Engineering a New Magnesium Binding Site in the Subunit Contact Region of Escherichia coli Inorganic Pyrophosphatase
A. N. Parfenyev1, A. Salminen2, A. A. Baykov1*, and R. Lahti2
1Belozersky Institute of Physico-Chemical Biology and School of Chemistry, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: baykov@genebee.msu.su2Department of Biochemistry, University of Turku, Turku, FIN-20500, Finland; fax: (358-2) 633-6860; E-mail: reila@utu.fi
* To whom correspondence should be addressed.
Received October 12, 1999
Three Gln-80 residues belonging to different subunits of homohexameric Escherichia coli pyrophosphatase are separated by only one water molecule to which they are hydrogen bonded. Substitution of Glu for Gln-80 stabilizes quaternary structure of the enzyme but has only a small effect on enzyme activity. The substitution stimulates Mg2+ binding and changes the appearance of the Mg2+ concentration dependence of the rate constant for the trimer --> hexamer transition. These data suggest that a new Mg2+ binding site is formed in the intersubunit contact region as a result of the substitution. Three-dimensional modeling of the mutated protein showed that a chelate complex might form involving two of the three Glu-80 residues.
KEY WORDS: pyrophosphatase, site-directed mutagenesis, magnesium
Inorganic pyrophosphatase (EC 3.6.1.1; PPase) catalyzes the interchange between pyrophosphate and orthophosphate and is essential for life [1, 2]. Because of its relative simplicity and high catalytic efficiency (kcat/Km . 109 M-1·sec-1), PPase has become a paradigm for mechanistic and structural studies of enzymatic phosphoryl transfer from phosphoric acid anhydrides to water. The best-studied PPases are those from Escherichia coli and Saccharomyces cerevisiae [3, 4].
A molecule of E. coli PPase (E-PPase) is formed by six identical subunits, 20 kD each, arranged with D3 symmetry in two layers of trimers [5, 6]. There are two different interfaces between trimers. The smaller one, of 140 Å2, is just a Tyr-77-Asn-24´ hydrogen bond. The larger, 640 Å2, chiefly involves an ion-triple formed between His-140, Asp-143, and His-136´. Another important interaction occurs through Mg2+ bound at the inter-trimeric interface [6, 7]. The Mg2+ ion is octahedrally associated with six water molecules, which in turn hydrogen bond to the side chains of Asn-24, Ala-25, and Asp-26 of two subunits. Substitution of Asp-26 with Asn or Ser eliminates Mg2+ binding to the inter-trimeric site and somewhat decreases hexamer stability, but hardly affects catalysis [8]. Hexamer dissociation into trimers greatly decreases the Michaelis constant for PPi but has no effect on the catalytic constant [8, 9].
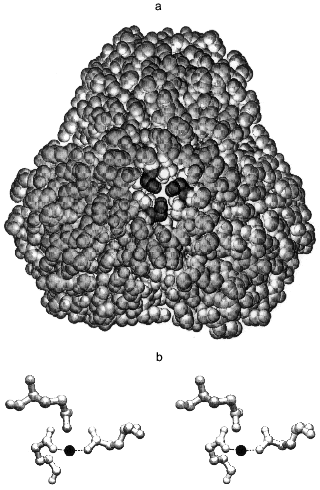
The intra-trimer contacts are of a circular “head-to-tail” type, meaning that each monomer has two different intra-trimer contact regions, 1300 Å2 in total [5, 6]. These involve a mixture of hydrophilic and hydrophobic interactions. Gln-80 participates in two interactions: its backbone NH forms a hydrogen bond with the Tyr-77 hydroxyl and its side chain interacts through a water molecule with the Gln-80 of two other monomers constituting the trimer [10]. The side chains of Gln-80 are quite close to one another (Fig. 1a), such that a metal binding site could form upon Gln to Glu substitution. The aim of the present work was to test this possibility.
