REVIEW: Polyphosphate in Bone
H. C. Schröder*, L. Kurz, W. E. G. Müller, and B. Lorenz
Institut für Physiologische Chemie, Universität, Duesbergweg 6, D-55099 Mainz, Germany; fax: +49-6131-3925243; E-mail: hschroed@mail.uni-mainz.de* To whom correspondence should be addressed.
Received December 20, 1999
Human bone-forming osteoblasts are an excellent model to investigate the multiple functions of inorganic polyphosphates (polyP) for the following reasons: 1) they contain relatively high amounts of polyP and polyP-dependent enzymes; 2) they allow the study of both general and specific functions of these polymers, and 3) medically relevant results can be expected from these studies.
KEY WORDS: polyphosphate, pyrophosphate, bisphosphonates, exopolyphosphatase, alkaline phosphatase, pyrophosphatase, mineralization, bone, osteoblasts, SaOS-2 cells
Abbreviations: 1,25(OH)2D3) 1alpha,25-dihydroxyvitamin D3; EGF) epidermal growth factor; Pi) orthophosphate; PPi) inorganic pyrophosphate; polyPn) inorganic polyphosphate with chain length of n residues; PBMC) peripheral blood mononuclear cells.
Inorganic linear polyphosphates (polyP) are polymers of orthophosphate
(Pi) residues linked by energy-rich phosphoanhydride bonds.
They may range in size up to several hundred Pi residues [1]. In the past these polymers have been studied
mainly in bacteria and yeast (for reviews, see [2,
3]), but they are also present in higher plants and
animals [4-6]. More recently,
polyP was found also in human cells and tissues [7-10].
The concentrations measured for “soluble” long-chain polyP (mainly polymers of 10-50 Pi residues) and “insoluble” long-chain polyP (mainly polymers of .50 Pi residues) in different cells and extracellular fluids from human are summarized in Table 1. Extraction of “soluble” long-chain polyP and “insoluble” long-chain polyP was performed as described by Clark et al. [11, 12]. In general, the concentration of soluble long-chain polyP is higher than that of insoluble long-chain polyP. PolyP concentrations in the same range as those shown in Table 1 have been detected in different mammalian cell lines (total polyP, 25 to 120 µM; expressed in Pi residues) [8]. PolyP is also present extracellularly in human blood plasma (Table 1) and serum [9]. However, the concentration of insoluble long-chain polyP in the cell-free blood fractions is much lower than that in human peripheral blood mononuclear cells (PBMC) and erythrocytes. It is not yet known whether the plasma polyP is synthesized within this body fluid or is due to the lysis of erythrocytes, as suggested by its smaller size. The highest amounts of polyP in humans were found in bone-forming osteoblasts [10] (Table 1).
Table 1. Concentration of long-chain polyP
in human cells and blood plasma*

*Extraction of “soluble” long-chain polyP and
“insoluble” long-chain polyP was performed as described by
Clark et al. [11]. Concentrations of
“soluble” (10-50 Pi residues) and
“insoluble” long-chain polyP (>50 Pi residues)
are presented (data from [10]).
**Values are the means (± SD) from three independent
determinations. Concentrations are based on the volume of the pelleted
cells.
***Unstimulated osteoblasts.
PolyP is found in nearly all cellular compartments. It is enriched in the nucleus [8, 13, 14], mitochondria [1], lysosomes [7], and plasma membrane [15]. The nuclear polyP mainly consists of long polyP chains [14].
In many mammalian cell lines, often two or more populations of polyP with a narrow range of chain lengths are found, e.g., two polyP classes with ~150 Pi residues and 25 to 45 Pi residues exist in human leukemic HL-60 cells [14], and with 500 to 800 Pi residues and 5 to 15 Pi residues in NIH3T3, PC12, and Jurkat cells [8]. PolyP shows a high turnover rate [8]. Metabolic labeling studies revealed that 32Pi taken up by HL-60 cells appears first in the low-molecular-weight polyP fraction (25 to 45 residues) before it is incorporated into high-molecular-weight polyP (~150 residues) [14].
Many functions have been proposed for polyP (for a review, see [16]). These functions may be different in different compartments of the cell. PolyP is assumed to act as: 1) storage molecule for energy-rich phosphate; 2) chelator for Ca2+ or other divalent cations; 3) counterion for basic amino acids; 4) regulator of intracellular level of adenylate nucleotides; 5) component of complexes with poly-beta-hydroxybutyrate and Ca2+ in cell membranes; 6) donor of Pi for certain sugar kinases, and 7) modulator of cellular stress response.
