Human Recombinant Laminin-Binding Protein: Isolation, Purification, and Crystallization
A. V. Sorokin1, A. M. Mikhailov2, A. V. Kachko1, E. V. Protopopova1, S. N. Konovalova1, M. E. Andrianova2, S. V. Netesov1, A. N. Kornev3, and V. B. Loktev1*
1Institute of Molecular Biology, State Research Center for Virology and Biotechnology “Vector”, Koltsovo, Novosibirsk Region, 630559 Russia; fax: (3832) 36-7409; E-mail: loktev@vector.nsc.ru2Shubnikov Institute of Crystallography, Russian Academy of Sciences, Leninsky pr. 59, Moscow, 117333 Russia; fax: (095) 135-1011; E-mail: amm@biostr.crystal.msk.ru
3Institute of Cell Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; E-mail: amm@biostr.crystal.msk.ru
* To whom correspondence should be addressed.
Received August 19, 1999; Revision received December 21, 1999
The mRNA of the precursor of laminin-binding protein (LBP) was isolated from a human embryo kidney cell line and cloned. The determined sequence of the LBP gene showed complete identity with the LBP genes isolated from human lung and large intestine cells. The human LBP was expressed by E. coli cells, and it was purified using Ni-NTA-Sepharose chromatography. The mobility of the homogeneous recombinant human laminin-binding protein on SDS-PAGE was 43 kD. A mixture of eight murine monoclonal antibodies, the MPLR Pool against LBP, reacted with the recombinant LBP in Western blot. The interaction of the antiidiotypical antibodies 10H10 and E6B provided evidence that the epitope binding to protein E of the tick-borne encephalitis (TBE) virus is also preserved on the human recombinant LBP. Enzyme immunoassay confirmed the ability of the recombinant LBP to interact with protein E of TBE virus. The biological activity of the recombinant LBP allowed us to perform X-ray analysis of the spatial arrangement of the LBP molecule using the recombinant protein. For this purpose, crystals of the human LBP were obtained by the standing drop version of the pore diffusion technique. The crystals appropriate for X-ray structural analysis were 0.3 × 0.1 × 0.05 mm in size. The X-ray diffraction field of the crystal extended to 2.5 Å.
KEY WORDS: human laminin-binding protein, LBP, cloning, crystal
Laminin is an extracellular matrix glycoprotein that plays an important role in a wide variety of biological processes including development, organ and tissue differentiation, growth of neoplasms, and their metastatic invasion [1-3]. Several cell membrane proteins have been reported to interact with laminin and to play a major role in these events [4-6]. Among those characterized of importance is the 67-kD laminin-binding protein (LBP) that interacts with laminin with high affinity [7-9]. LBP is present on the surface of normal and cancer cells, and it is a mediator of a strong interaction of cells with laminin. Its expression is considerably increased in highly metastatic cells [10-14].
Recent data have demonstrated that several alphaviruses and flaviviruses utilize the LBP as a cell receptor for virion attachment and penetration into cells [15-17]. The interaction of 37-kD LBP with prion protein was shown, and this opened a discussion on role of LBP in the development of prion diseases in animals and humans [18].
To better understand the biological activity of LBP, attempts have been made to clone the human and murine LBP [11, 19, 20]. The full-length gene was found to encode a 295-amino acid polypeptide with a calculated molecular weight 32 kD. Additional study of maturation of a 67-kD form of LBP was stimulated by the small size of the precursor-polypeptide. The experimentally determined amino acid composition of the affinity isolated 67-kD LBP demonstrated that it is identical with the precursor protein.
