Modeling of Substrate-Binding Region of the Active Site of Monoamine Oxidase A
A. V. Veselovsky*, A. E. Medvedev, O. V. Tikhonova, V. S. Skvortsov, and A. S. Ivanov
Institute of Biomedical Chemistry, Russian Academy of Medical Sciences, Pogodinskaya ul. 10, Moscow, 119832 Russia; fax: (095) 245-0768; E-mail: veselov@ibmh.msk.su* To whom correspondence should be addressed.
Received March 1, 2000; Revision received April 10, 2000
The mold of the substrate-binding region of the active site of monoamine oxidase A (MAO A) was designed using data of the enzyme interaction with reversible competitive inhibitors and the analysis of their three-dimensional structures. The superposition of ligands in biologically active conformations allowed determination of the shape and dimension of the active site cavity accommodating these compounds. The correctness of this approach was validated by the analysis of HIV protease interaction with its inhibitors using three-dimensional structures of HIV protease-inhibitor complexes. The mold of the substrate/inhibitor-binding site can be used for searching for new ligands in molecular databases and the development of a new generation of MAO inhibitors using lead structures that have not been employed for this purpose yet.
KEY WORDS: monoamine oxidase A, reversible inhibitors, active site, computer modeling, three-dimensional structure, molecular database
Spatial structure of proteins is usually evaluated by means of X-ray analysis or NMR-spectroscopy. These methods have been successfully employed for the determination of almost countless structures of water-soluble proteins. However, they have limited applicability for analysis of the numerous hydrophobic membrane-bound proteins. Lack of precise information on three-dimensional (3D) structure complicates investigation of mechanism of enzyme or receptor functioning and search for their effective ligands.
Monoamine oxidase (MAO; EC 1.4.3.4) plays an important role in the metabolism of important monoamine neurotransmitters. Changes in the activity of this enzyme have been observed in numerous neuropsychiatric disorders, and the employment of MAO inhibitors often produces a therapeutic effect [1]. In mammals MAO exists in two forms, MAO A and MAO B, which are encoded by two different genes [2], and characterized by different substrate specificity and sensitivity to selective inhibitors [1]. Both enzymes are integral proteins of the outer mitochondrial membrane, and they readily aggregate during purification procedures [3]. This feature explains the lack of information on their spatial structures. The possible use of computer modeling studies for evaluation of ternary structure is also complicated by the absence of proteins with known ternary structure which would be characterized by reliable homology of amino acid sequences with MAO A and B.
Thus, there are a few directions in modern analysis of structures of MAO A and B. The first is focused on studies of microbial soluble (mono)amine oxidases [3]. However, from our viewpoint the biochemical and molecular properties of these soluble enzymes differ from those of MAO A and B so drastically that they cannot be considered as evolutionary predecessors of mammalian MAOs [4].
The other direction is focused on the analysis of regions of amino acid sequences underlying differences of MAO A and B. Tsugeno and Ito identified “key amino acids” Phe-208 in MAO A and Ile-199 in MAO B responsible for the sensitivity of these enzymes to “diagnostic” substrates and inhibitors [5]. However, our analysis revealed that these amino acids cannot explain all differences in substrate specificity and inhibitor selectivity of MAO A and B [6].
Using a few series of reversible inhibitors of MAO A and B, we found steric differences of the active sites of these enzymes. Measurement of geometric sizes of rigid competitive (and selective) inhibitors suggests that the “length” of substrate/inhibitor-binding region of MAO A does not exceed 13-14 Å, whereas the “width” and “height” are 7.0 and 4.4 Å, respectively [6, 7]. According to these calculations, the substrate/inhibitor-binding region of MAO B is “shorter” (8.5 Å) and “narrower” (5.0 Å) [7, 8].
For subsequent analysis of structural features of the active sites of MAO A and MAO B and their visualization, we have used a method of spatial superposition of selective reversible competitive MAO inhibitors. In our view, such approach provides a “mold” of the substrate/inhibitor-binding region and estimates its dimensions and other characteristic features. For the independent evaluation of the correctness of this method, we have used HIV (human immunodeficiency virus) proteinase; this is an enzyme with known ternary structure.
MATERIALS AND METHODS
Our own and literature data on the inhibition of MAO A and MAO B by reversible competitive inhibitors were used.
The computer modeling was run on workstation Indigo 2 and Origin 200 server (Silicon Graphics, Inc.) using SYBYL 6.4 program (TRIPOS Inc.) [9]. Spatial alignment of protein-inhibitor complexes was carried out using MULTIFIT program. Three-dimensional models of the enzyme inhibitors were optimized using standard Tripos force field. Partial atom charges were calculated by semi-empirical quantum-mechanical method AM1. Conformations of molecules were calculated using program RandomSearch.
