REVIEW: Thrombin as a Regulator of Inflammation and Reparative Processes in Tissues
S. M. Strukova
Department of Human and Animal Physiology, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; E-mail: strukova@mail.ru
Received May 30, 2000; Revision received August 19, 2000
Activation of blood coagulation and thrombin formation accompany inflammation, wound healing, atherogenesis, and other processes induced by endothelial injury. Systems of hemostasis and inflammation play an important role in the pathogenesis of acute coronary syndromes. This paper reviews thrombin functions involved in its interaction with PAR family receptors, activation of platelets, endothelial cells, leukocytes, smooth muscle cells, and mast cells. Mechanisms of regulatory effects of thrombin on mast cells associated with nitric oxide release are discussed.
KEY WORDS: thrombin, receptor, agonist peptide, mast cells, nitric oxide, inflammation, wound healing
Abbreviations: ICAM-1) intracellular adhesion molecule-1; IL-1) interleukin 1; MAP kinase) mitogen-activated protein kinase; MCP-1) monocyte chemoattractant protein-1; L-NAME) methyl ether of N-nitro-L-arginine; NO) nitric oxide; cNOS) constitutive NO-synthase; iNOS) inducible NO-synthase; PAF) platelet activating factor; PAR) protease-activated receptor; PRP) platelet rich plasma; TGF-beta1) transforming growth factor beta; TNF-alpha) tumor necrosis factor alpha; TRAP) thrombin receptor agonist peptide; VCAM-1) vascular cell adhesion molecule-1.
Thrombin (EC 3.4.21.5), a serine protease of the trypsin family, is a
key enzyme of the blood clotting system. It converts fibrinogen to
fibrin and participates in the regulation of numerous physiological and
pathophysiological processes such as blood clotting and
anticoagulation, thrombus formation and fibrinolysis, regulation of
vascular tone, developmental processes, and also inflammation, tissue
reparative processes, atherogenesis, carcinogenesis, and Alzheimer's
disease [1-6].
The polyfunctionality thrombin arises from its structure: besides the “classical” active site, it contains several additional sites called exo-sites or sub-sites of the additional recognition centers for substrate and receptors. They are responsible for specific binding with substrates and receptors and for high selectivity in catalytic cleavage of peptide bond and substrate selection [7-9]. Structural-functional features of thrombin and mechanisms of its proteolytic activity with respect to fibrinogen and other substrates, proteins of the blood clotting system, are well characterized [7-10]. The involvement of thrombin in the inflammatory reactions and tissue reparative processes, which are always accompanied by the activation of the blood clotting system, thrombin formation, and its concentration in the a vascular lesion and surrounding tissue, still requires detailed investigation.
Until recently thrombin was considered mainly as an anti-inflammatory factor; its regulatory effects on cell functioning were not taken into consideration [11-13]. So, there is now interest in studies on the role of thrombin in reactions limiting the inflammatory processes (blockade of cell adhesion to the damaged vascular site, inhibition of mast cell secretion of inflammatory transmitters). Thrombin formed at a site of vascular damage activates numerous cells involved in the inflammatory and reparative response, including monocytes, blood T-lymphocytes, and connective tissue mast cells located along the blood vessels [1-5]. Mast cells are involved in the activation of endothelial cells, leukocyte migration, edema formation, and other processes related to tissue repair. Mast cells activated by immune and non-immune liberators release inflammatory transmitters and modulators [14-16]. Immune activation of mast cells and regulatory mechanisms responsible for secretion of these products and their functions are intensively investigated, whereas mechanisms of non-immune activation of these cells by thrombin are less studied.
Here we consider mechanisms of the action of thrombin as a regulator of inflammatory and proliferative phases of reparative processes of tissues.
MECHANISMS OF FORMATION OF THROMBIN
The exposure of the tissue factor on the surface of endothelial cells and activated monocytes is the key step during the development of the inflammatory response and subsequent blood clotting. It is provoked by endothelial lesions induced by balloon angioplasty, bacterial toxins, viral infection, cytokines (TNF, IL-1, etc.), oxidized lipoproteins, and certain growth factors [17-19]. The tissue factor is a transmembrane glycoprotein; it belongs to the family of cytokine receptor class II. The tissue factor can activate cells via two mechanisms. Complex formation between the tissue factor and factor VII/VIIa (VII active) of the blood clotting system initiates a proteolytic mechanism in which the tissue factor operates as a cofactor and modulator of factor VII/VIIa. Binding of factor VIIa with the tissue factor causes an increase in intracellular Ca2+ and phosphorylation of mitogen-activated protein kinases (MAP kinases) Erk-1, Erk-2, p38, and Jnk; this results in transcription of Egr-1 (early growth response) gene, usually inducible by cytokines and growth factors. The non-proteolytic mechanism involves the cytoplasmic domain of the tissue factor, which participates in intracellular signaling leading to cell adhesion and migration [20-22].
