryotic organisms [77].
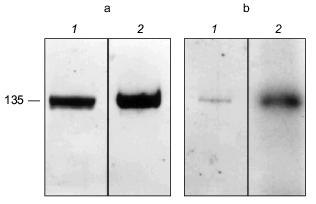
The erythrocyte Ca2+-ATPase was purified to homogeneity by calmodulin affinity chromatography [78-80]. The presence of PHB in the Ca2+-ATPase was demonstrated by a positive reaction to anti-PHB IgG on a Western blot (Fig. 13a). PHB was then measured by a chemical assay in which the polyester is converted via beta-elimination to its unique degradation product, crotonic acid, by heating in concentrated sulfuric acid [11, 81, 82]. The crotonic acid is then extracted, separated by HPLC chromatography, and quantitated by comparison of peak area with that of standards. Under the conditions of the assay, no amino acids or homopolymers of amino acids produce significant amounts of crotonic acid [82]. The presence of polyP in the protein was signaled by its reaction to the cationic dye, o-toluidine blue [83]. PolyP causes a shift in the absorption maximum of o-toluidine blue towards shorter wavelengths, i.e., from a maximum at 507 nm (blue) to 530 nm (violet-red). The absorption shift is specific for inorganic polyphosphates inasmuch as RNA and DNA are stained blue. The identity of polyP in the Ca2+-ATPase was confirmed, and its quantity determined by an enzymatic assay in which the conversion of [ 14 C]ADP by polyP to [14C]ATP is measured in the presence of E. coli polyphosphate kinase [84, 85]. Though only small quantities of the polymers were found (0.5 µg polyP/mg protein and 1.3 µg PHB/mg protein), the amounts are likely to be greatly understated, since both PHB and polyP are subject to extensive chemical and enzymatic hydrolysis during the isolation and purification procedures. The results have importance in that they place these two polymers, with proven capacities to facilitate ion transport, in a human calcium-transporting structure.
The Ca2+-ATPase protein was also shown to have polyP-ATP transferase and polyP-ADP phosphotransferase activities. Niggli et al. [78] demonstrated that the purified Ca2+-ATPase could be phosphorylated by [gamma-32P]ATP. An acyl phosphoenzyme is generated which can transfer the phosphoryl group back to ADP and regenerate ATP [86]:Fig. 13. a) Reaction of Ca2+-ATPase protein to anti-PHB IgG [74]. Purified Ca2+-ATPase protein (8.5 µg per lane) was separated by electrophoresis on a 10% polyacrylamide gel. 1) Coomassie blue stain of Ca2+-ATPase protein; 2) Western blot of Ca2+-ATPase protein probed with rabbit anti-PHB IgG. b) Phosphorylation of the Ca2+-ATPase by [32P]polyP [74]. Purified Ca2+-ATPase protein (2 µg) was phosphorylated at room temperature by [32P] polyP as described in [74] and separated by electrophoresis on a 10% polyacrylamide gel. 1) Coomassie blue stain of phosphorylated Ca2+-ATPase; 2) autoradiogram of phosphorylated Ca2+-ATPase.
[gamma-32P]ATP + Ca2+-ATPase <--> ADP + Ca2+-ATPase·(32P-PO3-).
The ability of polyP to directly phosphorylate the Ca2+-ATPase was demonstrated by autoradiography using [32P](polyP) (Fig. 13b):
[32P](polyP) + Ca2+-ATPase --> Ca2+-ATPase·(32P-PO3-).
Then the capacity of the phosphorylated Ca2+-ATPase to transfer phosphate to ADP and to generate [32P]ATP or to extend a polyP chain was confirmed (Fig. 14, a and b):
Ca 2+ -ATPase·(32P-PO3-) + ADP --> [32P]ATP,
Ca 2+ -ATPase·(32P-PO3-) + (polyP)n --> [32P](polyP)n+1.