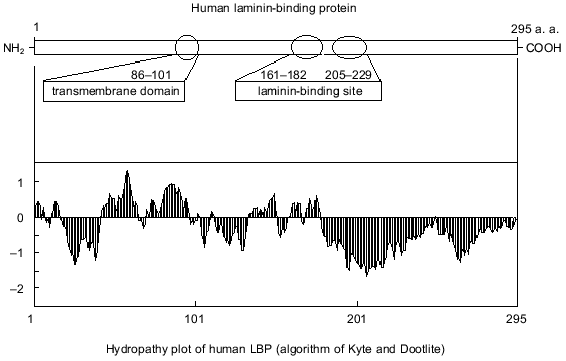
It is suggested that the 67-kD LBP molecule consists of two polypeptide chains of the precursor molecule, and formation of the mature form of LBP involves the acylation of the precursor [21]. Inhibition of 67-kD LBP formation by cerulenin indicates that fatty acids are involved in the processing of the LBP [22]. The antigenic identity of native 67- and 32-kD precursor was demonstrated using monoclonal and polyclonal antibodies [23, 24]. The active centers of LBP and its precursor that interact with laminin were also found to be similar [25]. A model of the structural-functional organization of the 32-kD laminin precursor is shown in Fig. 1. An organizational feature of the precursor is the absence of potential glycosylation sites. The expressed hydrophobic domain is located approximately one third from the N-terminal. Two binding sites for laminin are located in the 161-182 and 205-229 amino acid regions [25, 26].
There is no precise information regarding the spatial organization of LBP and its relationships with its 32-kD precursor. This prompted us to undertake a structural analysis of the laminin-binding protein. The reason why we focused our attention on LBP was the current information that a clearer understanding of the laminin-binding protein-laminin interaction would make it possible to search for new agents blocking metastatic tumor invasion. In fact, the key role of this protein in cell interaction with a number of viruses highly pathogenic for humans provides hitherto not fully exploited possibilities for finding new antiviral drugs blocking the interaction of virions with laminin-binding protein.
The stages of the present study were to clone the 32-kD LBP gene from human embryo kidney cells, to express the gene in E. coli cells, the preparation of highly purified, electrophoretically homogeneous recombinant LBP, the search for conditions of its crystallization, and the production of crystals appropriate for X-ray examination.
MATERIALS AND METHODS
Cell culture and virus. The culture of RH cells (human embryo kidney cells) was from the cell culture bank at the State Research Center of Virology and Biotechnology “Vector” (Koltsovo, Novosibirsk Region, Russia). The cells were cultured in Eagle's minimum essential medium with 10% fetal calf serum and 80 µg/ml gentamycin sulfate. The 205 strain of the tick-borne encephalitis (TBE) virus was from a viral collection at the State Research Center of Virology and Biotechnology “Vector”. TBE virus was purified from infected RH cells with subsequent centrifugation on a sucrose density gradient [27]. The homogeneous purified viral suspension was used for immunological experiments at concentration 0.75 mg/ml.
Molecular-biological method. Total RNA was isolated from RH cells by the standard method using a lysing buffer containing 4 M guanidine-isothiocyanate, 25 mM sodium citrate, and 0.5% sodium N-lauryl-sarcosylate [28]. Oligo-dT primer was used to construct cDNA. The cDNA construct was used to amplify the LBP genes in polymerase chain reaction (PCR) with specific primers calculated on the basis of the mRNA sequence of the LBP [29]. The following primers were used: LN1, 5´-TTCGGATCCAACTTTCACAA-3´; LN2, 5´-CCGGATCCAGGCAGCCTT-3´. LN3, 5´-CCAGATCTGGTTAGTGAAGGTTC-3´; LN4, 5´-TTCGGCCGTAAGAGCCTATGC-3´ (the LBP specific sequences are underlined). Restriction of the LBP amplified fragment and cloning were performed by standard methods [30]. The pQ-f35 vector [31], a derivative of the expressing pQE31 vector (Qiagen, Germany), was used as the initial vector for cloning and expressing the LBP gene. The E. coli strain was JM103 (endA1, hsdR, supE, sbcB15, thi-1, strA, Delta(lac-proAB), [F´, traD36, proAB, lac1q .DeltaM.5]). The nucleotide sequence of the cloned gene was determined according to Sanger utilizing a T7 Sequenase version 2.0 DNA sequencing kit (Amersham Life Science Inc., USA). The expression of the recombinant LBP (recLBP), the procedure for obtaining cell lysates of the producer, and its purification under denaturing conditions complied with Qiagen's protocol. The proteins were subjected to SDS-PAGE according to Laemmli [32]. Protein was determined using the Bio-Rad Protein Assay (USA). Restriction endonucleases and enzymes were products of the Sibenzyme (Novosibirsk, Russia) and Fermentas (Vilnius, Lithuania).