OSTEOBLAST MODEL
Osteoblasts are bone-forming cells which are derived from pluripotent mesenchymal stem cells. They synthesize and secrete most of the molecules of the bone matrix, e.g., type I collagen and proteoglycans. They possess a high content of alkaline phosphatase (a marker enzyme for osteoblasts), which is involved in mineralization.
Osteoblast differentiation can be subdivided into the following phases that are associated with the sequential expression of genes involved in the biosynthesis, organization, and mineralization of the bone extracellular matrix (reviewed in [17], Fig. 1): proliferation phase, matrix maturation phase, mineralization phase, and apoptosis phase.
Proliferation phase. In this phase, genes required for proliferation (e.g., c-fos) and the progress of the cell cycle (e.g., cyclins, histones) as well as the genes associated with the regulation of the biosynthesis of the extracellular matrix (e.g., type I collagen, transforming growth factor beta) or encoding cell adhesion proteins (e.g., fibronectin) are expressed.Fig. 1. Osteoblast development. Time-dependent expression of growth- and differentiation-dependent proteins. H4, histone H4; Col, collagen; Fn, fibronectin; Op, osteopontin; AP, alkaline phosphatase; MGP, matrix Gla protein; Oc, osteocalcin; PHB, polyhydroxybutyrate; HA, hydroxyapatite; Apopt, apoptosis.
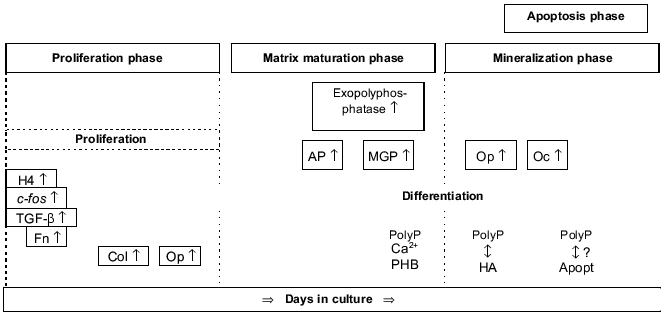
Matrix maturation phase. In this phase the expression of genes involved in maturation and organization of the bone extracellular matrix (e.g., alkaline phosphatase) is upregulated, allowing mineralization.
Mineralization phase. In this phase, the expression of genes associated with the ordered deposition of hydroxyapatite (osteopontin and osteocalcin) occurs.
Apoptosis phase. An increase in type I collagen and collagenase gene expression, apoptotic cell death, and compensatory proliferative activity are observed.
The osteoblast model is a suitable cell system to study the most likely multiple functions of polyP; using this model it is possible to investigate: differential gene expression of polyP metabolic enzymes; effect of modulators on these enzymes like vitamin D, corticosteroids, or cytokines; possible function of polyP in signal transduction and membrane transport; extracellular function(s) of polyP (involvement in the mineralization process); and pathophysiology of polyP metabolism (pathogenesis of bone diseases).
MODULATION OF PolyP METABOLISM IN OSTEOBLASTS
It is known that osteoblasts respond to 1alpha,25-dihydroxyvitamin D3 (1,25(OH)2D3), glucocorticoids, and growth factors. They possess high levels of alkaline phosphatase, which participates in mineralization. 1,25(OH)2D3 is a stimulator of bone resorption; in mature osteoblasts it increases the expression of genes that are associated with the mineralization process, e.g., osteocalcin, resulting in an enhanced mineralization. On the other hand, expression of osteocalcin is suppressed by dexamethasone. However, combined treatment of osteoblasts with 1,25(OH)2D3 and dexamethasone has been shown to result in a synergistic increase in transcriptional activity [17].