It is now well recognized that the so-called “extrinsic pathway” of blood clotting triggered by the tissue factor is the main mechanism of thrombinogenesis in the blood stream [23-25]. In the blood stream, ~1% of factor VII exists in the double-stranded form VIIa cleaved at Arg152; however, its conformation prevents contacts with plasma factors and their activation [23]. Binding of the tissue factor changes inactive conformation of factor VIIa into the active one and activates factors IX and X. Formation of small quantities of factor Xa stimulates the appearance of trace amounts of thrombin sufficient for activation of cofactors (factors V and VIII) into their active forms (factors Va and VIIIa). Activation of these two cofactors is important for the formation of the effective catalysts of blood clotting (bound with membrane phospholipids and Ca2+): the factor IXa-factor VIIIa complex (also called tenase) activating factor X and the factor Xa-factor Va complex (prothrombinase) converting prothrombin into thrombin. These complexes are ~105-106 times more active than their serine proteases. Activation of factor X by tenase is ~50 times more effective than by the factor VIIa-tissue factor complex.
Plasma, platelets, and the vascular wall contain an inhibitor of the extrinsic pathway, TFPI (tissue factor pathway inhibitor). Its initial binding with high affinity of factor Xa and then complex factor VIIa-tissue factor limits factor Xa formation. The discovery of TFPI led to the suggestion that the effective formation of factor Xa in vivo requires not only generation of the VIIa-the tissue factor complex but also generation of the factor IXa-factor VIIIa complex [23, 25]. Data on the heavy clinical course of hemophilia A caused by defects in the gene encoding factor VIII support this hypothesis.
During the inflammation process, thrombin may also be formed on the surface of activated monocytes via tissue factor- and factor VII-independent mechanisms. Monocyte activation is accompanied by exposure of adhesion proteins on the cell surface. This includes exposure of integrins alphaMbeta2, which is also called Mac-1 or CD 11b/CD 18. They belong to the beta2 family, the members of which can bind (among other proteins) factor X. Cathepsin G released from activated leukocytes induces proteolytic activation of membrane-bound factor X (into Xa), and the latter can convert prothrombin to thrombin [26].
POLYFUNCTIONALITY OF THROMBIN
The best-known function of thrombin is the conversion of fibrinogen into fibrin. Thrombin regulates blood clotting and exhibits both procoagulant and anticoagulant activities. Thrombin induces proteolytic activation of factors V, VIII, XI, and platelet aggregation, and it therefore accelerates its own formation. However, binding of endothelial thrombomodulin to thrombin paradoxically induces anticoagulant activity and changes the substrate specificity of thrombin. The latter cannot convert fibrinogen to fibrin but is able to catalyze the conversion of protein C into an active protease. The activated protein C blocks thrombin formation by inactivating factors Va and VIIIa. In the heavy chain of factor Va, activated protein C cleaves three peptide bonds--those at Arg306, Arg506, and Arg679; the resultant form cannot operate as a cofactor of factor Xa [25, 27, 28]. Thrombomodulin bound thrombin can block fibrinolysis. It activates TAFI (thrombin activated fibrinolysis inhibitor), and the resultant TAFIa (carboxypeptidase) cleaves the C-terminal lysine and arginine residues from the fibrin molecule; this reduces its affinity for plasminogen and the rate of formation of thrombin, the enzyme cleaving fibrin [29].
Thrombin stimulates cells involved in inflammatory processes and tissue repair: monocytes, T-lymphocytes, endothelial and smooth muscle cells, fibroblasts, and mast and nervous cells [1-6].
Thrombin stimulates the synthesis and exposure on the endothelial cell surface of adhesion proteins, activators and inhibitors of fibrinolysis, and activators and inhibitors of adhesion and aggregation of blood cells [1, 6, 30].
Under various conditions thrombin exerts opposite effects on vascular tone [31, 32]. It can stimulate vasodilatation by releasing nitric oxide and prostacyclin (PGI2) from endothelial cells. However, thrombin can also promote vasoconstriction by releasing endothelin-1 from endothelial cells and thromboxane A2 from platelets [31, 32].