Immunological assay. Monoclonal antibodies MAb MluC5 against 67-kD LBP and MPLR Pool (a mixture of monoclonal antibodies of 8 types) against recLBP [23, 24] were kindly provided by Dr. S. Menard (National Cancer Institute, Milan, Italy). We have described elsewhere rabbit polyclonal antiidiotypical antibodies 10H10 and E6B that model the protein E receptor regions of the tick-borne encephalitis virus and interact with the 67-kD laminin-binding protein [27, 33]. Rabbit anti-recLBP antibodies were obtained from three times immunized rabbits with two-week intervals. The first immunization was done by subcutaneous injection of 5 mg of affinity-purified recombinant LBP in a mixture with an equal volume of complete Freund's adjuvant. Other immunizations were carried out by intramuscular injections of 2.5 mg of affinity purified recLBP in a mixture with an equal volume of incomplete Freund's adjuvant. Immunoenzyme assay (ELISA) was conducted using 96-well plates and antispecies peroxidase conjugate against mouse and rabbit IgG [34]. Immunoblot analysis was carried out following Towbin [35, 36]. Immunoblot experiments were performed using monoclonal antibodies against LBP and antiidiotypical antibodies 10H10 and E6B. The formed immune complex was detected by incubation with horseradish peroxidase conjugated with antispecies IgG.
Interaction of recombinant LBP and protein E of TBE virus. Purified TBE virus was sorbed overnight at 4°C on a polystyrene 96-well microplate (200 ng/well). The plate was blocked by PBS (phosphate-buffered saline, pH 7.4) containing 0.5% casein. The purified recombinant LBP diluted in PBS containing 0.25% casein was added to wells and then incubated for 1 h at 37°C. Then the plate was washed five times in PBS and incubated with rabbit anti-LBP serum diluted in PBS with 0.25% casein for 1 h at 37°C. Immune complex was detected by incubation with horseradish peroxidase conjugated with goat anti-rabbit IgG.
Crystallization. Crystallization conditions were studied by the sitting drop version of the pore diffusion technique. We used the crystallization cuvettes we previously employed for crystal growth of neurotoxin from cobra toxin [37]. The X-ray qualitative control of the resulting crystals and determination of crystallographic parameters were performed on Laue's and precession snaps from an automated universal camera in Fourier-spectrum mode. A GX-20 generator (England) with rotating copper anode was used as the source of X-ray emission. The increase in intensities and focusing of X-ray beams were performed using a focusing set up of four bent quartz mirrors installed in pairs in two mutually perpendicular planes [38]. The determined unit-cell parameters on snaps were checked on a KARD-8 automated diffractometer with two-dimensional CD-1000 detectors [39].
RESULTS
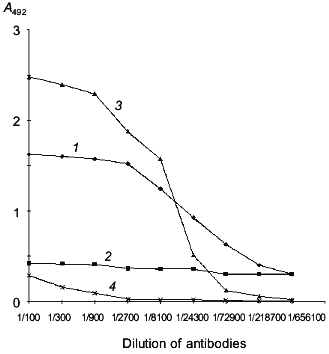
Cloning of the human laminin-binding protein gene from human embryo kidney cells. To isolate the LBP gene (see model of LBP organization on Fig. 1), a culture of human embryo kidney cells (RH) highly sensitive to the tick-borne encephalitis virus was chosen. According to previously data, the virus utilizes the LBP to penetrate into cells [17, 27, 33]. The presence of the LBP on the RH cell surface was demonstrated by enzyme-linked immunosorbent assay (ELISA) using MAb MluC5 and antiidiotypical antibodies 10H10 and E6B (Fig. 2). The presence of LBP at high concentration on RH cell surface suggested that the level of mRNA encoding the protein would be high.
Fig. 1. A model of the functional organization of LBP.