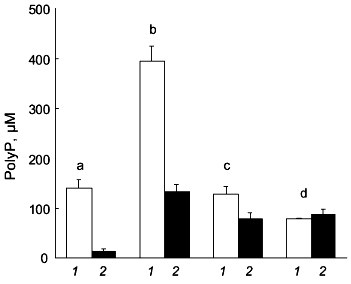
We found that polyP metabolism in human osteoblasts is modulated by stimulators of osteoblast proliferation and differentiation [10]. Combined treatment of the cells with dexamethasone, beta-glycerophosphate, epidermal growth factor (EGF), and ascorbic acid results in a strong decrease in polyP content. This decrease in polyP content is mainly caused by a decrease in the amount of soluble long-chain polyP (Fig. 2). The amount of this polyP fraction but not the amount of insoluble long-chain polyP, further decreases after additional treatment of the cells with 1,25(OH)2D3 (Fig. 2). The decrease in polyP content during treatment with dexamethasone, beta-glycerophosphate, EGF, and ascorbic acid is accompanied by a decrease in exopolyphosphatase activity [10]. However, additional treatment with 1,25(OH)2D3 results in a significant increase in the enzyme activity [10].
Similar effects of dexamethasone and 1,25(OH)2D3 were observed for osteoblast inorganic pyrophosphatase [10], alkaline phosphatase [10], and ectonucleoside triphosphate pyrophosphatase activity [18, 19]. The activities of inorganic pyrophosphatase and alkaline phosphatase are markedly reduced after combined treatment of the cells with dexamethasone, beta-glycerophosphate, EGF, and ascorbic acid, paralleling the decrease in exopolyphosphatase activity. Again, additional treatment with 1,25(OH)2D3 causes an increase in enzyme activity [10].Fig. 2. Changes in polyP content of human osteoblasts after treatment with stimulators of osteoblast proliferation and differentiation. The amount of soluble long-chain polyP (1) and insoluble long-chain polyP (2) was determined in unstimulated osteoblasts (b) and in osteoblasts subjected to combined treatment with dexamethasone, beta-glycerophosphate, EGF, and ascorbic acid for 6 days (c), or to combined treatment with the same additives for 3 days in the absence and then for 3 days in the presence of 1,25(OH)2D3 (d). For comparison, the polyP content of gingival fibroblasts (a) is shown. Results are means ± SD of three independent experiments.
The ectonucleoside triphosphate pyrophosphatase, which is like alkaline phosphatase located on the cell surface, is assumed to serve in the generation of extracellular PPi in bone [20]. The activity of this enzyme is modulated by dexamethasone and 1,25(OH)2D3. Dexamethasone causes a decrease in enzyme activity, while 1,25(OH)2D3 and transforming growth factor-beta (TGF-beta) induce an increase in activity [19]. On the other hand, TGF-beta1 and TGF-beta2 have been shown to decrease alkaline phosphatase activity. The increase in ectonucleoside triphosphate pyrophosphatase activity and decrease in alkaline phosphatase activity induced by TGF-beta may cause an increase in extracellular PPi and a decrease in extracellular Pi concentration, resulting in a lower mineralization in bone tissue.
POTENTIAL ROLE OF PolyP IN MINERALIZATION
In bone, extracellular PPi has been identified as a calcification inhibitor. PPi has been shown to inhibit both the formation [21, 22] and dissolution of calcium phosphate crystals [23], and might therefore act as physiological regulator of calcification and decalcification [23]. It is assumed that such inhibitors must be removed from the site of mineralization before calcification can occur [22]. PPi is hydrolyzed by alkaline phosphatase and inorganic pyrophosphatase activities. These enzymes can locally degrade PPi in bone, thus allowing formation of hydroxyapatite crystals. The increase in both enzyme activities induced by 1,25(OH)2D3 would be expected to result in an increased production of Pi and a decrease in PPi concentration, thus modulating mineralization during bone formation. Indeed, bone mineralization has been found to be associated with changes in pyrophosphatase activity [24].
Long polyP chains inhibit hydroxyapatite precipitation more strongly than PPi [22]. The efficiency of polyP11 and polyP27 to suppress calcium phosphate precipitation is ~10-fold higher than that of PPi and is ~103-fold higher than that of ATP [22]. Our studies showed that polyP is present in bone tissue [10]. Therefore, it is reasonable to assume that polyP may act as a further regulator of calcification and decalcification in bone tissue, in addition to PPi.