The pharmacophore model was built up using program DISCO and the following parameters: use of all compounds tested; the number of possible pharmacophore points varied from 2 to 6; possible deviation between pharmacophore points varied from 0.5 to 5.0 Å (with 0.5 Å interval).
The docking procedure was carried out using the original program of geometrical docking DockSearch developed in the Institute of Biomedical Chemistry, Russian Academy of Medical Sciences [10]. The program has the following parameters: angle of ligand rotation along each axis, 15°; standard deviation of distances between ligand atoms in complexes within one cluster, 0.25 Å.
For independent evaluation of the correctness of the method used, we also analyzed 3D-structures of complexes of HIV-proteinase taken from the KeyLock molecular database [11].
Molecular databases Comprehensive Medicinal Chemistry (CMC) and MDL Drug Data Report (MDDR) from MDL Information Systems, Inc. were also used in this study.
RESULTS
The interaction of substrates and reversible competitive inhibitors with an enzyme requires molecular complementarity between the ligand structure and its binding region at the active site, which also includes steric complementarity. In the case of an enzyme with wide substrate specificity, its interaction with compounds from various chemical classes may involve various functional groups located in different parts of the active site. So it is reasonable to expect that the sum of surfaces of these ligands would reflect the structure of the substrate/inhibitor-binding region of the active site. If this approach is correct, superposition of ligands may well describe the surface of this region of the active site.
For validation of this approach, we analyzed the active site of HIV-proteinase. This enzyme is characterized by known 3D-structures of its complexes with various inhibitors. Earlier it was demonstrated that inhibitors of this enzyme occupy a certain region of the active site and interact with the same functional groups [12].
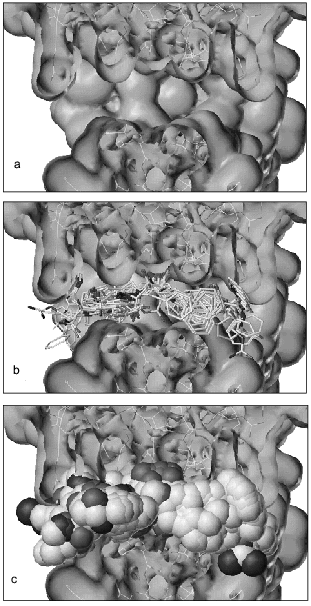
Figure 1a shows a cross-section of the 3D-structure of HIV-proteinase providing an open view on the surface of the active site, which represents an extended cavity. Figure 1b shows the active site of HIV-proteinase containing known inhibitor molecules in exact positions that they occupy in 3D-structures of enzyme-inhibitor complexes. This figure demonstrates that the inhibitor molecules are well superimposed and the main functional groups involved into protein molecule-binding coincide. Figure 1c shows the active site structure of HIV-proteinase with superimposed inhibitors whose atoms are shown as spheres with van der Waals radii. The set of inhibitors used represents a mold satisfactorily reflecting the structure of the active site cavity of this enzyme.
However, if the enzyme structure remains unknown, 3D-superposition of a ligand set becomes a real problem. In such case, determination of probable relative interposition of each ligand requires the design of an appropriate pharmacophore model. The pharmacophore model of HIV-proteinase inhibitors consists of two pharmacophore points: oxygen atom as an acceptor and phenolic ring (Fig. 2). These elements are found in all inhibitors; moreover, their interposition is tightly fixed in the inhibitors and they determine inhibitor binding at the active site of the HIV-proteinase [12]. In some compounds other functional groups closely neighbor each other. For example, many HIV-proteinase inhibitors contain a second phenol ring that has a less tightly fixed spatial location than the pharmacophore element. Such groups may also include some heteroatoms (oxygen or nitrogen). Thus, besides obligate (pharmacophore) elements the analysis of inhibitor structures revealed structural elements required for manifestation of high inhibitory activity; however, they are not ultimately essential for binding at the active site. These elements may be used for prediction of side chain ligand conformations during conformational analysis. The latter may also require additional data on stereoselectivity, results of 3D-QSAR analysis, etc.Fig. 1. Cross-section of the active site of HIV-proteinase: a) surface of the active site; b) the active site surface with superimposed inhibitors of HIV-proteinase; c) the active site surface with superimposed HIV-proteinase inhibitors shown as van der Waals spheres.