Serine protease inhibitors, serpines (antithrombin III, heparin cofactor II), the protease nexin 1, and alpha2-macroglobulin protease inhibitor regulate thrombin activity. Proteoglycans, endothelial thrombomodulin, and heparin (synthesized by mast cells) significantly potentiate thrombin inhibition by antithrombin III and heparin cofactor II [33].
The polyfunctionality of thrombin and its role as pro- and anticoagulant is determined by features of its structure and allosteric regulation of its activity.
STRUCTURAL-FUNCTIONAL FEATURES OF THROMBIN
Thrombin, a trypsin-like serine protease, is characterized by high specificity with respect to the cleaved substrates and peptide bonds. This high specificity is determined by the existence of not only its “classical” active site, but also additional recognition centers for substrates and receptors. Sub-sites of this recognition center include anion-binding exo-site 2 (also called the heparin-binding site) and anion-binding exo-site 1 (also called the fibrinogen recognition site). The latter interacts with complementary sites of specific substrates, inhibitors, and receptors and thus is responsible for the high proteolytic specificity of thrombin and its regulatory functions [7-9]. X-Ray analysis, site-directed mutagenesis, and chemical modification have revealed the important role of anion-binding exo-site 1 in the interaction of thrombin with fibrinogen, fibrin, thrombomodulin, heparin cofactor II, protease activated receptor (PAR), and hirudin (a highly selective exogenous thrombin inhibitor). These methods also revealed the role of exo-site 2 in heparin binding. Besides positively charged groups, binding of hirudin and other ligands also required a hydrophobic site on the thrombin molecule [34].
Binding of thrombomodulin [1] and Na+ [35-37] causing conformational changes in the thrombin molecule are responsible for the allosteric regulation of this protease. The effect of monovalent cations on the kinetics of substrate hydrolysis by thrombin was initially demonstrated more than two decades ago [38, 39]. Recent X-ray analysis revealed conformational changes that occur in the thrombin molecule during Na+ binding and that result in an increased rate and specificity of fibrinogen cleavage [37]. Two Na+-binding sites have been identified. Binding of Na+ induces an allosteric conformational change of the thrombin molecule and a structural transition from the “slow” (Na+-free) to “fast” (Na+-bound) form. Both forms exist under physiological conditions (145 mM NaCl) [40]. The transition from the slow to the fast form is a key stage in the molecular recognition of substrates. The fast form is characterized by higher affinity and catalytic activity with respect to fibrinogen than the slow form. The latter is more specific in the activation of the anticoagulant protein C. The point mutation Trp60-->Asp reduces Na+ binding and causes a shift to the slow form accompanied by an increase in the anticoagulant activity of thrombin [41]. This modified form of thrombin might be employed in clinical practice for anticoagulant therapy together with widely used dicoumarins and other vitamin K antagonists, reducing the rate of prothrombin activation. Thus, allosterically induced conformational changes may determine both the pro- and anticoagulant properties of thrombin.
THROMBIN RECEPTORS
The effects of thrombin on cells are mediated by membrane-bound thrombin receptors. Some serine proteases not only cleave proteins but also act as signaling molecules that interact with cell receptors and regulate cell functions [42]. Receptors for factor Xa, protein C, and urokinase, which regulate cell functions without cleavage of receptors, have been recognized [43-46]. In contrast to these proteases, thrombin regulates cell activity by cleaving and activating PAR receptors [30, 42, 47-51]. All members of PAR family are related to the superfamily of integral membrane single-domain G-protein-coupled receptors. Receptors of catecholamines, acetylcholine, serotonin, histamine, angiotensins, tachykinins, endothelins, PAF, etc. also belong to this superfamily. Four main members of the PAR family have been recognized: PAR-1, -3, and -4 are thrombin receptors, PAR-2 is a receptor for trypsin and mast cell tryptase [30, 47-53] (table).