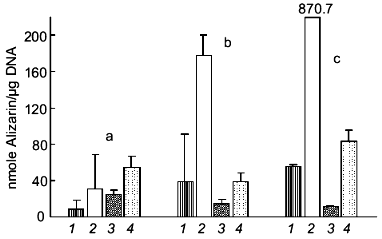
Recently, we demonstrated that treatment of cultures of human osteosarcoma cell line SaOS-2 with polyP causes a strong suppression of mineralization induced by beta-glycerophosphate (Kurz et al., to be published). As shown in Fig. 3, cells grown in the presence of ascorbic acid and in the absence of beta-glycerophosphate showed a small but progressive increase in calcium deposition as measured by Alizarin Red S staining. Addition of beta-glycerophosphate caused a strong enhancement of the mineralization process. This increase was abolished in the presence of polyP, indicating that this polymer may act as a mineralization inhibitor.
Fig. 3. Effect of polyP on mineralization of SaOS-2 cells. Human osteosarcoma SaOS-2 cells were grown to confluency (day 0) and then incubated for 4 (a), 8 (b), and 10 days (c) in the presence of 0.25 mM ascorbic acid (1), 0.25 mM ascorbic acid and 10 mM beta-glycerophosphate (2), 0.25 mM ascorbic acid and polyP35 (1 mM, expressed as Pi) (3), and 0.25 mM ascorbic acid, 10 mM beta-glycerophosphate, and polyP (1 mM Pi) (4). Calcium deposition was quantified by Alizarin Red S staining. Means ± SD (n = 3).
PolyP-DEPENDENT ENZYMES IN OSTEOBLASTS
The presence of polyphosphatases, which locally destroy polyP (an inhibitor of calcium phosphate precipitation), might be necessary for bone tissue to mineralize, in addition to PPi-degrading pyrophosphatase and alkaline phosphatase activities [10]. In addition, polyphosphatases may be involved in generation of Pi required for formation of hydroxyapatite crystals.
Exopolyphosphatase. PolyP degradation is known to be catalyzed by several endo- and exopolyphosphatases, which differ in substrate specificity and product [5, 25-30]. Exopolyphosphatase activity is also present in human osteoblasts [10]. The specific activity of the enzyme in unstimulated osteoblasts is much higher than that in other mammalian cells and tissues tested (Table 2). Exopolyphosphatase activity has also been found extracellularly, e.g., in synovial fluid (see Fig. 4) as well as in human blood plasma and serum [9].
Table 2. Exopolyphosphatase activities in
different cells, tissues, and extracellular fluids from mammals*

*Data are from [9, 10, 14].
**Mean values from three independent experiments; polyP35 was used as substrate at 37°C.
***Adult animals.
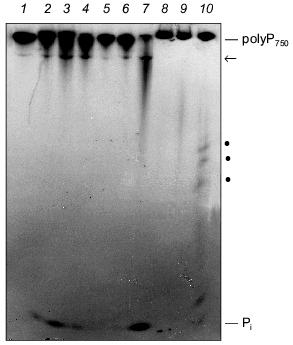
The degradation of long-chain polyP by extracts from SaOS-2 cells and human synovial fluid is shown in Fig. 4. Both the membrane fraction (Fig. 4, lanes 2-4) and cytosolic fraction (Fig. 4, lanes 5-7) from SaOS-2 cells contain exopolyphosphatase activity cleaving long-chain polyP750 under formation of Pi. In addition, a further high-molecular-weight polyP band became visible during incubation, possibly due to an endopolyphosphatase activity (see below). Exo- and endopolyphosphatase activity is also present extracellularly in synovial fluid (Fig. 4, lanes 8 and 9).Fig. 4. Degradation of long-chain polyP by cell extracts from human SaOS-2 osteosarcoma cell line and human synovial fluid. The substrate, [32P]polyP750 (1), was synthesized by recombinant E. coli polyphosphate kinase (a kind gift from Prof. A. Kornberg). The reaction products obtained after incubation of the membrane fraction (lanes 2-4) and the cytosolic fraction (lanes 5-7) from SaOS-2 cells or synovial fluid from human (lanes 8-10) for 2 h (lanes 2, 5, and 8), 4 h (lanes 3, 6, and 9), and 12 h (lanes 4, 7, and 10) were analyzed by electrophoresis on urea/polyacrylamide gel followed by autoradiography. Pi, orthophosphate; arrow and dots, polyP of intermediate chain lengths.
Moreover, we found that in non-proliferating (confluent) SaOS-2 cells higher levels of exopolyphosphatase activity were present than in proliferating cells. More than 50% of the exopolyphosphatase activity in SaOS-2 cells is membrane-bound (Kurz et al., to be published).