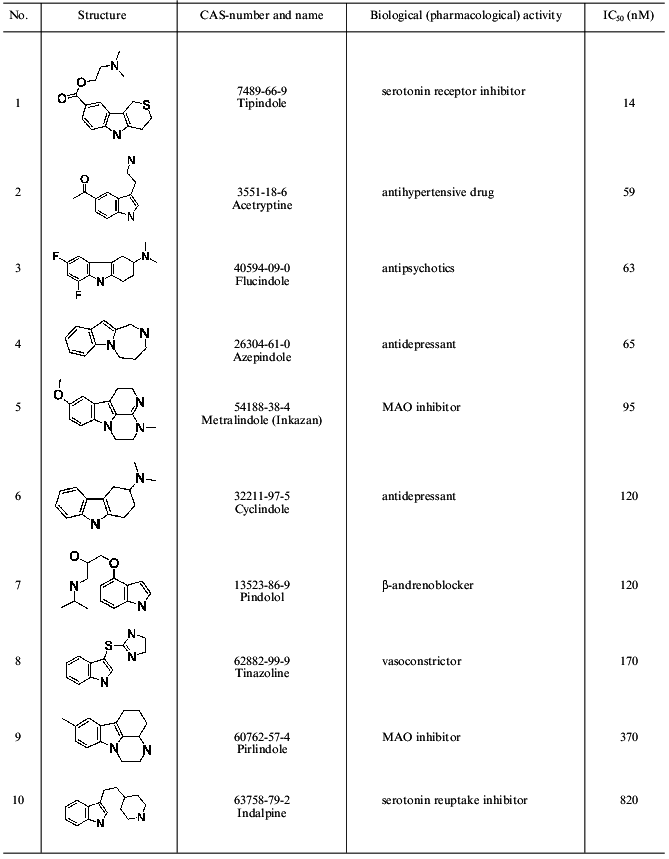
Molding of the substrate/inhibitor-binding region of the active site of MAO A was “facilitated” by the existence of a few series of selective inhibitors characterized by rigid basic structure (derivatives of indole, isatin, pyrazinocarbazoles) [13]. Since rigid structures have a very limited number of possible conformers, compounds possessing a rigid moiety were used for molding (Table 1). Comparative analysis revealed the following common elements present in all inhibitors: phenolic ring with neighboring negatively charged nitrogen or oxygen atom. This is consistent with our previous pharmacophore model proposed for MAO A inhibitors [14]. The presence of a phenol ring is an ultimate precondition for the manifestation of MAO A inhibitory activity. Substitution of this phenol ring for a non-aromatic cycle sharply reduces MAO A inhibitory activity [15]. So inhibitor molecules were aligned by these pharmacophore elements. Molecules possessing a rigid nucleus (compounds 18-20) were used for the backbone of the mold.Fig. 2. Pharmacophore model of HIV-proteinase inhibitors. AA, acceptor atom; DS, donor site; HR, centroid of hydrophobic ring.
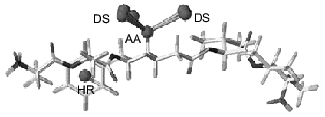
Table 1. Structures of MAO A inhibitors used
for the active site molding

Prediction of the position of side radicals was based on two approaches. The first employed models of the MAO A active site developed using 3D-QSAR + CoMFA models, which were built up on the basis of indole and isatin [14] or pyrazinocarbazole [7] MAO inhibitors. These models allowed prediction of favorable and unfavorable regions for substituents and to construct the scheme of the active sites of MAO A and B. The data obtained predict the existence of some cavity in the active site of MAO A accommodating a substituent at the 8th position of pyrazinocarbazole (compounds 20-22, Table 1). Such cavity is absent in the active site of MAO B [7].
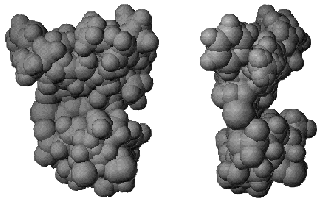
The second approach included conformational analysis of side radicals of inhibitors and search of those conformers of these inhibitors that are characterized by close interposition of functional groups potentially involved in binding to the protein. So radical phenol rings of compounds 2, 13, and 22 from one side and 8 and 19 from another were superimposed. Oxygen atoms of carboxy-groups of compounds 8 and 6, nitrogen atoms of cyano-groups of compounds 2 and 4, nitrogen atoms of compounds 11, 12, 16 (and also some other groups) were also superimposed. Figure 3 shows the resultant mold of the substrate/inhibitor-binding region of MAO A.