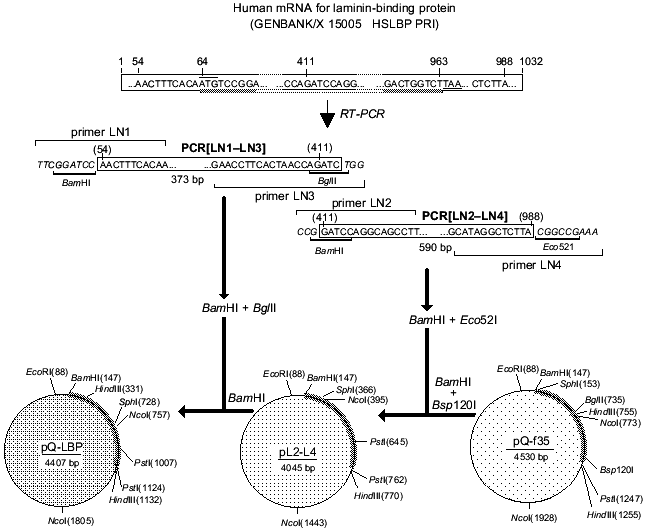
Total RNA was isolated from RH cells and oligo-dT was used to construct cDNA. The cDNA was utilized to amplify the LBP genes by PCR with specific primers. The full-size LBP gene was cloned using the two overlapping amplified fragments. Figure 3 is a schematic representation of cloning of the amplified fragments and assembly of the full-size gene of LBP. The amplified product [LN2-LN4] was digested with BamH I and Eco52 I (Xma III) and cloned into BamH I-Bsp120 I sites of the pQ-f35 vector. The amplified product [LN1-LN3] was BamH I and Bgl II digested, inserted into an intermediate plasmid, and obtained by the first cloning of pQ-CLBP into the BamH I site. The generated clones contained the pQ-LBP plasmid carrying the cloned fragment in the correct orientation. The nucleotide sequence of the LBP gene fragments contained in the pQ-CLBP and pQ-LBP plasmids was analyzed in six independent clones for each plasmid. No difference in sequence was found between the clones. The sequence was identical (data not shown) with those previously described for the LBPs from human lung [40] and human colon carcinoma cells [11].Fig. 2. Interaction of antibodies with human laminin-binding protein within the membrane fraction of RH cells in ELISA (membranes of RH cells in dilution 1:10 were immobilized on plastic plates (20 µl per well), then fixed with ethanol): 1) interaction of MAb MluC5 with the membrane fraction of RH cells; 2, 4) negative control (normal mouse and rabbit sera, respectively); 3) interaction of antiidiotypical antibodies 10H10 with the membrane fraction of RH cells.
Purification of recombinant laminin-binding protein. The pQ-LBP plasmid we constructed contains an open reading frame for the recombinant LBP gene formed by the sequence of the vector part of the plasmid and by the sequence encoding the LBP gene. The generated LBP contains all the 295 amino acid residues of the full-size protein precursor. These include 16 amino acid residues at the N-terminus in addition to the natural LBP formed by the sequence of the vector part of the plasmid and the nucleotide sequence encoding a block of six consecutive histidine residues facilitating the isolation of the recombinant protein using Ni-NTA-Sepharose resin.Fig. 3. Scheme for construction of the plasmid pQ-LBP.
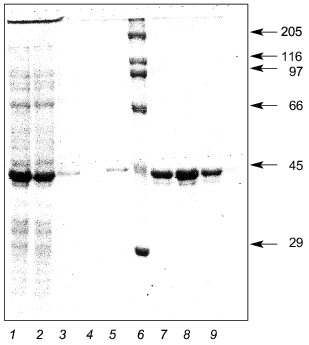
E. coli JM103 clones transformed by the plasmid pQ-LBP were used to express the recombinant LBP. The data indicate that during the induction with isopropyl beta-D-thiogalactoside of culture JM103[pQ-LBP] a recombinant protein with an electrophoretic mobility of about 43-kD is intensely synthesized, although the calculated molecular weight of the LBP is 34.8 kD. The biosynthesis level of the recombinant LBP was not less than 80 mg/liter of culture medium. Purification of the recombinant LBP from cell lysates by Ni-NTA-Sepharose resin yielded virtually homogeneous 43-kD recombinant protein (Fig. 4).
Thus, large quantities of highly purified LBP were easily obtained. This enabled us to perform immunochemical studies and to undertake experiments in which LBP crystals were generated for subsequent X-ray analysis.Fig. 4. Expression and purification of recombinant LBP: 1) initial cell lysate of E. coli JM103[pQ-LBP]; 2) lysate after sorption on Ni-NTA resin; 3, 4, 5) passage through column; 6) molecular weight markers (their molecular weights in kD are shown on the right); 7, 8, 9) purified recombinant LBP.
