REVIEW: Cardiovascular Effects of Carnosine
P. R. Roberts1* and G. P. Zaloga2
1Department of Anesthesiology, Wake Forest University School of Medicine, Medical Center Blvd., Winston-Salem, NC 27157-1009, USA2Department of Medicine, Washington Hospital Center, 110 Irving St., NW, Washington, DC 20010, USA
* To whom correspondence should be addressed.
Received December 17, 1999
Carnosine (beta-alanyl-L-histidine) is an endogenous dipeptide found in various cells at millimolar concentration with its specific function(s) largely unknown. Our interests in therapeutic peptides led to the discovery that carnosine dramatically increases contractility when perfused into isolated rat hearts. Carnosine's effects are not mediated by histaminic or beta-adrenergic receptors or by increasing cyclic AMP, but carnosine does cause a rise in myoplasmic Ca2+ concentration. In chemically skinned cardiac cells, carnosine releases calcium, produces contracture, and alters the contractile protein's tension response to calcium. Carnosine also acts directly on the ryanodine receptor calcium release channel producing large increases in open state probability and dwelltime. In this manuscript, we will review studies which provide a basis for considering carnosine a modulator of calcium-regulated proteins in cardiac muscle cells and consequently an important determinant of contractility and cardiac function.
KEY WORDS: carnosine, isolated rat heart, calcium ion, calcium channels, ryanodine receptor
The failing heart represents an enormous clinical problem and is a major cause of death throughout the world. For example, 400,000 new cases are diagnosed in the United States each year and account for $10.2 billion in direct medical costs [1]. New therapies [2] are needed to treat heart failure because current treatment has only a limited impact on survival and annual costs. We have taken a novel approach to the failing heart by investigating a role for cytoplasmic peptides in the regulation of cardiac contractility. Small peptides are known to act as regulators of metabolic activity in cells. Examples include thyrotropin releasing hormone and endogenous opioid peptides, which act primarily via extracellular receptor systems. The dipeptide carnosine (beta-alanyl-L-histidine) occurs in cardiac muscle at concentrations of 2 to 10 mM [3-7], but an exact physiologic function for carnosine in muscle is unknown. Carnosine is postulated to participate in the maintenance of intracellular pH and may act as an antioxidant [8-10]. Tissue carnosine levels are decreased in animals with chronic infection [11] or following trauma [12], pathologic states often associated with impaired cardiac contractility [13]. Consequently, we performed a series of studies which tested the hypothesis that carnosine is an endogenous regulator of cardiac contraction.
Carnosine was perfused through the aorta of isolated rat hearts to determine effects on cardiac function [14]. Figure 1 shows sample traces from one of these experiments. In a concentration dependent manner, carnosine increased dP/dtmax and -dP/dtmax values, left ventricular systolic pressures, and heart rate (Table 1, Fig. 2). Carnosine's cardiotonic actions occurred at a perfused threshold concentration of 1 mM. Contractility, as assessed by dP/dtmax, was increased 440% above the pre-carnosine baseline level. When the carnosine perfusate was exchanged for control solution, the average time for dP/dtmax to decrease by 50% was 2.6 ± 0.6 min. Carnosine's cardiotonic actions were similar to those of isoproterenol (Table 1, Fig. 3) but lacked some of the undesirable side effects of isoproterenol. Three of six hearts perfused with isoproterenol >0.2 µM developed ventricular fibrillation while no hearts perfused with carnosine (up to 20 mM) developed this arrhythmia. From these studies we concluded that carnosine, at concentrations approximating intracellular levels (i.e., 2-10 mM), possessed potent inotropic effects in the isolated heart, increasing dP/dtmax from 206 ± 66 to 909 ± 81 mm Hg/sec. In an additional series of experiments, we demonstrated that neither the carnosine structural analogs beta-alanyl-L-phenylalanine, N-acetyl-L-histidine, and beta-alanyl-L-glycine nor the two, individual, component amino acids (i.e., beta-alanine and histidine) combined altered contractility when perfused into the isolated heart. Thus, the ability of carnosine to increase cardiac contractility was specific to its peptide structure.