Endopolyphosphatase. Endopolyphosphatase activities cleaving long-chain polyP have been identified in a variety of eukaryotic organisms including mammals [30]. The enzyme catalyzes the degradation of long-chain polyP with formation of shorter chains up to a chain length of about 60 residues; tripolyphosphate is produced in a ratio of polyP>=60 to polyP3 of about 3 (in terms of polymer concentration) [30]. Preliminary results indicate that this enzyme is also present in bone-forming osteoblasts (see Fig. 4).
Alkaline phosphatase. Alkaline phosphatase is a well-known enzyme which is widely used in clinical diagnosis of various diseases including bone disorders. This enzyme cleaves phosphate groups from monophosphate ester substrates; in addition, it exhibits phosphotransferase and protein phosphatase activity [32]. In human, four isoforms of alkaline phosphatase exist: intestinal, placental, placenta-like (germ cell type) and tissue-nonspecific [33]. The tissue-nonspecific isoform of alkaline phosphatase is present in bone, liver and kidney; it differs from the other forms by posttranslational modification [34, 35].
The physiological function of alkaline phosphatase is still uncertain. It is assumed that the enzyme is involved in bone mineralization [36], dissolution of calcium pyrophosphate crystals [37, 38], phosphate and calcium transport [39, 40], and intestinal phosphate absorption [41]. The natural substrates of the enzyme include PPi, beta-glycerophosphate, phosphoethanolamine, and pyridoxal-5´-phosphate.
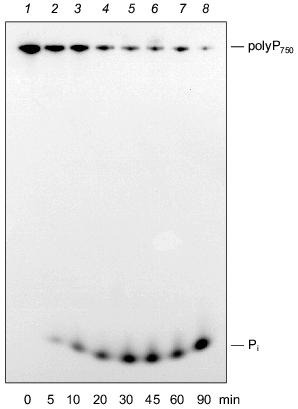
We could demonstrate that the intestinal isoform of alkaline phosphatase is able to degrade polyP molecules with a wide range of chain lengths, in addition to PPi [42]. As shown in Fig. 5, alkaline phosphatase from calf intestine cleaves polyP750 with formation of Pi, apparently by a processive mechanisms. No formation of intermediate chain lengths was found in the course of the reaction. The pH optimum of the enzyme for polyP hydrolysis is in the alkaline range like that for the synthetic substrate, p-nitrophenylphosphate. The enzyme is also active in the absence of divalent cations, in contrast to other exopolyphophatases [16, 42], except for exopolyphosphatase I from Tethya lyncurium [5].
The placenta-type and tissue nonspecific isoenzymes of mammalian alkaline phosphatase were found to be unable to degrade polyP under the tested conditions [42]. Therefore, exopolyphosphatase activity in osteoblasts [10, 43] cannot be due to tissue nonspecific alkaline phosphatase activity present in these cells. However, it cannot be excluded that the assay conditions used were not appropriate to allow polyP hydrolysis by this isoform.Fig. 5. Degradation of long-chain polyP by purified alkaline phosphatase (calf intestine). Reaction products obtained after incubation of [32P]polyP750 in 50 mM Tris-HCl buffer (pH 7.8) at 22°C for different time periods (0 (1), 5 (2), 10 (3), 20 (4), 30 (5), 45 (6), 60 (7), 90 min (8)) were analyzed on urea/polyacrylamide gel.
EFFECT OF BISPHOSPHONATES USED IN THERAPY OF BONE DISEASES ON
PolyP METABOLISM
Bisphosphonates are synthetic analogs of PPi containing a P-C-P bond instead of P-O-P bond [44]. These molecules are stable under physiological conditions and cannot be degraded by pyrophosphatase, alkaline phosphatase or exopolyphosphatase. Bisphosphonates are effective inhibitors of bone resorption. They are clinically used in the treatment of disorders characterized by excessive bone loss, including osteoporosis, Paget's disease, tumor-induced hypercalcemia, and metastatic bone disease [45]. Bisphosphonates bind to hydroxyapatite crystals and inhibit, like PPi and polyP, both precipitation and dissolution of calcium phosphate [46-48].
We found that osteoblast exopolyphosphatase activity is inhibited by the bisphosphonates, etidronate and, to a lesser extent, clodronate and pamidronate [10]. Etidronate inhibits the degradation of both long and short polyP chains by the exopolyphosphatase [10]. These results indicate that polyP metabolism in bone tissue may be a further target for these drugs.