MATERIALS AND METHODS
Wild-type E-PPase and its Q80E variant (Q80E-PPase) were prepared as described elsewhere [12, 13]. The final preparations were homogeneous according to SDS-PAGE. Enzyme concentrations are expressed in terms of monomer and were determined spectrophotometrically, using the molecular weight of 20 kD and A1cm, 2801% of 11.8 [14].
The initial rate of PPi hydrolysis was measured continuously with an automatic phosphate analyzer [15]. The assay medium contained, except as noted, 22.5 µM PPi, 20 mM Mg2+, 0.15 M Tris-HCl buffer (pH 7.2), and 40 µM EGTA. The reaction was initiated by adding enzyme and was carried out for 3-4 min at 25°C. The values for the catalytic constant (kcat) and Michaelis constant (Km) were determined from the dependences of initial rate of hydrolysis on substrate (Mg2PPi complex) concentration, which was varied over the 0.2 Km-50 Km range at each level of [Mg2+]. Mg2PPi and free Mg2+ concentrations were calculated as described previously [16].
Trimeric E-PPase was prepared by incubating hexameric E-PPase (0.2 mg/ml) for 10 min at 2°C in medium containing 0.1 M acetic acid-KOH buffer (pH 4.1), 1 mM MgCl2, 0.5 mM dithiothreitol, 40 µM EGTA, and bovine serum albumin (1 mg/ml). To follow hexamer reassociation, an aliquot of the solution was diluted 100-fold at 25°C with medium containing 83 mM TES-KOH buffer (pH 7.2), 17 mM KCl, 0.5 mM dithiothreitol, 50 µM EGTA, bovine serum albumin (1 mg/ml) and MgCl2 at varied concentration, and incubated for 1-7 h at 25°C. Aliquots were withdrawn during the incubation and assayed for enzyme activity. As trimer activity (AT) is approximately one tenth of hexamer activity (AH) [17], enzyme specific activity is increased upon hexamer reformation. Under the conditions used, the dissociation and subsequent reassociation of the enzyme were virtually complete [17], allowing determination of the rate constant for the association (ka) with the equation for a second-order irreversible reaction:
In this equation, A is specific activity at time t, [E]0 is enzyme concentration in terms of monomer, and 1.5 is the factor used to convert monomer concentration into trimer and hexamer concentrations. Numerical values of the parameters were obtained with the SCIENTIST program for nonlinear regression analysis (MicroMath Scientific Software, USA).
The procedure for equilibrium dialysis measurements of Mg2+ binding to the enzyme was described previously [18].
All the incubations, except as noted, were at 25°C.
The structure of the Mg2+ complex of the mutant PPase was modeled with the programs MM2 and MOPAC from CS Chem3D Pro, version 5.0 (Cambridge Soft Corporation, USA). The basic structure was 2.15 Å wild-type E-PPase structure (1FAJ in Protein Data Bank) [5] in which Gln-80 amides were changed to oxygens.
RESULTS AND DISCUSSION
Modeling the magnesium complex of Q80E-PPase. The middle part of the hexameric E-PPase molecule contains a cavity with two triples of Gln-80 residues belonging to different trimers on its opposite sites, about 22 Å from each other (Fig. 1a). Upon Gln-80-Glu substitution, two triples of close carboxylates appear in the cavity, each being able to form a chelate complex with metal ions.