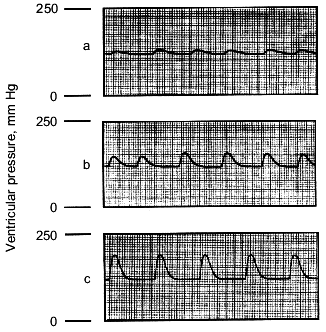
Table 1. Isolated rat heart left-ventricular systolic and diastolic pressures and heart rate response to increasing doses of carnosine (n = 6) and isoproterenol (n = 6) (data are mean ± SEM)Fig. 1. Representative traces of ventricular contraction showing the effect of carnosine on ventricular pressures in an isolated rat heart. Hearts were treated with 0 (a), 5 (b), and 10 mM (c) carnosine.
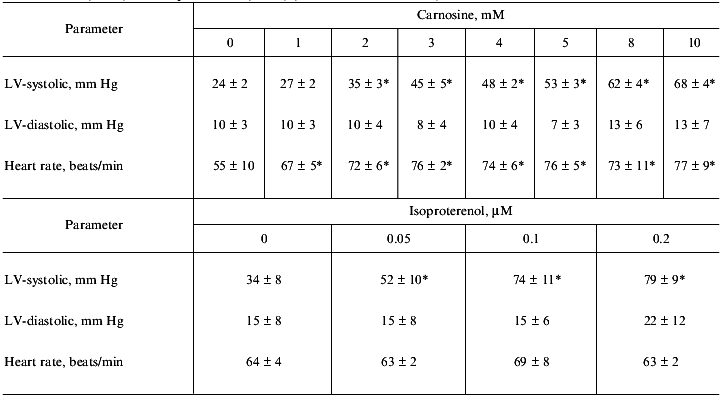
*p < 0.05 compared to baseline (ANOVA plus Tukey's multiple comparison test).
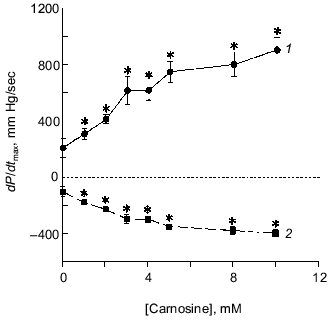
Fig. 2. Concentration-dependent effects of carnosine on dP/dtmax in isolated heart studies. From 1 to 10 mM, carnosine produced an increase in dP/dtmax (1). As measured by -dP/dtmax (2), rate of relaxation was also increased by increasing concentrations of carnosine (lower part of graph). All post-carnosine values (+dP/dtmax and -dP/dtmax) are statistically, significantly different (*) from the pre-carnosine control values.
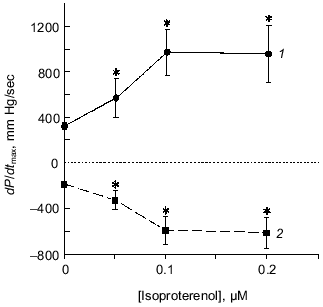
We also investigated possible pharmacologic pathways for carnosine's inotropic effects [14]. Histamine, perfused at 10-7 to 10-2 M, failed to increase contractility in 5 isolated hearts and at the highest concentration it depressed contraction, suggesting that carnosine did not mediate its cardiotonic actions via conversion to histamine [12, 15]. Following beta-adrenergic receptor blockade by perfusion of isolated hearts with propranolol (0.1 µM), carnosine administration increased contractility. beta-Adrenergic blockade was confirmed by lack of response to isoproterenol before and after carnosine perfusion. Thus, carnosine did not appear to depend upon the beta-adrenergic receptor for its cardiotonic effects. cAMP production by carnosine's action was ruled out by studies on isolated myocardial cell suspensions in which epinephrine increased [cAMP] from 0.62 ± 0.05 to 1.13 ± 0.14 pg/ml (p < 0.05) and forskolin increased the levels to 3.53 ± 0.48 pg/ml (p < 0.05). Carnosine failed to elevate cAMP levels in the myocardial cells, with concentrations of 0.76 ± 0.11 and 0.70 ± 0.08 pg/ml measured in the presence of 5 and 10 mM carnosine, respectively.Fig. 3. The effect of isoproterenol on ventricular contraction (dP/dtmax) in the isolated rat heart. Isoproterenol increased dP/dtmax (1) and -dP/dtmax (2) in a dose-dependent manner. All post-isoproterenol values (+dP/dtmax and -dP/dtmax) are statistically, significantly different (*) from pre-isoproterenol control values.