Immunochemical study of recombinant LBP. To identify LBP, we used monoclonal antibodies of the MPLR Pool (a mixture of MAbs of 8 types) against LBP [23, 24] and rabbit antiidiotypical antibodies 10H10 and E6B that interact with the receptor epitope on the 67-kD human LBP for TBE virus [27, 33]. The reactivity of monoclonal antibodies, the MPLR Pool, was tested using immunoblots. Figure 5 shows that the antibodies react with the recombinant LBP. We therefore concluded that the human recombinant LBP weisolated and characterized is identical with the preparation of the protein previously generated by others [23, 24]. Antiidiotypical antibodies 10H10 and E6B that model the receptor region for protein E of TBE virus and interact with the native 67-kD LBP in ELISA revealed strong interaction with the recombinant LBP (Fig. 5). This meant that recombinant LBP retains the surface epitope for interaction with the surface protein E of TBE virus and that it is quite appropriate for studying the mechanism of TBE virus virions-LBP interaction.
As shown in Fig. 5, antiidiotypical antibodies 10H10 and E6B interact with two proteins with molecular masses of 43 and 67 kD, respectively. The presence of a polypeptide with a molecular mass of 67 kD in the purified preparation indicated that a small part of the recLBP dimerizes, although electrophoresis was conducted under denaturing conditions. This indirectly supported the literature data that the native laminin-binding protein consists of two molecules of the precursor protein [21].Fig. 5. Immunoblotting of purified recombinant LBP: 1) antiidiotypical antibodies 10H10; 2) normal rabbit serum; 3) antiidiotypical antibodies E6B; 4) normal mouse serum; 5) MAb MPLR Pool against LBP. Molecular weights of protein markers in kD are shown on the right.

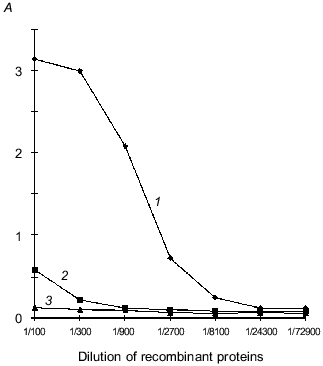
Interaction of recLBP with protein E of TBE virus. The solid-phase variant of enzyme immunoassay for study of interaction of recLBP and protein E was developed for checking the availability of receptor site on recLBP. The sucrose gradient-purified virions of the TBE virus were immobilized on a polystyrene plate. A complex of these proteins was formed on surface of the virions after adding a solution of recLBP to the wells. The protein E-recLBP complex was detected by incubation with rabbit anti-LBP antibodies and secondary antibody labeled with horseradish peroxidase. As shown in Fig. 6, recLBP effectively interacts with protein E of TBE virus. This suggested that the receptor site of recLBP is intact. The biological activity of the recombinant LBP and immunochemical similarity with the native 67-kD LBP suggested that structural studies may be furthered by using the recombinant LBP as a good model of native human laminin-binding protein.
Crystallization of laminin-binding protein. LBP crystals of plate form with size 0.3 ×_0.1 ×_0.05 mm were the most suitable for X-ray analysis. The crystal growth time was 2 weeks at 4°C. Crystallization conditions consisted of 40 µl protein (at concentration 2 mg/ml) in 0.05 M Tris-maleate/NaOH buffer (pH 7.5), 2 mM CaCl2, 0.6% beta-octylglucoside, 20% 2-methyl-2,4-pentadiol, and 0.02% NaN3. The counter solution contained 40% 2-methyl-2,4-pentadiol in 0.1 M Tris-maleate/NaOH buffer, 4 mM CaCl2, 1.2% beta-octylglucoside; the pH of the counter solution was in the range from 5.3 to 7.5. The concentration of precipitant in the counter solution was gradually raised to a maximum value. The packing of LBP molecules in the crystal is described by space group P21212 with unit-cell parameters a = 88.1 Å, b = 75.0 Å, c = 50.9 Å on the basis of X-ray diffraction investigations. The asymmetric part of the unit-cell contains one LBP molecule (Matthews coefficient Vm = 2.42 Å3 per dalton [41]). Thus, 50% of the volume of the asymmetric unit cell is occupied by crystallization solution. This value varies in the range from 27 to 65%. The X-ray diffraction field extends to 2.5 Å.Fig. 6. Interaction of protein E of TBE virus with recombinant LBP in ELISA: 1) interaction of purified recLBP with protein E of TBE virus; 2) interaction of purified recombinant VP40 protein of Marburg virus with protein E of TBE virus (negative control, VP40 has similar molecular weight, polyhistidine domain, and method for purification as used for recLBP); 3) negative control with normal rabbit serum at dilution 1:2000. The initial concentration of the recombinant protein is 5 mg/ml.