This possibility was tested by computer modeling of the structure of the mutant protein by a modified Ellinger molecular mechanics method (MM2 program). It became clear already at the initial stages of the modeling that gross changes in protein structure are required to include all three carboxylates in the metal coordination sphere. Therefore, all further attempts considered only two groups in a trans position as metal ligands in the inner coordination sphere. The optimal Mg-O bond length was taken as 2.05 Å, and Mg-O-C and O-Mg-O angles as 143 and 180°, respectively. These values were calculated by the MNDO-d semiempirical molecular method (MOPAC) for the interaction of Mg2+ with low-molecular-weight carbonic acids and agree well with the coordination of Mg2+ in the E-PPase active site [6, 7]. Energy minimization for a trimer with bound Mg2+ ion, allowing changes in positions of only those atoms which are closer than 10-15 Å to the new binding site, led to the structure shown in Fig. 1b. The structure is strained, as indicated by the value of 128° for the O-Mg-O angle instead of 180°, which should clearly decrease complex stability. The values of Mg-O bond length (1.95 Å) and the Mg-O-C angles were 140 and 145°, i.e., close to the optimal values. The overall geometry of the site is such that the third carboxylate can also interact with Mg2+, but only through a water molecule, rather than directly. Thus, the computer modeling has indicated that two of the three new Glu-80 residues can coordinate Mg2+ with some distortions from the ideal octahedral configuration.Fig. 1. Stereo drawing showing positions of three Gln-80 residues in wild-type PPase as determined by X-ray crystallography [5] (a) and of three Glu-80 residues in Q80E-PPase as predicted by three-dimensional modeling (b). a) View along the 3-fold axis with the trimer which is closer to the viewer removed. The Gln-80 residues (shown in black) are located at the bottom of the central cavity. b) Dashed lines show predicted bonds with the Mg2+ ion (black sphere). Both figures were produced with the program Swiss-PdbViewer [11].
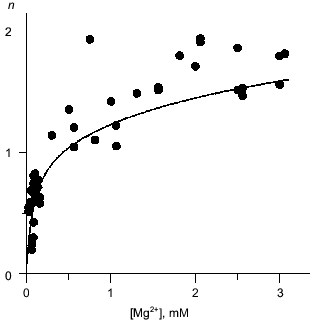
Mg2+ binding. Equilibrium dialysis was used to study the interaction of Q80E-PPase with Mg2+. In this method, the enzyme solution is equilibrated with a Mg2+ solution separated by a semipermeable membrane and the Mg2+ content of both compartments is measured by atomic absorption spectroscopy [18]. E-PPase can bind 2.5 Mg2+ ions per monomer--two in each active site and one per two monomers in the inter-trimer interface [7, 19]. The appearance of a new binding site in Q80E-PPase should increase the binding stoichiometry by 0.33 mole/mole. This is not much; therefore, the binding measurements were conducted at pH 7.2, i.e., under conditions where the active site exhibits low affinity to the second Mg2+ ion and wild-type E-PPase binds <1.5 moles/mole at Mg2+ concentration <3 mM [20]. Despite rather large scatter, the data do indicate a statistically significant increase in the binding stoichiometry in Q80E-PPase (Fig. 2). For most points, binding was greater than with wild-type PPase.
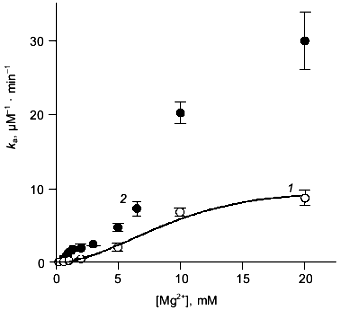
Mg2+ effect on the rate of hexamer formation. A much greater effect was observed in measurements of the Mg2+ effect on the rate of hexamer formation from trimers. As previously shown [8, 21], both wild-type E-PPase and Q80E-PPase can be dissociated into trimers by incubation in acidic medium and the hexameric structure reforms with a measurable rate at pH 7.2. Hexamer reformation can be followed by measuring activity at low substrate concentration because the Michaelis constant is increased by 2-3 orders of magnitude in the trimeric enzyme [8, 9]. The dependences of the rate constant for the association on Mg2+ concentration are shown in Fig. 3. The value of ka is increased gradually with [Mg2+] because of Mg2+ binding to the “half-site” in the potential inter-trimer interface (the complete site is formed only in the hexamer). Q80E-PPase exhibits, in addition, a sharp rise at [Mg2+] > 2 mM, indicating occurrence of a new Mg2+ binding site with a dissociation constant of <1 mM.Fig. 2. Equilibrium dialysis measurements of Mg2+ binding to Q80E-PPase (n is the number of bound Mg2+ ions per monomer). The line shows Mg2+ binding to wild-type E-PPase [20]. Conditions: 0.1 M Tris-HCl (pH 7.2), 40 µM EGTA, 0.7-0.8 mM enzyme.