We next investigated carnosine's effect on free intracellular calcium utilizing experiments with myocyte suspensions loaded with the fluorescent indicator Fura-2 acetoxymethyl ester [14]. Following baseline determinations of myoplasmic [Ca2+], the effects of carnosine (4 to 20 mM), thapsigargin (10-5 M), and BayK (1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]pyridine-3-carboxylic acid methyl ester) (10-5 M) on [Ca2+] were tested. Carnosine increased free intracellular [Ca2+] in a dose-dependent manner (Table 2). A concentration of 20 mM carnosine increased [Ca2+] to levels comparable to that produced by the Ca-ATPase inhibitor thapsigargin but to significantly less than the levels produced by the calcium channel agonist BayK (Table 2).
Table 2. Effect of carnosine on myocyte free
intracellular calcium levels (values are means ± SEM)
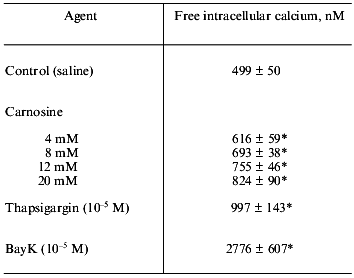
*p < 0.05 compared to baseline by ANOVA and Tukey's
multiple comparison test.
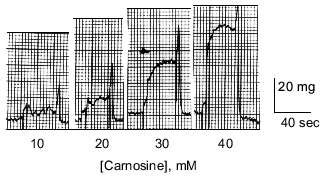
The effects of carnosine on contractility of the isolated heart and on free intracellular calcium levels in isolated cardiac myocytes prompted us to investigate the involvement of carnosine in myocardial calcium regulation. Chemically skinned rat ventricle cells were mounted for a calcium loading/releasing paradigm with tension measurements to assess carnosine's effect on calcium regulation in this model [14]. Carnosine, in a concentration dependent manner (5 to 40 mM), produced calcium release and tension responses in the skinned ventricular muscle cells (Fig. 4). Carnosine was less potent than caffeine (a releaser of intracellular calcium) for tension development, with 40 mM carnosine producing 80% of the tension produced by 20 mM caffeine. Procaine, a local anesthetic that blocks calcium release from the ryanodine receptor channel [16], effectively blocked the tension response to 40 mM carnosine, suggesting that carnosine acted by opening this calcium release channel.
A previous study by Lamont and Miller [17] reported that carnosine increased both skeletal and cardiac muscle contractile protein response to calcium. Investigation of detergent-treated (sarcoplasmic reticulum-deprived), chemically skinned rat ventricle cells by our colleagues showed that the effect of carnosine on contractile protein tension response to [Ca2+] was influenced by the [Ca2+] environment [14]. The small amount of tension generated at pCa = 6.4 (or [Ca2+] = 0.398 µM) was not altered by carnosine while at pCa = 6.0 (or [Ca2+] = 1.0 µM) carnosine reduced the tension response by 50%. At pCa = 5.4 (or [Ca2+] = 3.98 µM) carnosine was without effect, and at pCa = 4.8 (or [Ca2+] = 15.85 µM) the tension response was increased by 120%. We concluded from these tension studies that carnosine altered cardiac contractility by acting on two different mechanisms: the major intracellular calcium release channel and the contractile proteins. Furthermore, carnosine's effect on these processes may be dependent upon the existing myoplasmic [Ca2+], which can change markedly from nanomolar to micromolar concentrations during excitation-contraction coupling.Fig. 4. Carnosine produces tension in chemically skinned cardiac muscle cells. Increasing concentrations of carnosine produced increased tension responses up to 60 mg as carnosine was increased from 10 to 40 mM. These tension responses are a direct consequence of calcium release from the sarcoplasmic reticulum stores.