This mold reflects the structure and shape of the active site. However, correct characterization of the substrate/inhibitor-binding region requires creation of the surface limiting the volume of the mold. In this case, “removal” of mold-forming molecules yields a cavity mimicking the active site cavity. The resultant model of the active site of an enzyme with unknown 3D-structure may be employed for searching for new ligands in molecular databases by means of docking programs.Fig. 3. Orthogonal projection of the mold of substrate/inhibitor-binding region of MAO A active site.
Applicability of this method for characterization of the active site structure of MAO A was analyzed using indole, isatin [14], and pyrazinocarbazole derivatives [7] as the enzyme inhibitors. Molecules from the CMC database (containing information about 6,300 structures of known drugs) were docked into the resultant cavity after pre-processing of this database. The latter included selection of molecules containing an indole moiety in their structures. For molecules which were accommodated within this cavity, we have calculated IC50 values using a 3D-QSAR + CoMFA model developed for pyrainocarbazole derivatives [7]. Table 2 shows molecules with the lowest IC50 values predicted. Two of ten compounds with the highest predicted inhibitory activity are MAO A inhibitors.
Table 2. Compounds of the CMC database,
their known biological (pharmacological) activity, and MAO A inhibitory
activity predicted by the 3D-QSAR + CoMFA model
DISCUSSION
The rather high interest in monoamine oxidases is generally explained by the use of some MAO inhibitors in clinical practice. However, 3D-structures of both MAO A and MAO B remain unknown. Attempts of several laboratories to elucidate structural elements of these enzymes by such experimental methods as construction of chimeric molecules from MAO A and MAO B [16-18] or site-directed mutagenesis [5, 19, 20] did not result in exhaustive information on the structure and characteristic features of these enzymes. Lack of such information stimulated active use of computer methods for investigation of structural features of MAO A and MAO B [7, 14, 21-23].
Our approach for active site molding is based on two assumptions: 1) compounds bind at the active site of the enzyme provided that they can be accommodated within its cavity; 2) ligand (substrate or potent competitive inhibitor) binding at the active site requires shape complementarity. In this case, a set of effective ligands may well characterize the spatial structure of the active site.
Earlier several approaches for modeling of the active site spatial structure for enzymes with unknown 3D-structure were proposed. The first approach includes pseudoreceptor modeling [24-26]; the second is the so-called active analogs approach [27]. Pseudoreceptor modeling visualizes the active site structure by “covering” a single ligand (or aligned set of ligands) with a polypeptide shell. This method uses general notions on favorable interposition of various functional groups of the protein and the ligand. For example, taking into consideration the possibility of hydrogen bond formation, a histidine residue of the protein is usually positioned near the hydroxy group of ligand phenyl ring, whereas an asparagine residue usually neighbors an amino group of the ligand. Ligand removal yields some cavity in the protein structure that is subsequently used for prediction of active site properties and searching for new ligands. However, the resultant cavity is far from the actual active site structure [26].
The active analogs approach uses a set of ligands containing both active and inactive compounds. Prediction of the active site cavity by this method involves spatial alignment of active and inactive analogues followed by comparison of their volumes [27].
In contrast to the active analogs approach, our method employs only active ligands that readily fit into the active site. This yields a cavity accommodating known active ligands. Cutting unfavorable or unknown regions occurs along the border of the region occupied by effective ligands. In contrast to the pseudoreceptor method, our active site molding method does not predict the structure and active site properties at the level of individual amino acid residues.
Our approach reflects the 3D-dimensions of the active site cavity of MAO A. This cavity together with the method of geometric docking can be used for searching for new ligands in molecular databases. This significantly accelerates analysis of molecular databases and selection of potential candidates for subsequent investigation. Pilot use of this method for screening the CMC database revealed its effectiveness. For example, Table 2 includes Pirlindole (compound 9) and Inkazan (5). They are effective MAO A inhibitors employed clinically [28]. Selection of Pirlindole in this database was not unexpected because this compound had been used in molding and the 3D-QSAR model. Inkazan was not used in the previous analysis, and its selection demonstrates the correctness of our method.
Thus, our method visualizes the 3D-structure of the substrate/inhibitor-binding region, which may promote subsequent characterization of the active site of MAO A. Besides clarifying basic aspects underlying structural features of this important enzyme, our “mold” can be used to search for new inhibitors in molecular databases. We believe that this approach significantly extends modern tools for the development of a new generation of effective MAO inhibitors.
This work was supported by the Russian Foundation of Basic Research (grant No. 99-04-48822) and INTAS (grant No. 99-00433).