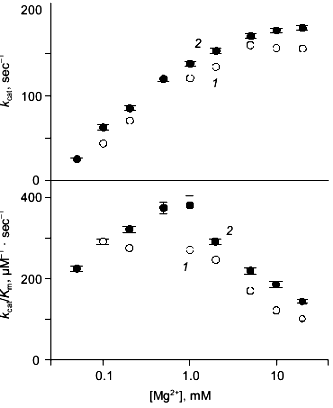
Kinetics of PPi hydrolysis. To estimate the effect of the metal ion bound in the new site on catalysis by hexameric E-PPase, values of the catalytic constant and the Michaelis constant were measured for wild-type PPase and Q80E-PPase over a wide range of Mg2+ concentrations (Fig. 4). Values of kcat were somewhat greater for Q80E-PPase, but the dependences of kcat on [Mg2+] were similar. The same refers to the kcat/Km dependences, except for a small difference observed at 0.5-1 mM Mg2+.Fig. 3. Mg2+ dependence of the rate constant for hexamer formation from trimers: 1) wild-type E-PPase; 2) Q80E-PPase. The curve for wild-type PPase was obtained with Eq. (4) from Efimova et al. [8].
The results confirm experimentally the occurrence of a new metal binding site in Q80E-PPase, as predicted from the structure of the wild-type enzyme. The measured value of the respective dissociation constant for the Mg2+ complex (<1 mM) is characteristic of bidentate complexes formed by this ion, which also agrees with the modeled structure.Fig. 4. Mg2+ dependence of kcat and kcat/Kmfor PPi hydrolysis: 1) wild-type E-PPase; 2) Q80E-PPase. Conditions: 83 mM TES-KOH (pH 7.2), 17 mM KCl, 50 µM EGTA.
The inter-subunit interactions are appreciably affected upon metal binding to the new site. This site is in a solvent-assessable inner cavity located on a threefold axis. In the wild-type enzyme, the Gln-80 residues belonging to three monomers are in contact with each other through a water molecule, which is replaced by a Mg2+ ion linking two of the three monomers in the mutated enzyme. The intra- and inter-trimer contact zones are quite close and interdependent [17], which may explain the effect of the new site on the rate of hexamer formation from trimers. The increase in ka may also explain stabilization of hexameric Q80E-PPase in acidic medium [8]. Mg2+ binding appears to require some changes in enzyme structure optimizing the metal coordination sphere. Such changes were also reported for Mg2+ binding in E-PPase active site [22]. Inter-monomer interactions with trimer are also likely to be affected by the Mg2+ ion bound in the new site, but this is difficult to prove experimentally. The structural changes are localized in the vicinity of Glu-80 and are not spread over the distant active site, explaining minor effects of the new site on catalysis. Similarly, no effect on catalysis results from Mg2+ binding to the sites located between trimers [8].
The assistance of Dr. V. N. Kasho in this work is gratefully acknowledged. This work was supported by the Russian Foundation for Basic Research, projects No. 97-04-48487 and 96-15-97969, and the Academy of Finland, project No. 35736.
REFERENCES
1.Chen, J., Brevet, A., Formant, M., Leveque, F.,
Schmitter, J.-M., Blanquet, S., and Plateau, P. (1990) J.
Bacteriol., 172, 5686-5689.
2.Lundin, M., Baltscheffsky, H., and Ronne, H. (1991)
J. Biol. Chem., 266, 12168-12172.
3.Cooperman, B. S., Baykov, A. A., and Lahti, R.
(1992) Trends Biochem. Sci., 17, 262-266.
4.Baykov, A. A., Cooperman, B. S., Goldman, A., and
Lahti, R. (1999) Progr. Mol. Subcell. Biol., 23,
127-150.