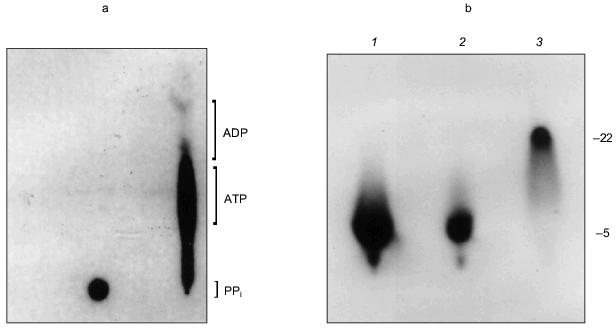
The manner in which the PHB and polyP interact with the protein is not known and it remains uncertain whether they play an active role in the export of Ca2+; however, the presence of these highly conserved polymers and enzymatic activities in the erythrocyte Ca2+-ATPase provides the basis for a hypothetical molecular mechanism for polyphosphate-aided Ca2+ transmembrane transport in which the pump transports Ca2+ out of the cytoplasm by catalyzing the transfer of phosphoryl groups to polyP. The added units attract both Mg2+ and Ca2+; however, Ca2+ is preferentially selected over Mg2+ as the hydration shells of the cations are replaced by the ester carbonyl oxygens of PHB within the bilayer, perhaps aided by oxygens of strategically placed amino acid side chains. Factors in the selection of Ca2+ include its larger ion diameter, coordination geometry, tolerance for variability in bond lengths and angles, and more rapid hydration kinetics [48]. The net effect is the coexport of Ca2+ and PO42- from the cytoplasm fueled by ATP. Strict adherence to this mechanism would suggest that two ATP molecules are required to transport one Ca2+, yet the Ca2+-ATPase pumps one Ca2+ per one ATP. This suggests that other factors such as Ca2+/proton exchange must contribute to the process.Fig. 14. a) Formation of [32P]ATP from [32P]polyP and ADP by Ca2+-ATPase [74]. The [ 32 P]polyP, with average chain length >400 residues, was incubated with 1 mM ADP, 100 mM KCl, 4 mM MgCl2, 0.1 mM CaCl2 in 10 mM Hepes-KOH (pH 7.4) and Ca2+-ATPase for 30 min at 37°C. ADP and ATP (15 nmoles each) were added as carriers, and ADP, ATP, and polyP were resolved by thin-layer chromatography on PEI cellulose, visualized by UV, and autoradiographed. Brackets indicate regions of the chromatogram occupied by long chain polyP, ATP, and ADP standards after development. Right lane, reaction in the presence of Ca2+-ATPase. Over 80% of the radioactive phosphate in the product co-migrated with ATP. The remainder, which appears to be short-chain [32P]polyP resulting from incomplete reaction, formed a band between the origin and the ATP band. Left lane, reaction in the absence of Ca2+-ATPase. b) Elongation of polyP 5 by [gamma-32P]ATP [74]. PolyP 5 (10 µg) was incubated with 2 µg Ca 2+ -ATPase, [gamma-32P]ATP (30 Ci/mmole), 50 mM Hepes-KOH, pH 7.4, 130 mM KCl, 8 mM MgCl 2 , 0.15 mM C 12 E 8 , 20 µM 1-palmitoyl-2-oleoyl-phosphatidylcholine, 1 mM EGTA, 1 mM CaCl2. Electrophoresis was performed on 20% acrylamide, 7 M urea minigels. PolyP 5 was selected for this assay because increases in chain length are more noticeable on gels for short-chain polyphosphates. Standards were polyP 5 , [ 32 P]Pi, xylene cyanol, and bromophenol blue. The position of the dye markers was calibrated against short-chain polyPs that had been prepared and measured as described by Clark and Wood [74]. Visualization was with o-toluidine blue and autoradiography. 1) PolyP 5 and [gamma-32P]ATP in the presence of Ca2+-ATPase and CaCl2 as above; 2) same as lane (1) except CaCl2 was absent and solution contained 10 mM EGTA; 3) same as lane (1) except Ca2+-ATPase was absent.
PolyP and PHB in the Streptomyces lividans potassium channel. It is also of interest to determine whether polyP and/or PHB have been conserved as components of protein ion channels. The potassium channel of the Gram-positive soil bacterium, Streptomyces lividans, (KcsA) was selected for this study since it is the first ion channel to have its structure analyzed by X-ray crystallography [87]. Although KcsA is a prokaryotic channel, the structural elements responsible for selectivity and gating are reasonably conserved among prokaryotes and eukaryotes. Accordingly, the pore structure and extracellular entryway of KcsA are comparable to complimentary regions of eukaryotic K+ channels such as the Shaker K+ channel from Drosophila and vertebrate voltage-gated K+ channels [88].