However, it should be noted that more than one mechanism is most likely involved in the mode of action of bisphosphonates. These drugs inhibit the activity, recruitment, and adhesion of the bone-resorbing osteoclasts and reduce osteoclast lifespan [49].
PolyP IN DENTISTRY
PolyP is often used as a component of toothpaste. It has been shown to possess anticaries activity [50]. This effect of polyP can be explained by two mechanisms: polyP can reduce both acid solubility [23, 51, 52] and mineralization of hydroxyapatite [53]. Topical administration of bisphosphonates decreases formation of dental calculus. Bisphosphonates were also found to inhibit experimental periodontitis characterized by excessive bone resorption in rats and monkeys [54-56].
CONCLUSION AND FUTURE ASPECTS
Our studies suggest that besides PPi, polyP is involved in the mineralization process in bone tissue. This assumption is supported by the following findings: 1) in osteoblasts higher concentrations of polyP exist than in other mammalian cells [10]; 2) long-chain polyP molecules inhibit hydroxyapatite precipitation in vitro stronger than PPi [22]; 3) polyP also affects mineralization in cultured cells; 4) osteoblasts possess comparably high polyP-degrading exopolyphosphatase activity [10]; 5) this enzyme can eliminate the mineralization inhibitor polyP allowing mineralization to occur. Moreover, Pi required for calcium precipitation is generated during the enzyme reaction; 6) in osteoblasts, exopolyphosphatase activity and polyP content respond to modulators of osteoblast growth and function [10]; and 7) exopolyphosphatase is inhibited by bisphosphonates [10], indicating that polyP metabolism in bone tissue represents a further target for these drugs.
However, there are a number of questions, which should be addressed in future studies, using the osteoblast model. These studies should include more detailled investigations of the effects of modulators of osteoblast proliferation/differentiation on polyP metabolism. In particular, it might be interesting to investigate both the independent effects and the combined effects of modulators. For example, it is known that 1,25(OH)2D3 increases the expression of the osteocalcin gene, whereas dexamethasone suppresses the expression of this gene. However, the combination of both agents causes a synergistic effect.
During osteoblast development, a sequential expression of genes occurs, possibly including genes for polyP-dependent enzymes. Therefore, the effect of some modulators on polyP metabolism could depend on the specific phase of osteoblast differentiation. Osteoblasts may also represent a suitable model to investigate the role of polyP in apoptosis due to their high polyP content [14]. Apoptotic osteoblasts are observed in heavily mineralized cultures [57].
Further studies have to clarify the intracellular localization of polyP and polyP-dependent enzymes in osteoblasts. It is not yet known whether polyP molecules are present in a free state or associated with some other cellular components in these cells.
One important question concerns the mechanism of formation of extracellular polyP in bone tissue. PolyP might be released into the extracellular space by osteoblasts or synthesized extracellularly. It might be interesting to investigate the effect of polyP on dissolution of the organic bone matrix (polyP is an inhibitor of cathepsin) and the effect of polyP binding to collagen fibrils on the mineralization process. Collagenases and cathepsin are the most important catabolic enzymes of the organic bone matrix; cathepsin is inhibited by polyP [58].Furthermore, polyP might influence the signal transduction processes in osteoblasts. Thus, it is interesting to investigate the role of polyP in regulation of intracellular free calcium concentration and intracellular pH in osteoblasts. PolyP has been shown to act as a modulator of enzymes involved in signal transduction; e.g., polyP is able to stimulate the rate of formation of the ras protein-GTP complexes (regeneration of the GTP form from the GDP form) [59]. PolyP is an inhibitor of the cAMP phosphodiesterase [60] and phospholipase A2 [61].
This work was supported by a grant from the European Community (INCO-Copernicus program ERB IC 15-CT98-019).
REFERENCES
1.Kornberg, A. (1995) J. Bacteriol.,
177, 491-496.
2.Kulaev, I. S. (1979) Biochemistry of Inorganic
Polyphosphates, J. Wiley & Sons, N. Y.
3.Wood, H. G., and Clark, J. E. (1985) Ann. Rev.
Biochem., 57, 235-260.
4.Offenbacher, S., and Kline, E. (1954) Arch.
Biochem. Biophys., 231, 114-123.
5.Lorenz, B., Batel, R., Bachinski, N., Müller,
W. E. G., and Schröder, H. C. (1995) Biochim. Biophys.
Acta, 1245, 17-28.