DISCUSSION
It is becoming increasingly apparent that LBP is importantly involved in many processes: the formation of organs and tissues, tumor growth and metastasis, and, possibly, in the penetration of pathogenic viruses into human cells. Clearly, more thorough studies of LBP are needed. The data would clarify the mechanisms of LBP interaction with the laminin, with various pathogenic viruses, and would hopefully show how LBP might participate in the formation of malignancies and in their metastatic spread. Quite recently, monomeric LBP has been identified in high concentration in cell cytoplasm [42]. The possible involvement of LBP in the formation of ribosomal complex and in the interaction with cell chromosomal DNA made the need for its further structural study more obvious.
The highly conserved primary structure of LBP indirectly supports the idea that LBP is importantly involved in many events at the cell and organ levels. So far, LBP gene has been sequenced for more than 20 organisms. LBP has been detected in organisms such as microorganisms, yeasts, helminths, insects, and mammals [43-51]. The homology of the LBP structure in these organisms is quite high, thereby indicating that LBP is functionally significant. The homology degree of LBP is more than 95% for mammals. However, analysis of the spatial structure of a typical representative of laminin-binding protein may give a good idea of what the entire family may be.
LBP is a structural component of membranes, ribosomes, and chromosomes; it possesses a distinct hydrophobic region so that detergents are needed to isolate it from cells. It is undesirable to use detergents for producing LBP crystals suitable for structural X-ray analysis. It is also difficult to isolate LBP from cells because of its presence in relatively low concentrations and the lack of standard methods for its purification and isolation. LBP may be present in different oligomeric forms in cells, it may be subjected to posttranslational modification, and its primary structure may be variable. All this introduces uncertainties when first undertaking its structural study. We chose another strategy to circumvent this difficulty. We cloned the human LBP gene and then derived a producer of recombinant LBP in E. coli cells. This made highly purified LBP in sufficiently high quantities available for X-ray analysis. The molecular size of recLBP is 9 kD more than that calculated on the basis of its amino acid sequence. Similar data were earlier observed for the monomer form of LBP [21-26]. Electrophoretic evaluation of the molecular weigh of LBP ranged up from 37 to 43 kD. In our opinion, the anomalous electrophoretic mobility of recLBP is a result of its particular physicochemical properties.
Based on the results of immunochemical assays of the recombinant LBP, using monoclonal and antiidiotypical antibodies, we concluded that the antigenic structure of the recombinant laminin-binding protein is preserved. The interaction with antiidiotypical antibodies was indirect evidence that the receptor region for the interaction with TBE virus was retained. The ELISA confirms the ability of recombinant LBP to interact with protein E on the surface of TBE virus. The results suggest that the isolated recombinant LBP can serve as an adequate model for both monomeric and dimeric LBPs and that structural studies would provide better understanding of the fundamental role of the LBP domains and also clarify the possible mechanism of ligand-receptor interaction of the protein. It is hoped that the spatial structure of LBP will be determined relying on crystallization experiments and X-ray data. A defined three-dimensional LBP structure is a necessary condition for proper understanding of the structural-functional aspect of the mechanism action of the protein.
The authors are grateful to Dr. S. Menard (National Cancer Institute, Milan, Italy) for kindly supplying monoclonal MAb MluC5 and MPLR Pool antibodies against human laminin-binding protein.
This research was supported by a grant from the National Aeronautics and Space Administration of the USA and the Program of the Ministry of Science and Technologies of Russia for SRC VB “Vector”.
REFERENCES
1.Timpl, R., Rohde, P. G., Rennard, J. M., and
Martin, G. R. (1979) J. Biol. Chem., 254, 9933-9937.