KcsA is a 160 amino acid polypeptide that oligomerizes in a tetramer to form a highly selective potassium channel [89-92]. It has been cloned, overexpressed, purified to homogeneity, and its stability and electrophysiological behavior have been examined [89-96]. Its small size, high levels of expression, and stability in detergent solutions have made it an excellent candidate for structural and biochemical studies. Recently, Doyle et al. [87] determined the structure of truncated S. lividans KcsA by X-ray crystallography to 3.2 Å resolution. Analysis of the data revealed that four identical subunits create an inverted teepee structure in which a wide vestibule lined with hydrophobic amino acids leads to a narrow pore at the outer end.
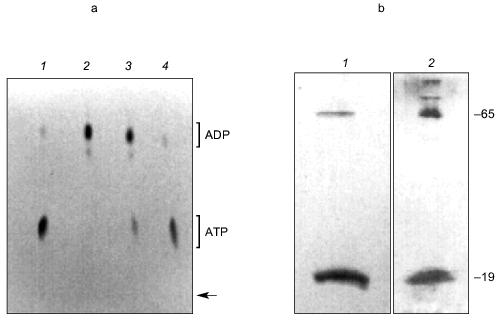
Examination of KcsA channels by Reusch [97] revealed the presence of both polyP and PHB. PolyP was detected in KcsA tetramers, but not in monomers, by its metachromatic reaction on SDS-PAGE gels to o-toluidine blue stain [83]. A band of free polyP was also visible, suggesting that polyP is released when tetramers dissociate. The exopolyphosphatase of Saccharomyces cerevisiae [98] degraded the free polyP, but tetramer-associated polyP was not affected, indicating it was inaccessible to the enzyme. PolyP in KcsA was estimated by enzymatic assay, using E. coli polyphosphate kinase to transfer phosphates from polyP to [14C]ADP yielding [14C]ATP [83, 84] (Fig. 15a), as ~17 µg/mg KcsA or ~15 polyP monomer units per tetramer. Loss of polyP during purification of KcsA could not be estimated.
PHB was detected in both tetramer and monomer species of KcsA by reaction to anti-PHB IgG on Western blots (Fig. 15b), and estimated by chemical assay [82] as 34 µg/mg protein or 28 monomer units PHB per KcsA tetramer, presumably 7 units per subunit. PHB is uncharged and does not itself migrate on electrophoretic gels, hence its co-migration with protein indicates it is tightly bound. Moreover, although PHB is soluble in chloroform, it was not removed from the protein by repeated extraction with warm chloroform, suggesting the bonding is covalent.Fig. 15. a) Autoradiogram showing formation of [14C]ATP from [14C]ADP and polyP in KcsA [72]. Samples were incubated with 50 mM K-Hepes, pH 7.2, 40 mM (NH4)2SO4, 4 mM MgCl2, 0.5 mM [14C]ADP, 2,000 units polyphosphate kinase, at 37°C for 45 min. ADP and ATP (5 mM each) were added as carriers, and ADP and ATP were resolved by thin layer chromatography on PEI cellulose and visualized by UV and autoradiography. Brackets indicate regions of the chromatogram occupied by ADP and ATP standards after development with 1 M LiCl, 1 M HCOOH. 1) 0.1 µg polyP (type 45); 2) as in lane (4) but without polyphosphate kinase; 3) native KcsA; 4) polyP isolated from KcsA as described in [72]. Arrow indicates origin. b) Presence of PHB in KcsA tetramers and monomers: 1) 12% SDS-PAGE gel of partially dissociated KcsA visualized with Coomassie Brilliant Blue stain; 2) Western blot of a similar gel probed with anti-PHB IgG. Second antibody was conjugated to alkaline phosphatase. Samples were heated for 1 min at 70°C before loading. Molecular masses of marker proteins in kD on the right are indicated. c) Analytical isoelectric focusing of KcsA. Focusing was carried out on 0.4 mm gels of 5% acrylamide/bis (33.7:1), 20 mM n-dodecyl-beta-D-maltoside, 5% pH 3-10 ampholytes formed on gel support film. The gel was stained with Coomassie Brilliant Blue R-250, crocein scarlet and destained with 40% methanol, 10% acetic acid. 1) pI standards; 2) unheated KcsA; 3) KcsA heated for 5 min at 90°C. Molecular masses of marker proteins in kD on the right are indicated.