The ryanodine receptor (calcium release channel), a homotetrameric protein, is considered the main pathway for calcium release in both skeletal and cardiac muscle. It is a ligand-gated channel with naturally occurring modulators, which regulate open and closed states of the channel (activators include ATP, low calcium concentration; inactivators include magnesium, high calcium concentration, calmodulin). The cardiac ryanodine receptor protein isoform (RYR2) was isolated by our colleagues for investigation of carnosine's effects on radioligand binding and on the gating properties of single, purified, and reconstituted channels [14]. Binding of [3H]ryanodine to RYR is increased by modulators that increase the open state conformation of the RYR channel [18]. [3H]Ryanodine binding to RYR is optimized by the non-hydrolyzable ATP analog AMP-PCP (1 mM) and by 100 µM Ca2+, and in the absence of AMP-PCP the binding affinity is reduced by half. Carnosine affected [3H]ryanodine binding in a way similar to AMP-PCP by increasing binding affinity. In contrast, the dipeptide beta-alanyl-L-glycine at 20 mM had no effect on binding. The activation of [3H]ryanodine binding to RYR2 caused by carnosine was consonant with the effects observed when a single, cation-conducting RYR2 molecule was exposed to carnosine in electrophysiologic, planar lipid bilayer experiments [14]. Increasing concentrations of carnosine (1 to 5 mM) applied to the cytoplasmic (CIS) chamber produced increases in the channel's open state probability (Po), reaching 6-fold the control Po at 5 mM carnosine (Fig. 5). At 10 and 20 mM, carnosine caused a decline in activation down to 3-fold control Po levels. Carnosine (10 mM) produced a 2-fold increase in the longer open-state dwelltime constants of the channel while no statistically significant effect of carnosine on the closed dwelltime constants was found. The 6-fold increase in open state probability and the 2-fold increase in open dwelltime states produced by carnosine suggest that this dipeptide regulates the major calcium release channel in cardiac muscle.
In a separate set of experiments, we evaluated the effect of carnosine on vascular tone of isolated thoracic aortic rings from rats [19]. Aortic rings were preconstricted with phenylephrine. Carnosine (0.625-20 mM) produced dose-dependent vascular relaxation that was independent of endothelium. This effect was not produced by L-histidine and/or beta-alanine. The soluble cyclic-GMP guanylate cyclase inhibitor methylene blue significantly decreased the relaxation produced by carnosine suggesting that carnosine's vasodilatory actions were mediated by induction of cGMP in vascular smooth muscle.Fig. 5. The dose dependent effects of carnosine on the opening/closing of a single ryanodine receptor calcium release channel incorporated into a lipid bilayer. Traces show highest open state probability at 5 mM carnosine, and as carnosine was increased to 20 mM, the potentiation effect was diminished.

Ischemia is a common cause of myocardial damage. Carnosine possesses antioxidant properties and has been shown to scavenge oxygen free radicals, protect mitochondrial membranes from free radical damage, decrease lipid peroxidation of cell membranes, and has inotropic properties. Thus, it is reasonable to propose that carnosine may protect the heart from injury and improve function following cardiac ischemia. Prokop'eva et al. [20] evaluated the effect of carnosine (15 mM) in isolated rat hearts after hypoxia and reoxygenation. Carnosine increased coronary blood flow, diminished release of lactate dehydrogenase, and decreased the extent of contracture. In other studies, Rusakov and Dolgikh [21] studied the effect of carnosine on myocardial integrity in rats following hemorrhage-induced myocardial ischemia. Rats were hemorrhaged until cardiac arrest, and 4-6 min later the animals were resuscitated with blood with or without carnosine, closed cardiac massage, and artificial ventilation. Resuscitated rats that received carnosine had decreased coronary levels of two cardiac enzymes (aspartic aminotransferase and malic dehydrogenase). The carnosine treated animals also had decreased levels of lipid peroxidation products (diene conjugates and malonic dialdehyde). Lee et al. [22] assessed the relative ability of carnosine or histidine to scavenge singlet oxygen (a reactive oxygen species) in isolated rat hearts. Hearts were isolated and after an equilibration period were perfused with buffer containing carnosine (1 mM) or histidine (1 or 10 mM) for 20 min, then subjected to 40 min of global ischemia. Then the hearts were reperfused for 30 min. Carnosine perfused hearts had significantly better functional recovery as assessed by improvement in left ventricle diastolic pressure and dP/dt than histidine. Carnosine did not improve coronary flow or heart rate following injury. These data suggest that carnosine protects the myocardial cell from ischemic injury. Gercken and colleagues [23] reported beneficial cardiac effects of carnosine when administered as part of cardioplegic solutions. Unfortunately, controlled clinical trials have not compared carnosine-containing cardioplegic solutions with non-carnosine solutions.