Receptors of the PAR family and their agonists and producing cells
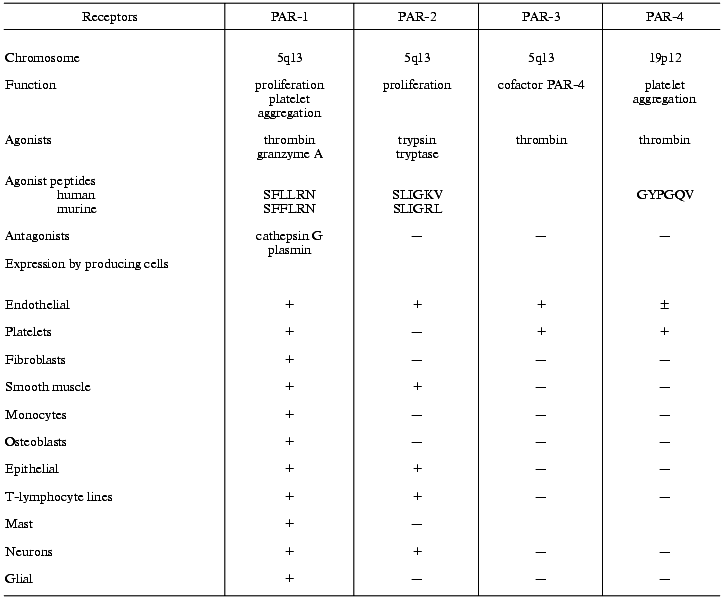
PAR receptors expressed by numerous cell types are involved in various processes such as growth, development, mitogenesis, atherogenesis, and inflammation [30, 42-50, 53].
Proteases cleave one peptide bond in the extracellular domain of PAR, and the new N-terminal site (so-called “bound ligand”) interacts with the extracellular domain of the cleaved receptor and activates it. This is a characteristic feature of the PAR family.
Mechanisms of PAR-1 activation by thrombin have been studied using gene engineering--chimera and point mutations. Thrombin cleaves the Arg41-Ser bond in the domain LDPR41SFLLRN located before the negatively charged cluster D51KYEPF, which is complementary to the anion-binding exo-site 1 of the recognition center of the thrombin molecule. Specific binding of thrombin with this cluster is necessary for effective cleavage of the receptor [30, 48, 49].
Synthetic peptide SFLLRN corresponding to the N-terminus of the bound ligand is a thrombin receptor agonist peptide (TRAP). It activates cells by a receptor cleavage-independent mechanism. Analysis of SFLLRN analogs and site directed mutagenesis of the bound ligand revealed that Phe2, Leu4, and Arg5 residues are crucial for the activity of the ligand [42, 54]. PAR-1 agonist peptide interacts with the receptor at the site of the second extracellular loop [55]. Thus, PAR-1 can be activated by both irreversible and reversible (ligand binding) mechanisms. The former represents the physiological mechanism. The second requires high concentrations of TRAP analogs, significantly (~3 orders of magnitude) exceeding effective concentrations of thrombin.
Thrombin functions related to inflammation, tissue repair, and wound healing involve the activation of PAR-1. The expression of the gene encoding PAR-1 changes under certain pathological conditions. Increased PAR-1 expression was noted in atheromas, in areas enriched by macrophages and smooth muscle cells, and also in synovial membrane fibroblasts in rheumatoid arthritis [13, 30, 42].
Release of TGF-beta1 and PDGF (platelet derived growth factor) from aggregated platelets stimulates the expression of PAR-1 mRNA. This stimulates the mitogenic activity of thrombin with respect to smooth muscle cells at lesion sites on the vascular wall [56]. Recently, expression of PAR-1 (but not PAR-2) mRNA was found in rat peritoneal mast cells; however, contradictory data exist on thrombin-induced histamine secretion by these cells [57, 58]. A new mechanism of mouse platelet activation by thrombin was found in Coughlin's laboratory [59]: PAR-3 is not activated by thrombin but functions as a cofactor required for cleavage and activation of PAR-4 by thrombin. This may explain the ineffectiveness of peptide analogs of PAR-3 agonist in activation of the receptor [42].
There are mechanisms that interrupt signal transduction and prevent uncontrolled receptor stimulation. They include cleavage of bound ligand and phosphorylation of the receptor followed by its internalization with subsequent recycling to the cell surface (after ligand cleavage and dephosphorylation) or lysosomal degradation [42, 60]. Some proteases can cleave PAR-1 at sites distinct from those required for receptor activation. This produces a partially degraded receptor form which is not activated by thrombin. For example, in addition to the Arg41-Ser bond, cathepsin G also hydrolyzes the Phe43-Leu and Phe55-Trp peptide bonds; this is accompanied by cleavage of the bound ligand and the resulting tolerance of PAR-1 to thrombin [42].