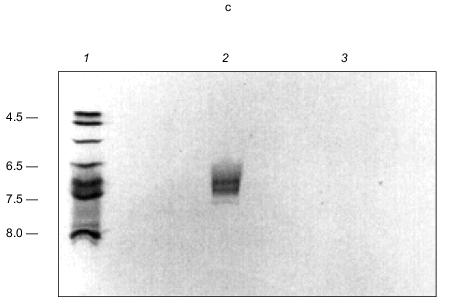
In transferring KcsA to nitrocellulose membranes, it was noted that the tetramer transferred in buffers of pH 8.6 whereas the monomer required buffers of much higher pH (11.3). This signaled a large difference in the isoelectric points (pI) of the subunits and the tetramer. The pI of KcsA subunits could not be determined experimentally because of its insolubility in nonionic detergents, but it was calculated using the ExPAsy Compute pI/Mw tool (http://www.expasy.ch/tools/pi_tool.html) [99, 100] as 10.3. This high pI is consistent with the excess of positively charged residues (19+, 13-), mainly arginines, in the protein. Yet it seems curious that four subunits, each with high positive charge, come together to form a very stable tetramer. The stability of the tetramer is demonstrated by its ability to remain undissociated in nonionic detergent for over a month at room temperature, at least an hour in the ionic detergent, sodium dodecyl sulfate [91, 92], and on exposure to heat up to ~60°C for at least 30 min [92]. Also incongruous is the experimentally determined pI of the KcsA tetramer, which is substantially more acidic, i.e., 6.5-7.5, than that of the subunits (Fig. 15c). These perceived electrostatic anomalies are explained by the presence of the polyanion, polyP, in the tetramer. PolyP can attract and bind together the four basic monomers in a stable tetramer. At the same time, it converts the net charge of the channel interior from positive to negative, and provides an attractive entryway and staircase of negative charges for K+ transport. By drawing the basic residues inward, it reduces the positive charge at the protein surface.
Neither polyP nor PHB are discernible in the electron density maps of KcsA recently reported by Doyle et al. [87]; however, only 60% of the channel residues, comprising the most hydrophobic region (residues 23-119), were imaged. Most of the intracellular entryway (residues 126-158) had been excised and its remaining residues were disordered. Much of the PHB and polyP may have been eliminated by this excision, which included the most highly charged region of the protein (15 of 23 residues, 7+, 8-). Even if still present, both polyP and PHB are polymorphic and hence disordered and difficult to image.
It is postulated that KcsA protein creates an environment in which polyP/PHB is selective for K+. The polyP acts as a voltage sensor and attracts cations to the intracellular entryway. Strategic placement of basic residues of the protein (primarily arginines) attenuates the negative charge density of polyP, transforming its cation selectivity from divalent to monovalent. Discrimination between K+ and Na+ requires the cooperation of all three polymers. PolyP and PHB provide the oxygen ligands that form the binding cavities, but protein architecture defines the distance between ligands of polyP and PHB, and between PHB ligands on neighboring subunits. By attaching a chain of PHB to each subunit, the discriminating ligands are kept too far apart to solvate Na+. Transport of K+ then proceeds by stepwise movement up the polyP ladder.
According to the X-ray structure, there is a ~10 Å cavity lined with hydrophobic amino acids in the center of the channel [28]. Estimates of molecular size based on van der Waal radii of component atoms indicate that the width of a PHB chain is ~ 4 Å and that of polyP ~3 Å. The Pauling radius of K+ is 1.33 Å. This implies that a cavity of ~13.5 Å diameter is needed to accommodate a PHB/polyP/K+ complex, though considerably less space may be required if the methyl groups of PHB are nestled within hydrophobic pockets along the protein wall. Allowing for experimental error, the hydrophobic-lined cavity could contain a PHB/polyP/K+ complex with the polyP chain trailing down to an arginine-rich region of the intracellular entryway. It is also possible the polymers are located entirely in the intracellular portion that was not imaged.