Dolgikh et al. [24] assessed the effect of carnosine (25 mg/kg) on post-resuscitation arrhythmias in rats. Carnosine administered at the start of resuscitation promoted earlier recovery of cardiac contractions and respiratory and corneal reflexes and decreased cardiac rhythm disturbances. Carnosine also improved cardiac bioenergetics as indicated by increased ATP and phosphocreatine levels. In addition, carnosine also decreased lipid peroxidation products in heart tissue. In our studies in isolated rat hearts we also saw a decreased incidence of ischemia induced cardiac arrhythmias (especially ventricular fibrillation) compared to hearts receiving control buffer or beta-adrenergic agonists.
Carnosine has been reported to activate myofibrillar ATPase [25, 26]. Also, Simoniya et al. [27] evaluated the effect of carnosine on erythrocyte membrane Na,K-ATPase activity. The Na,K-ATPase activity of erythrocyte membranes (in vitro) was increased by carnosine. Interestingly, erythrocyte Na,K-ATPase activity was decreased in patients with congestive heart failure and acute myocardial infarction. The effect of carnosine on cardiac Na,K-ATPase activity is unknown. However, blockade of membrane Na,K-ATPase (i.e., digitalis) improves cardiac contractility. Thus, effects on this enzyme are unlikely to contribute to carnosine's inotropic actions. Carnosine was also reported to stimulate skeletal muscle sarcolemmal Na,K-ATPase1.
1Editor's note: The data were published by A. Boldyrev, V. Tkachuk, and V. Titanji (1974) Biochim. Byophys. Acta, 357, 319-324.
Our studies on isolated hearts did not determine if the inotropic effects of carnosine depend on the peptide acting intracellularly, extracellularly, or both. The skinned cell experiments identified two intracellular sites of action: carnosine-induced calcium release from the sarcoplasmic reticulum and an alteration of the contractile protein's tension-generating response to calcium. In the tension response of skinned muscle cells, carnosine effects were opposite at high and at low concentrations of calcium and consistent with carnosine's facilitation of contraction (at high calcium) and relaxation (low calcium) in the intact heart studies. Carnosine-induced calcium release in skinned muscle cells could be explained by an activation of the RYR2 calcium release channel. The blockage of carnosine-induced calcium release in skinned muscle cells by procaine and carnosine's effect to increase open state probability and dwelltimes in single RYR2 channel molecules are consistent with this notion.
Carnosine has been shown to have a variety of effects on the cardiovascular system. Carnosine improves contractility via calcium dependent mechanisms. It also decreases vascular tone, reducing the heart afterload. Finally, studies of carnosine during ischemic injury suggest that its antioxidant actions may offer a protective role for carnosine for maintenance of cardiac function with ischemic injury. Further in vivo studies in heart failure models are needed to determine the therapeutic potential of carnosine as a treatment for heart failure or ischemic injury.
Our investigations provide evidence that carnosine, an endogenous peptide, is involved in the regulation of calcium concentrations in cardiac muscle cells and also in the tension response of contractile proteins to changes in intracellular calcium concentrations. While these studies provide no information regarding how these carnosine effects are turned on and off in the muscle cell, they suggest new, exciting possible mechanisms by which contractility of the heart can be regulated by this unique endogenous peptide. We believe that this peptide is a previously unrecognized important intracellular messenger. Alterations in the levels and control of intracellular carnosine may play a role in the decreased contractility found in many disease states (i.e., congestive heart failure, sepsis). The discovery that the endogenous peptide carnosine may play a major multifactorial role in the regulation of cardiac muscle cell contraction offers new and exciting opportunities for increasing our knowledge regarding muscle physiology and for discovering new causes and therapeutic modalities for treating heart failure.
We wish to acknowledge technical assistance provided by Miyuki N. Shouse, Kimberly W. Black, Gisele Zapata-Sudo, Roberto T. Sudo, and Marina Lin in the experiments performed in our studies. Dr. Tom Nelson, a coinvestigator in our studies, continues to provide us with invaluable advice and encouragement to pursue answers to ongoing questions about the role of carnosine in cardiac function.