6.Niemeyer, R. (1999) in Inorganic Polyphosphates
- Biochemistry, Biology, Biotechnology (Schröder, H. C., and
Müller, W. E. G., eds.) Progr. Mol. Subcell. Biol., Vol.
23, Springer-Verlag, Berlin-Heidelberg-New York, pp. 83-100.
7.Pisoni, R. L., and Lindley, E. R. (1992) J.
Biol. Chem., 267, 3626-3631.
8.Kumble, K. D., and Kornberg, A. (1995) J. Biol.
Chem., 270, 5818-5822.
9.Lorenz, B., Leuck, J., Kohl, D., Müller, W. E.
G., and Schröder, H. C. (1997) J. Acq. Immune Defic. Synd.
Human Retrovir., 14, 110-118.
10.Leyhausen, G., Lorenz, B., Zhu, H., Geurtsen, W.,
Bohnensack, R., Müller, W. E. G., and Schröder, H. C. (1998)
J. Bone Miner. Res., 13, 803-812.
11.Clark, J. E., Beegen, H., and Wood, H. G. (1986)
J. Bacteriol., 168, 1212-1219.
12.Lorenz, B., and Schröder, H. C. (1999) in
Inorganic Polyphosphates - Biochemistry, Biology, Biotechnology
(Schröder, H. C., and Müller, W. E. G., eds.) Progr. Mol.
Subcell. Biol., Vol. 23, Springer-Verlag, Berlin-Heidelberg-New
York, pp. 217-236.
13.Penniall, R., and Griffin, J. B. (1984)
Biosci. Rep., 4, 957-962.
14.Lorenz, B., Munkner, J., Oliveira, M. P.,
Kuusksalu, A., Leitao, J. M., Müller, W. E. G., and Schröder,
H. C. (1997) Biochim. Biophys. Acta, 1335, 51-60.
15.Reusch, R. N. (1989) Proc. Soc. Exp. Biol.
Med., 191, 377-381.
16.Schröder, H. C., Lorenz, B., Kurz, L., and
Müller, W. E. G. (1999) in Inorganic Polyphosphates -
Biochemistry, Biology, Biotechnology (Schröder, H. C., and
Müller, W. E. G., eds.) Progr. Mol. Subcell. Biol., Vol.
23, Springer-Verlag, Berlin-Heidelberg-New York, pp. 45-81.
17.Stein, G. S., Lian, J. B., Stein, J. L., van
Wijnen, A. J., and Montecino, M. (1996) Physiol. Rev.,
76, 593-629.
18.Oyajobi, B. O., Caswell, A. M., and Russell, R.
G. (1994) J. Bone Miner. Res., 9, 99-109.
19.Oyajobi, B. O., Russell, R. G., and Caswell, A.
M. (1994) J. Bone Miner. Res., 9, 1259-1266.
20.Caswell, A. M., and Russell, R. G. (1988)
Biochim. Biophys. Acta, 966, 310-317.
21.Fleisch, H., and Bisaz, S. (1962) Am. J.
Physiol., 203, 671-675.
22.Fleisch, H., and Neuman, W. F. (1961) Am. J.
Physiol., 200, 1296-1300.
23.Fleisch, H., Russell, R., and Straumann, R.
(1966) Nature, 212, 901-903.
24.Meyer, J. L., and Reddi, A. H. (1985) Arch.
Biochem. Biophys., 242, 532-539.
25.Akiyama, M., Crooke, E., and Kornberg, A. (1993)
J. Biol. Chem., 268, 633-639.
26.Andreeva, N. A., and Okorokov, L. A. (1993)
Yeast, 9, 127-139.
27.Wurst, H., and Kornberg, A. (1994) J. Biol.
Chem., 269, 10996-11001.
28.Kowalczyk, T. H., and Phillips, N. F. (1993)
Anal. Biochem., 212, 194-205.
29.Lorenz, B., Müller, W. E. G., Kulaev, I. S.,
and Schröder, H. C. (1994) J. Biol. Chem., 269,
22198-22204.
30.Kumble, K. D., and Kornberg, A. (1996) J.
Biol. Chem., 271, 27146-27151.
31.Nakamura, T., Nakamura, K., and Stinson, R.
(1988) Arch. Biochem. Biophys., 265, 190-196.