This hypothetical mechanism may not satisfy all experimental data acquired for this channel; certainly, more experimental work is needed to clarify the role(s) of PHB and polyP, and it may be necessary to reevaluate earlier experimental results in the light of this new information.
Evolutionary aspects and concluding remarks. The ubiquity of these simple homopolymers suggests they are fundamental constituents of biological cells and have physiological role(s) fundamental to life. It is likely that both polymers were components of the earliest cells and preceded the development of the RNA world. PolyPs were present in the prebiotic environment. They are formed when phosphate salts are subjected to high temperatures or high pressures; accordingly, they are found in volcanic condensates and near deep-sea thermal vents [2, 101]. Synthesis of PHBs is more complex and probably was a later development, but only acetate and reducing agents are required and both were available in the primordial milieu.
It is intriguing to speculate that the association between polyP and PHB formed early in cell evolution in response to the need for a system to control internal Ca2+, as Ca2+ would tend to precipitate organic phosphates that were essential to the development of RNAs. PolyPs could be employed to sequester divalent cations, but this would also remove Mg2+, a critical cation for many biosynthetic reactions, and would require large internal concentrations of polyP. The association of PHB with polyP makes it possible to specifically sequester Ca2+ and transport it out of the cell. Once positioned across a bilayer, polyP/PHB complexes are versatile structures and it is possible they were used not only to store or expel Ca2+, but also to transfer informational polyphosphates, such as DNA [14]. The complexes also provide a means for rapid import of Ca2+, controlled by potential or concentration gradients, and set the stage for cell signaling later in evolution.
Simpler, more efficient, simpler to produce, easier to assemble, calcium-selective channels than polyP/PHB are difficult to imagine. Every atom of both polymers is critical to the tasks of ion selection and transport. No deletions or substitutions are conceivable that would improve the effectiveness of the complexes. As peptides and proteins evolved, they may have associated with the channels to support and regulate their activity. At first, this association may have been noncovalent, and in time developed into a more secure covalent attachment to PHB. In this view, many of the channels and pumps of prokaryotes and eukaryotes may be supramolecular structures in which polyP, PHB, and protein have joined together for efficient regulation of transmembrane ion transport.
REFERENCES
1.Harold, F. M. (1967) Bacteriol. Rev.,
30, 772-794.
2.Kulaev, I. S. (1979) in The Biochemistry of
Inorganic Polyphosphates (Brookes, R. F., ed.) John Wiley &
Sons, N. Y., pp. 1-248.
3.Kulaev, I. S., and Vagabov, V. M. (1983) Adv.
Microb. Phys., 24, 83-171.
4.Kumble, K. D., and Kornberg, A. (1995) J. Biol.
Chem., 270, 5818-5822.
5.Schröder, H. C., Lorenz, B., Kurz, L., and
Müller, W. E. (1999) Prog. Mol. Subcell. Biol., 23,
45-81.
6.Kornberg, A. (1999) Prog. Mol. Subcell.
Biol., 23, 1-18.
7.Lemoigne, M. (1927) Ann. Inst. Pasteur,
41, 148.
8.Dawes, E. A., and Senior, P. J. (1973) Adv.
Microb. Physiol., 10, 135-206.
9.Wood, H. G., and Clark, J. E. (1988) Ann. Rev.
Biochem., 57, 235-260.
10.Doi, Y. (1990) Microbial Polyesters, VCH,
N. Y.
11.Reusch, R. N. (1989) Proc. Soc. Exp. Biol.
Med., 191, 377-381.
12.Anderson, A. J., and Dawes, E. A. (1990)
Microbiol. Rev., 54, 450-472.
13.Reusch, R. N., Sparrow, A. W., and Gardiner, J.
(1992) Biochim. Biophys. Acta, 1123, 33-40.
14.Reusch,R. N. (1992) FEMS Microbiol. Rev.,
103, 119-130.
15.Seebach, D., Brunner, A., Burger, H. M.,
Schneider, J., and Reusch, R. N. (1994) Eur. J. Biochem.,
224, 317-328.
16.Müller, H. M., and Seebach, D. (1994)
Angew. Chem., 32, 477-502.