REFERENCES
1.Williams, R. S. (1995) N. Engl. J. Med.,
332, 817-818.
2.Milano, C. A., Allen, L. F., Rockman, H. A.,
Dolber, P. C., McMinn, T. R., Chien, K. R., Johnson, T. D., Bond, R.
A., and Lefkowitz, R. J.(1994) Science, 264, 582-586.
3.Chan, W. K. M., Decker, E. A., Chow, C. K., and
Boissonneault, G. A. (1994) Lipids, 29, 461-466.
4.Flancbaum, L., Fitzpatrick, J. C., Brotman, D. N.,
Marcoux, A. M., Kasziba, E., and Fisher, H.(1990) Agents
Actions, 31, 190-196.
5.Crush, K. G. (1970) Comp. Biochem.
Physiol., 34, 3-30.
6.O'Dowd, J. J., Robins, D. J., and Miller, D. J.
(1988) Biochim. Biophys. Acta, 967, 241-249.
7.House, J. R., Miller, D. J., and O'Dowd, J. J.
(1989) J. Physiol., 417, 162P.
8.Davey, C. L. (1960) Arch. Biochem. Biophys.,
89, 303-308.
9.Boldyrev, A. A. (1993) Int. J. Biochem.,
25, 1101-1107.
10.Boldyrev, A. A., Koldobski, A., Kurella, E.,
Maltseva, V., and Stvolinski, S. (1993) Mol. Chem.
Neuropathol., 19, 185-192.
11.Fitzpatrick, D., Amend, J. F., Squibb, R. L., and
Fisher, H.(1980) Proc. Soc. Exp. Biol. Med., 165,
404-408.
12.Fisher, D. E., Amend, J. F., Strumeyer, D. H.,
and Fisher, H.(1978) Proc. Soc. Exp. Biol. Med., 158,
402-405.
13.Parrillo, J. E., Parker, M. M., Natanson, C.,
Suffredini, A. F., and Danner, R. L.(1990) Ann. Int. Med.,
113, 227-242.
14.Zaloga, G. P., Roberts, P. R., Black, K. W., Lin,
M., Zapato-Sudo, G., Sudo, R. T., and Nelson, T. E. (1997) Am. J.
Phys., 272, H462-H468.
15.Fitzpatrick, J. C., Fisher, H., and Flancbaum, L.
(1991) Nephron, 59, 299-303.
16.Ohnishi, S. T., and Endo, M. (1981) The
Mechanism of Gated Calcium Transport Across Biological Membranes,
Academic Press, N. Y., pp. 257-264.
17.Lamont, C., and Miller, D. J. (1992) Am. J.
Physiol., 454, 421-434.
18.Chu, A., Diaz-Munoz, M., Hawkes, M. J., Brush,
K., and Hamilton, S. L. (1990) Mol. Pharmacol., 37,
735-741.
19.Ririe, D., Roberts, P. R., Shouse, M. N., and
Zaloga, G. P. (2000) Nutrition, in press.
20.Prokop'eva, V. D., Laptev, B. I., and Afanas'ev,
S. A. (1992) Biokhimiya, 57, 1389-1392.
21.Rusakov, V. V., and Dolgikh, V. T. (1992)
Biokhimiya, 57, 1393-1397.
22.Lee, J. W., Miyawaki, H., Bobst, E. V., Hester,
J. D., Ashraf, M., and Bobst, A. M. (1999) J. Mol. Cell. Card.,
31, 113-121.
23.Gercken, V. G., Bischoff, H., and Trotz, M.
(1980) Arzneimmittelforschung, 30, 2140-2143.
24.Dolgikh, V. T., Rusakov, V. V., Korpacheva, O.
V., et al. (1992) Resuscitation, 23, 179-191.
25.Yun, J., and Parker, C. J. (1965) Biochim.
Biophys. Acta, 110, 212-214.
26.Bowen, W. J. (1992) Arch. Biochem.
Biophys., 112, 436-442.
27.Simoniya, G. V., Tatishvili, N. I., Sheliya, D.
S., Bakanidze, N. T., and Khachidze, M. V. (1992) Biokhimiya,
57, 1343-1347.