32.McCarthy, A. D., Cortizo, A. M., Gime'nez Segura,
G., Bruzzone, L., and Etcheverry, S. B. (1998) Mol. Cell.
Biochem., 181, 63-69.
33.Trowsdale, J., Martin, D., Bicknell, D., and
Campbell, I. (1990) Biochem. Soc. Trans., 18,
178-180.
34.Price, C. P. (1993) Ann. Clin. Biochem.,
30, 355-372.
35.Miura, M., Sakagishi, Y., Hata, K., and Komoda,
T. (1994) Ann. Clin. Biochem., 31, 25-30.
36.Tenenbaum, H. C., Torontali, M., and Sukhu, B.
(1992) Bone, 13, 249-255.
37.Caswell, A. M., All, S. Y., and Russell, R. G.
(1987) Biochim. Biophys. Acta, 924, 276-283.
38.Shinozaki, T., Xu, Y., Cruz, T. F., and Pritzker,
K. P. (1995) J. Rheumatol., 22, 117-123.
39.Moog, F., and Glazier, H. S. (1972) Comp.
Biochem. Physiol., A42, 321-336.
40.PetitClerc, C., and Plante, G. E. (1981) Can.
J. Physiol. Pharmacol., 59, 311-323.
41.Roubaty, C., and Portmann, P. (1988) Pflugers
Arch., 412, 482-490.
42.Lorenz, B., and Schröder, H. C. (2000)
submitted.
43.Lorenz, B., Munkner, J., Oliveira, M. P., Leitao,
J. M., Müller, W. E. G., and Schröder, H. C. (1997) Anal.
Biochem., 246, 176-184.
44.Fleisch, H. (1991) Drugs, 42,
919-944.
45.Fleisch, H. (1997) Bisphosphonates in Bone
Disease, 3rd ed., The Parthenon Publishing Group, London.
46.Francis, M. D. (1969) Calcif. Tissue Res.,
3, 151-162.
47.Fleisch, H., Russell, R. G. G., and Francis, M.
D. (1969) Science, 165, 1262-1264.
48.Russell, R. G. G., Muhlbauer, R. C., Bisaz, S.,
Williams, D. A., and Fleisch, H. (1970) Calcif. Tissue Res.,
6, 183-196.
49.Fleisch, H. (1999) in Inorganic Polyphosphates
- Biochemistry, Biology, Biotechnology (Schröder, H. C., and
Müller, W. E. G., eds.) Progr. Mol. Subcell. Biol., Vol.
23, Springer-Verlag, Berlin-Heidelberg-New York, pp. 197-216.
50.Harris, R. S., Nizel, A. E., and Walsh, N. B.
(1967) J. Dent. Res., 46, 290-294.
51.Brudevold, F., Amdur, B. H., Vogel, J. J., and
Spinelli, M. (1964) J. Dent. Res., 43, 1168-1176.
52.Navia, J. M., and Harris, R. S. (1969) J.
Dent. Res., 48, 183-191.
53.McGaughey, C., and Stowell, E. C. (1977) J.
Dent. Res., 56, 579-587.
54.Brunsvold, M. A., Chaves, E. S., Komman, K. S.,
Aufdemorte, T. B., and Wood, R. (1992) J. Periodontol.,
63, 825-830.
55.Weinreb, M., Quartuccio, H., Seedor, J. G.,
Aufdemorte, T. B., Brunsvold, M., Chaves, E., Komman, K. S., and Rodan,
G. A. (1994) J. Periodont. Res., 29, 3540.
56.Shoji, K., Horiuchi, H., and Shinoda, H. (1995)
J. Periodont. Res., 30, 277-284.
57.McCabe, L. R., Kockx, M., Lian, J., Stein, J.,
and Stein, G. (1995) Exp. Cell Res., 218, 255-262.
58.Ducastaing, A., Azanza, J. L., Raymond, J.,
Robin, J. M., and Creach, P. (1976) Biochimie, 58,
783-791.
59.De Vendittis, E., Zahn, R., and Fasano, O. (1986)
Eur. J. Biochem., 161, 473-478.
60.Speziali, G. A., and van Wijk, R. (1971)
Biochim. Biophys. Acta, 235, 466-472.
61.Sultana, G. N., Watanabe, Y., and Tama, Y. (1995)
Biotechnol. Appl. Biochem., 21 (Pt. 1), 101-110.