17.Reusch, R. N., and Gruhn, A. G. (1997) in 1996
Int. Symp. on Bact. Polyhydroxyalkanoates, NRC Research Press,
Ottawa, pp. 10-19.
18.Corbridge, D. E. C. (1985) Stud. Inorg.
Chem., 6, 170-178.
19.Eisenman, G., and Horn, R. (1983) J. Membr.
Biol., 76, 197-225.
20.Matheja, J., and Degens, E. T. (1971)
Structural Molecular Biology of Phosphates, Gustav Fischer
Verlag, Stuttgart, pp. 78-90.
21.Majling, J., and Hanic, F. (1980) Topics
Phosphorus Chem., 10, 341-502.
22.Hankermeyer, C. R., and Tjeerdema, R. S. (1999)
Rev. Environ. Contam. Toxicol., 159, 1-24.
23.Armand, M. B. (1987) in Polymer Electrolyte
Reviews 1 (MacCallum, J. R., and Vincent, C. A., eds.) Elsevier
Applied Science, N. Y., pp. 1-22.
24.MacCallum, J. R., and Vincent, C. A. (1987) in
Polymer Electrolyte Reviews 1 (MacCallum, J. R., and Vincent, C.
A., eds.) Elsevier Applied Science, N. Y., pp. 23-37.
25.Watanabe, M., and Ogatu, N. (1987) in Polymer
Electrolyte Reviews 1 (MacCallum, J. R., and Vincent, C. A., eds.)
Elsevier Applied Science, N. Y., pp. 39-68.
26.Gray, F. M. (1992)in Solid Polymer
Electrolytes, VCH, N. Y., pp. 1-4.
27.Seebach, D., Bürger, M., and Plattner, D. A.
(1993) Helv. Chim. Acta, 76, 2581-2601.
28. Bürger, H. M., and Seebach, D. (1993)
Helv. Chim. Acta, 76, 2570-2580.
29.Reusch, R. N., and Reusch, W. H. (1993) U. S.
Patent No. 5,266,422.
30.Fritz, M. G., Walde, P., and Seebach, D. (1999)
Macromolecules, 32, 574-580.
31.Seebach, D., and Fritz, M. G. (1999) J. Biol.
Macromol., 25, 217-236.
32. Miller, C. (1983) Physiol. Rev.,
63, 1209-1242.
33. Alvarez, O., Benos, D., and Latorre, R. (1985)
J. Electrophysiol. Tech., 12, 159-177.
34. Reusch, R. N., Huang, R., and Bramble, L. L.
(1995) Biophys. J., 69, 754-766.
35. Plattner, D. A., Brunner, A., Dobler, M.,
Müller, H.-M., Petter, W., Zbinden, P., and Seebach, D. (1993)
Helv. Chim. Acta, 76, 2004-2033.
36.Seebach, D., Bürger, H. M., Müller,
H.-M., Lengweiler, U. D., Beck, A. K., Sykes, K. E., Barker, P. J., and
Barham, P. J. (1994) Helv. Chim. Acta, 77, 1099-1123.
37.Seebach, D., Brunner, A., Burger, H. M., Reusch,
R. N., and Bramble, L. L. (1996) Helv. Chim. Acta, 79,
507-517.
38. Van Zutphen, H., Merola, A. J., Brierley, G. P.,
and Cornwell, D. G. (1972) Arch. Biochem. Biophys., 152,
755-766.
39.Tanaka, J. C., Furman, R. E., and Barchi, R. L.
(1986) in Ion Channel Reconstitution (Miller, C., ed.) Plenum
Press, N. Y., pp. 277-305.
40.Marsh, D., Watts, A., and Knowles, P. F. (1976)
Biochemistry, 15, 3570-3578.
41.Antonov, V. F., Petrov, V. V., Molnar, A. A.,
Predvoditelev, D. A., and Ivanov, A. S. (1980) Nature,
283, 585-586.
42.Cornibert, J., and Marchessault, R. H. (1975)
Macromolecules, 8, 296-305.
43.Seebach, D., Bürger, M., Müller, H.-M.,
Lengweiler, U. D., Beck, A. K., Sykes, K. E., Barker, P. A., and
Barham, P. J. (1994) Helv. Chim. Acta, 77, 1099-1123.