2.Kleinman, H. K., Cannon, F. B., Laurie, G. W.,
Hassell, J. R., Aumailley, M., Terranova, V. P., Martin, G. R., and
DuBois-Dalcq, M. (1985) J. Cell Biochem., 27,
317-325.
3.Liotta, L. A. (1986) Cancer Res.,46,
1-7.
4.Sonnenberg, A. (1993) Curr. Top. Microbiol.
Immunol., 184, 7-35.
5.Stewart, M., Thiel, M., and Hogg, N. (1995)
Curr. Opin. Cell Biol., 7, 690-696.
6.Striz, I., and Costadel, U. (1992)
Sarcoidosis,9, 88-94.
7.Rao, C. N., Barsky, S. H., Terranova, V. P., and
Liotta, L. A. (1983) Biochem. Biophys. Res. Commun., 111,
804-808.
8.Lesot, H., Kuhl, U., and von der Mark, G. (1983)
EMBO J.,2, 861-865.
9.Malinoff, H. L., and Wicha, M. S. (1983) J. Cell
Biol., 96, 1475-1479.
10.Gastronovo, V., Taraboletti, G., Liotta, L. A.,
and Sobel, M. E. (1989) J. Natl. Cancer Inst., 81,
781-788.
11.Yow, H., Wong, J. M., Chen, H. S., Lee, C.,
Steele, G. D., Jr., and Chen, L. B. (1988) Proc. Natl. Acad. Sci.
USA, 85, 6394-6398.
12.Gastronovo, V., Colin, C., Claysmith, A. P.,
Chen, P. H., Lifrange, E., Lambotte, R., Krutzsch, H., Liotta, L. A.,
and Sobel, M. E. (1990) Am. J. Pathol.,137,
1373-1381.
13.Cioce, V., Castronovo, V., Shmooker, M. B.,
Garbisa, S., Grigione, W. F., Liotta, L. A., and Sobel, M. E. (1991)
J. Natl. Cancer Inst.,83, 29-36.
14.Montuori, N., and Sobel, M. E. (1996) Curr.
Top. Microbiol. Immunol., 213, 205-214.
15.Strauss, J. H., Wang, K.-S., Schmaljohn, A. L.,
Kuhn, R. J., and Strauss, E. G. (1994) Arch. Virol. (Suppl.),
9, 473-484.
16.Ludwig, G. V., Kondig, J. P., and Smith, J. F.
(1996) J. Virol., 70, 5592-5599.
17.Protopopova, E. V., Konovalova, S. N., and
Loktev, V. B. (1997) Vopr. Virusol., 42, 264-268.
18.Rieger, R., Edenhofer, F., Lasmezas, C. I., and
Weiss, S. (1997) Nature Med., 3, 1383-1388.
19.Rao, C. N., Castronovo, V., Schmiitt, M. C.,
Wewer, U. W., Claysmith, A. P., Liotta, L. A., and Sobel, M. E. (1989)
Biochemistry,28, 7476-7486.
20.Makrides, S., Chipatima, S. T., Bandyopadhyayr,
C., and Braverman, G. (1988) Nucleic Acids Res., 16,
2349.
21.Landowski, T. H., Dratz, E. A., and Starkey, J.
R. (1995) Biochemistry, 34, 11276-11287.
22.Buto, S., Tagliabue, E., Ardini, E., Magnifico,
A., Ghirelli, C., van den Brule, F., Castronovo, V., Colnaghi, M. I.,
Sobel, M. E., and Menard, S. (1998) J. Cell. Biochem.,
69, 244-251.
23.Saleh, W., Delvenne, P., van den Brule, F. A.,
Menard, S., Boniver, J., and Castronovo, V. (1997) J. Pathol.,
181, 287-293.
24.Buto, S., Ghirelli, C., Aiello, P., Tagliabue,
E., Ardini, E., Magnifico, A., Montuori, N., Sobel, M. E., Colnaghi, M.
I., and Menard, S. (1997) Int. J. Biol. Markers,12,
1-5.
25.Castronovo, V., Taraboletti, G., and Sobel, M. E.