REFERENCES
1.Gorkin, V. Z., and Medvedev, A. E. (1995) in
Proteins and Peptides [in Russian], Vol. 1, Nauka, Moscow, pp.
83-88.
2.Shih, J. C., Chen, K., and Ridd, M. J. (1999)
Annu. Rev. Neurosci., 22, 197-217.
3.Singer, T. P., Yankovskaya, V. L., Bernard, S.,
Cronin, K., and Sablin, S. O. (1997) Vopr. Med. Khim.,
43, 440-456.
4.Veselovsky, A. V., Ivanov, A. S., and Medvedev, A.
E. (1997) Vopr. Med. Khim., 43, 527-536.
5.Tsugeno, Y., and Ito, A. (1997) J. Biol.
Chem., 272, 14033-14036.
6.Veselovsky, A. V., Ivanov, A. S., and Medvedev, A.
E. (1998) Biochemistry (Moscow), 63, 1441-1446.
7.Medvedev, A. E., Veselovsky, A. V., Shvedov, V. I.,
Tikhonova, O. V., Moskvitina, T. A., Fedotova, O. A., Axenova, L. N.,
Kamyshanskaya, N. S., Kirkel, A. Z., and Ivanov, A. S. (1998) J.
Chem. Inf. Comput. Sci., 38, 1137-1144.
8.Medvedev, A. E., Ivanov, A. S., Kamyshanskaya, N.
S., Kirkel, A. Z., Moskvitina, T. A., Gorkin, V. Z., Li, N. Y., and
Marshakov, V. Yu. (1995) Biochem. Mol. Biol. Int., 36,
113-122.
9.SYBYL 6.4. Tripos Inc.1699 South Hanley Road. St.
Louis. Missouri. 63144. USA.
10.Ivanov, A. S., Skvortsov, V. S., Tikhonova, O.
V., Veselovsky, A. V., and Archakov, A. I. (1998) 12 Int. Symp. on
Microsomes and Drug Oxidations, 20-24 July, 1998, Montpellier,
France, 259.
11.http://lmgdd.ibmh.msk.su/KeyLock.
12.Wlodawer, A., and Vondrasek, J. (1998) Annu.
Rev. Biophys. Biomol. Struct., 27, 249-284.
13.Cesura, A. M., and Pletscher, A. (1992) Progr.
Drug Res., 38, 171-297.
14.Medvedev, A. E., Ivanov, A. S., Veselovsky, A.
V., Skvortsov, V. S., and Archakov, A. I. (1996) J. Chem. Inf.
Comput. Sci., 36, 664-671.
15.Harfenist, M., Joyner, C. T., Mize, P. D., and
White, H. L. (1994) J. Med. Chem., 37, 2085-2089.
16.Gottowik, J., Malherbe, P., Lang, G., Daprada,
M., and Cesura, A. M. (1995) Eur. J. Biochem., 230,
934-942.
17.Chen, K., Wu, H. F., and Shih, J. C. (1996) J.
Neurochem., 66, 797-803.
18.Grimsby, J., Zentner, M., and Shih, J. C.
(1996) Life. Sci., 58, 777-787.
19.Zhou, B. P., Lewis, D. A., Kwan, S. W., Kirksey,
T. J., and Abell, C. W. (1995) Biochemistry,34,
9526-9531.
20.Hiro, I., Tsugeno, Y., Ogata, F., and Ito, A.
(1996) J. Biochem., 120, 759-765.
21.Maret, G., Tayar, N. E., Carrupt, P.-A., Testa,
B., Jenner, P., and Baird, M. (1990) Biochem. Pharmacol.,
40, 783-792.
22.Altomare, C., Carrupt, P.-A., Gaillard, P.,
Tayar, N. E., Testa, B., and Carotti, A. (1992) Chem. Res.
Toxicol., 5, 366-375.
23.Thull, U., Kneubuhler, S., Gaillard, P., Carrupt,
P. A., Testa, B., Altomare, C., Carotti, A., Jenner, P., and McNaught,
K. S. P. (1995) Biochem. Pharmacol., 50, 869-877.
24.Hahn, M. (1995) J. Med. Chem., 38,
2080-2090.
25.Hahn, M., and Rogers, D. (1995) J. Med.
Chem., 38, 2091-2102.
26.Zbinden, P., Dobler, M., Folkers, G., and Vedani,
A. (1998) Quant. Struct.-Act. Relat., 17, 122-129.
27.Kettmann, V., and Holtje, H. (1998) Quant.
Struct.-Act. Relat., 17, 91-101.
28.Mashkovsky, M. D. (1993) Drug Reference Book
[in Russian], Meditsina, Moscow.