44.Sykes, K. E., McMaster, T. J., Miles, M. J.,
Barker, P. A., Barham, P. J., Seebach, D., Müller, H.-M., and
Lengweiler, U. D. (1995) J. Mat. Sci., 30, 623-627.
45.Fettiplace, R., and Haydon, D. A. (1980)
Physiol. Rev., 60, 510.
46.Seebach, D., Brunner, A., Bachmann, B., Hoffmann,
T., Kühnle, F. N., and Lengweiler, U. D. (1996) Ernst Shering
Research Found., 28, 1-105.
47.Reusch, R. N., and Sadoff, H. L. (1988) Proc.
Natl. Acad. Sci. USA, 85, 4176-4180.
48.Martin, R. B. (1990) Metal Ions Biological
Systems, 26, 1-13.
49.Reusch, R. N., and Sadoff, H. L. (1983) J.
Bacteriol., 156, 778-788.
50.Reusch, R. N., Hiske, T. W., and Sadoff, H. L.
(1986) J. Bacteriol., 168, 553-562.
51.Reusch, R., Hiske, T., Sadoff, H., Harris, R.,
and Beveridge, T. (1987) Can. J. Microbiol., 33,
435-444.
52.Reusch, R. N. (1999) Prog. Mol. Subcell.
Biol., 23, 151-182.
53.Chang, C. F. (1986) J. Bacteriol.,
167, 935-939.
54.Gangola, P., and Rosen, B. P. (1987) J.
Biol.Chem., 26, 12570-12574.
55.Jones, H. E., Holland, I. B., Baker, H. L., and
Campbell, A. K. (1999) Cell Calcium, 25, 265-274.
56.Straley, S. C., Plano, G. V., Skrzypek, E.,
Haddix, P. L., and Fields, K. A. (1993) Mol. Microbiol.,
6, 1005-1010.
57.Tisa, L. S., Olivera, B. M., and Adler, J. (1993)
J. Bacteriol., 175, 1235-1238.
58.Tisa, L. S., and Adler, J. (1994) Proc. Natl.
Acad. Sci. USA, 89, 11804-11808.
59.Norris, V., Grant, S., Freestone, P., Canvin, J.,
Sheikh, F. N., Toth, I., Trinei, M., Modha, K., and Norman, R. I.
(1996) J. Bacteriol., 178, 3677-3682.
60.Lynn, A. R., and Rosen, B. P. (1987) in Ion
Transport in Procaryotes (Rosen, B. P., and Silver, S., eds.)
Academic Press, N. Y., pp. 181-201.
61.Ivey, D. M., Guffanti, A. A., Zemsky, J., Pinner,
E., Karpel, R., Padan, E., Schuldiner, S., and Krulwich, T. A. (1993)
J. Biol. Chem., 268, 11296-11303.
62.Matsushita, T., Hirata, H., and Kusaka, I. (1989)
Ann. N. Y. Acad. Sci., 560, 276-278.
63.Saimi, Y., Loukin, S. H., Zhou, X. L., Martinac,
B., and Kung, C. (1999) Meth. Enzymol., 294, 507-524.
64.Das, S., Lengweiler, U. D., Seebach, D., and
Reusch, R. N. (1997) Proc. Natl. Acad. Sci. USA, 94,
9075-9079.
65.Lengweiler, U. D., Fritz, M.G., and Seebach, D.
(1996) Helv. Chim. Acta, 79, 670-701.
66.Castuma, C., Huang, R., Kornberg, A., and Reusch,
R. N. (1995) J. Biol. Chem., 270, 12980-12983.
67.Hille, B. (1992) Ionic Channels of Excitable
Membranes, Sinauer Associates Inc, Sunderland, MA.
68.Das, S., and Reusch, R. N. (1999) J. Memb.
Biol., 170, 135-145.
69.Abel, E., Meadows, E. S., Suzuki, I., Jin, T.,
and Gokel, G. W. (1997) Chem. Commun., 1145-1146.
70.Akita, S., Elaga, Y., Miyski, Y., and Fujita, H.
(1976) Macromol., 9, 774-780.
71. Tzeng, C. M., and Kornberg, A. (1998) Mol.
Microbiol., 29, 381-382.