J. (1991) Biol. Chem., 266, 20440-20446.
26.Landowski, T. H., Uthayakumar, S., and Starkey,
J. R. (1995) Clin. Exp. Metastasis, 13, 357-372.
27.Protopopova, E. V., Khusainova, A. D.,
Konovalova, S. N., and Loktev, V. B. (1996) Vopr. Virusol.,
41, 50-53.
28.Puri, B., Henchal, E. A., Burans, J., Porter, K.
R., Nelson, W., Watts, D. M., and Hayes, C. G. (1994) Arch.
Virol.,134, 29-37.
29.Van den Ouweland, A. M., van Duijnhoven, H. L.,
Deichmann, K. A., van Groningen, J. J., de Leij, L., and van de Ven, W.
J. (1989) Nucleic Acids Res., 17, 3829-3843.
30.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor, N. Y.
31.Sorokin, A. V., Kazachinskaya, E. I., Kachko, A.
V., Ivanova, A. V., Bukreev, A. A., and Razumov, I. A. (1999) Vopr.
Virusol., 44, 206-213.
32.Laemmli, U. K. (1970) Nature (London),
227, 680-685.
33.Protopopova, E. V., Konovalova, S. N., and
Loktev, V. B. (1996) Zh. Infekts. Patol., 3, 60-63.
34.Razumov, I. A., Agapov, E. V., Pereboev, A. V.,
Protopopova, E. V., Lebedeva, S. D., and Loktev, V. B. (1991) Vopr.
Virusol., 36, 34-37.
35.Towbin, H., Staehelin, T., and Gordon, J. (1979)
Proc. Natl. Acad. Sci. USA, 76, 4350-4354.
36.Towbin, H., and Gordon, J. (1984) J. Immunol.
Meth., 72, 313-340.
37.Nikitenko, A. V., Trakhanov, S. D., and
Chetverina, E. V. (1990) Kristallografiya, 35,
1465-1474.
38.Kornev, A. N., and Mikhailov, A. M. (1990)
Kristallografiya, 35, 190-196.
39.Kheiker, D. M., Andrianova, M. E., Sul'yanov, S.
N., Zanevskii, Yu. V., Fateev, O. V., and Chernenko, S. P. (1996)
Kristallografiya, 41, 362-369.
40.Satoh, K., Narumi, K., Sakai, T., Abe, T.,
Kikuchi, T., Matsushima, K., Sindoh, S., and Motomiya, M. (1992)
Cancer Lett., 62, 199-203.
41.Matthews, B. W. (1968) J. Mol. Biol.,
33, 491-497.
42.Kinoshita, K., Kaneda, Y., Sato, M., Saeki, Y.,
Wataya-Kaneda, M., and Hoffmann, A. (1998) Biochem. Biophys. Res.
Commun., 253, 277-282.
43.Garrels, J. I., Futcher, B., Kobayashi, R.,
Latter, G. I., Schwender, B., Volpe, T., Warner, J. R., and McLaughlin,
C. S. (1994) Electrophoresis, 15, 1466-1486.
44.Norbeck, J., and Blomberg, A. (1997) J. Biol.
Chem., 272, 5544-5554.
45.Zhang, L., Leggatt, G. R., Kalinna, B. H., Piva,
T. J., and McManus, D. P. (1997) Mol. Biochem.
Parasitol.,87, 183-192.
46.Melnick, M. B., Noll, E., and Perrimon, N. (1993)
Genetics, 135, 553-564.
47.Yow, H. K., Wong, J. M., Chen, H. S., Lee, C. G.,
Davis, S., Steele, G. D., Jr., and Chen, L. B. (1988) Proc. Natl.
Acad. Sci. USA, 85, 6394-6398.
48.Satoh, K., Narumi, K., Sakai, T., Abe, T.,
Kikuchi, T., Matsushima, K., Sindoh, S., and Motomiya, M. (1992)
Cancer Lett., 62, 199-203.
49.Tohgo, A., Takasawa, S., Munakata, H., Yonekura,
H., Hayashi, N., and Okamoto, H. (1994) FEBS Lett., 340,
133-138.
50.Makrides, S., Chitpatima, S. T., Bandyopadhyay,
R., and Brawerman, G. (1988) Nucleic Acids Res., 16,
2349.
51.Grosso, L. E., Park, P. W., and Mecham, R. P.
(1991) Biochemistry, 30, 3346-3350.