5.Kankare, J., Salminen, T., Lahti, R., Cooperman,
B., Baykov, A. A., and Goldman, A. (1996) Acta Crystallogr.,
D52, 551-563.
6.Harutyunyan, E. H., Oganessyan, V. Yu., Oganessyan,
N. N., Avaeva, S. M., Nazarova, T. I., Vorobyeva, N. N., Kurilova, S.
A., Huber, R., and Mather, T. (1997) Biochemistry, 36,
7754-7760.
7.Kankare, J., Salminen, T., Lahti, R., Cooperman,
B., Baykov, A. A., and Goldman, A. (1996) Biochemistry,
35, 4670-4677.
8.Efimova, I. S., Salminen, A., Pohjanjoki, P.,
Lapinniemi, J., Magretova, N. N., Cooperman, B. S., Goldman, A., Lahti,
R., and Baykov, A. A. (1999) J. Biol. Chem., 274,
3294-3299.
9.Velichko, I. S., Mikalahti, K., Kasho, V. N.,
Dudarenkov, V. Yu., Hyytiä, T., Goldman, A., Cooperman, B. S.,
Lahti, R., and Baykov, A. A. (1998) Biochemistry, 37,
734-740.
10.Harutyunyan, E. H., Oganessyan, N. N., Terzyan,
S. S., Popov, A. N., Rubinskiy, S. B., Vainstein, B. K., Nazarova, T.
I., Kurilova, S. A., Vorobjeva, N. N., and Avaeva, S. M. (1996)
Kristallografiya, 41, 84-96.
11.Guex, N., and Peitsch, M. C. (1997)
Electrophoresis, 18, 2714-2723.
12.Lahti, R., Pohjanoksa, K., Pitkaranta, T.,
Heikinheimo, P., Salminen, T., Meyer, P., and Heinonen, J. (1990)
Biochemistry, 29, 5761-5766.
13.Salminen, T., Käpylä, J., Heikinheimo,
P., Kankare, J., Goldman, A., Heinonen, J., Baykov, A. A., Cooperman,
B. S., and Lahti, R. (1995) Biochemistry, 34,
782-791.
14.Wong, S. C. K., Hall, D. C., and Josse, J. (1970)
J. Biol. Chem., 245, 4335-4345.
15.Baykov, A. A., and Avaeva, S. M. (1981) Anal.
Biochem., 116, 1-4.
16.Volk, S. E., Baykov, A. A., Duzhenko, V. S., and
Avaeva, S. M. (1982) Eur. J. Biochem., 125, 215-220.
17.Salminen, A., Efimova, I. S., Parfenyev, A. N.,
Magretova, N. N., Mikalahti, K., Goldman, A., Baykov, A. A., and Lahti,
R. (2000) J. Biol. Chem., 274, 33898-33904.
18.Käpylä, J., Hyytiä, T., Lahti, R.,
Goldman, A., Baykov, A. A., and Cooperman, B. S. (1995)
Biochemistry, 34,792-800.
19.Baykov, A. A., Hyytiä, T., Turkina, M. V.,
Efimova, I. S., Kasho, V. N., Goldman, A., Cooperman, B. S., and Lahti,
R. (1999) Eur. J. Biochem., 260, 308-317.
20.Baykov, A. A., Hyytiä, T., Volk, S. E.,
Kasho, V. N., Vener, A. V., Goldman, A., Lahti, R., and Cooperman, B.
S. (1996) Biochemistry, 35, 4655-4661.
21.Borshchik, I. B., Magretova, N. N., Chernyak, V.
Ya., Sklyankina, V. A., and Avaeva, S. M. (1986) Biokhimiya,
51, 1484-1489.
22.Avaeva, S. M., Rodina, E. V., Vorobyeva, N. N.,
Kurilova, S. A., Nazarova, T. I., Sklyankina, V. A., Oganesyan, V. Yu.,
and Harutyunyan, E. G. (1998) Biochemistry (Moscow), 63,
592-599.