72. Kornberg, A., Kornberg, S., and Simms, E. (1956)
Biochim. Biophys. Acta, 20, 215-227.
73.Kulaev, I. S., Andreeva, N. A., Lichko, L. P.,
and Kulakovskaya, T. V. (1997) Microbiol. Res., 152,
221-226.
74.Reusch, R. N., Huang, R., and Kosk-Kosicka, D.
(1997) FEBS Lett., 412, 592-596.
75.Dunham, E. T., and Glynn, I. M. (1961) J.
Physiol., 156, 274-293.
76.Schatzmann, H. J. (1966) Experientia,
22, 364-368.
77.Penniston, J. T. (1983) in Calcium and Cell
Function, Academic Press, N. Y., pp. 99-149.
78.Niggli, V., Penniston, J., and Carafoli, E.
(1979) J. Biol. Chem., 254, 9955-9958.
79.Kosk-Kosicka, D., and Inesi, G. (1985) FEBS
Lett., 189, 67-81.
80.Kosk-Kosicka, D., Scaillet, S., and Inesi, G.
(1986) J. Biol. Chem., 261, 3333-3338.
81.Karr, D. B., Waters, J. K., and Emerich, D. W.
(1983) Appl. Environ. Microbiol., 46, 1339-1344.
82. Huang, R., and Reusch, R. N. (1996) J. Biol.
Chem., 271, 22196-22201.
83.Griffin, J. B., Davidian, N. M., and Penniall, R.
(1965) J. Biol. Chem., 240, 4427-4429.
84. Ahn, K., and Kornberg, A. (1990) J. Biol.
Chem., 265, 11734-11739.
85.Crooke, E., Akiyama, M., Rao, N. N., and
Kornberg, A. (1994) J. Biol. Chem., 269, 6290-6295.
86.Chiesi, M., Zurini, M., and Carafoli, E. (1984)
Biochemistry, 23, 2595-2600.
87. Doyle, A. D., Cabral, J. M., Pfuetzner, R. A.,
Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T., and MacKinnon, R.
(1998) Science, 280, 69-77.
88. MacKinnon, R., and Doyle, D. A. (1997) Nature
Struct. Biol., 4, 877-879.
89.Schrempf, H., Schmidt, O., Kümmerlen, R.,
Sinnah, D., Müller, M., Betzler, T., Steinkamp, T., and Wagner, R.
(1995) EMBO J., 14, 5170-5178.
90.Heginbotham, L., Odessey, E., and Miller, C.
(1997) Biochemistry, 36, 10335-10342.
91.Cordes, D. M., and Perozo, E. (1997)
Biochemistry, 35, 10343-10352.
92.Li, H. L., Sui, H. X., Ghanshani, S., Lee, S.,
Walian, P. J., Wu, C. L., Chandy, K. G., and Jap, B. K. (1998) J.
Mol. Biol., 282, 211-216.
93.Heginbotham, L., Kolmakova-Partensky, L., and
Miller, C. (1998) J. Gen. Physiol., 111, 741-749.
94.MacKinnon, R. S., Cohen, L., Kuo, A., Lee, A.,
and Chait, B. T. (1998) Science, 280, 106-109.
95.Cuello, L. G., Romero, J. G., Cortes, D. M., and
Perozo, E. (1998) Biochemistry, 37, 3229-3236.
96.Perozo, E., Cortes, D. M., and Cuello, L. G.
(1999) Science, 285, 73-78.
97.Reusch, R. N. (1999) Biochemistry, in
press.
98.Wurst, H., and Kornberg, A. (1994) J. Biol.
Chem., 269, 10996-11001.
99.Bjellqvist, B., Hughes, G. J., Pasquali, Ch.,
Paquet, N., Ravier, F., Sanchez, J.-Ch., Frutiger, S., and
Hochstrasser, D. F. (1993) Electrophoresis, 14,
1023-1031.
100.Wilkins, M. R., Gasteiger, E., Bairoch, A.,
Sanchez, J.-C., Williams, K. L., Appel, R. D., and Hochstrasser, D. F.
(1998) 2-D Proteome Analysis Protocols (Link, A. J., ed.) Humana
Press, N. Y.
101.Yamagata, Y., Watanabe, H., Saitoh, M., and
Namba, T. (1991) Nature, 352, 516-519.