MECHANISMS OF SIGNAL TRANSDUCTION DURING ACTIVATION OF PAR-1 BY
THROMBIN
The PAR-1 receptor of thrombin is coupled to various G-proteins (Galphaq/11, Go, Galpha12, Galpha13, Gi), so its activation triggers various intracellular signal transducing systems [42, 61]. In many cells, activation of PAR-1 by thrombin involves receptor binding to Galphaq/11 followed by activation of phospholipase C (PLC)beta. The latter results in the formation of phosphatidylinositol-4,5-bisphosphate (PIP2) and of the classic second messengers: inositol-1,4,5-trisphosphate (IP3) (which releases Ca2+ from intracellular stores) and diacylglycerol (DAG) (which activates protein kinase C, PKC) [1, 6, 42, 61]. In platelets, PKC phosphorylates Ser/Thr residues in myosin light chains (p20) and in plecstrin (p47). The latter can bind PIP2 and block its subsequent hydrolysis; this mechanism plays a role in the regulation of cytoskeleton actin [61]. PKC and Ca2+ activate platelets and promote the exposure of alphaIIbbeta3 integrins on the cell surface; these integrins bind fibrinogen and stimulate new signal transduction and aggregation in the later stages of platelet activation [61, 62].
Thrombin causes a dramatic increase of Tyr phosphorylation in many platelet proteins by activation of non-receptor tyrosine kinases such as Src (p60), Syk (p72), and FAK (p125). Activation of FAK kinase is related to the activation of alphaIIbbeta3 integrins and fibrinogen binding [63, 64]. Exposure on the platelet surface of fibrinogen receptor alphaIIbbeta3 and reorganization of cytoskeleton is controlled by GTP-binding proteins: Ras, Rho, and Rac [61, 62].
Since mechanisms of stimulation by thrombin of DNA synthesis and of cell proliferation and differentiation involve PAR-1 binding to various G-proteins, the activation of Ras and related proteins and also of MAP kinases can occur via several pathways [42]. In cultured fibroblasts thrombin activates non-receptor tyrosine kinases (such as Fyn and Src) which interact with receptor tyrosine kinases. Epidermal growth factor (EGF) receptor is the ultimate component for the PAR-mediated mitogenic signal of thrombin and other ligands (agonists of G-protein coupled receptors) [64]. Tyrosine phosphorylation in Shc adapter protein causes binding of complex Grb2 (growth factor receptor binding protein 2)-SOS (son of sevenless protein) that activates Ras protein (p21) [65]. This is the start of a serine/threonine protein kinase cascade: p21Ras phosphorylates Raf-1 kinase (or kinase of MAP kinase kinase). The latter phosphorylates MEK-1/-2 (kinase of MAP kinase), which in turn phosphorylates ERK-1/-2 (protein kinases regulated by the extracellular signal) (MAP kinases). This complicated (and not completely understood) mechanism of signal transduction from receptor tyrosine kinases to MAP kinases involves many regulators of Ras activity [66, 67]. Activation of this signaling pathways ends in the activation of transcription factors triggering early response genes [42, 65]. Such factors as NF-kappaB, Erg-1, Sp1, and AP-1 regulate transcription of the tissue factor and intracellular adhesion molecule 1 (ICAM-1) [22, 68-70]. Factors Sp1 and AP-2 also influence transcription of PDGF-B, PDGF-A, TGF-beta1, u-PA [68, 69]. For each of these proteins, regulatory genes contain binding sites that determine the potential interaction between the gene and the protein.
Cytoplasmic transcription factor NF-kappaB can be activated in various signaling pathways transducing effects of inflammatory cytokines, oxidized lipids, and factors present in atheromatous plaque; this factor can rapidly activate many genes [70]. In the cytosol of many cells, factor NF-kappaB exists as an inactive complex which contains Rel-related proteins including p65 (Rel A) and p50 (NF-kappaB1) bound to inhibitory proteins IkappaB [71, 72]. Members of the Rel family contain activating domains required for gene induction; they are all characterized by distinct DNA-binding properties [73]. Usually, an inactive heteromeric complex of NF-kappaB associated with one of the IkappaB inhibitors (IkappaBalpha, IkappaBbeta_or others) exists in the cytoplasm [73]. Activation of NF-kappaB occurs after phosphorylation of serine residues by ubiquitin-regulated serine kinases and subsequent degradation of phosphorylated IkappaB by a multi-catalytic proteosome complex [73-75].
Inactivation of IkappaB is a central event in the inflammatory process and leads to gene expression of adhesion molecules (ICAM, VCAM) and cytokines (TNF-alpha, IL-1, etc.). Transcriptional factors Sp1 and Egr-1 contain DNA binding motifs known as zinc fingers by which they bind to DNA and regulate transcription of numerous genes [76]. Egr-1 binding sites on the PDGF gene promoter overlap with Sp1 binding sites. Endothelial damage activates Egr-1, which can replace Sp1 on the PDGF gene promoter and thus modulate transcription [77].
Thrombin can directly influence the activity of the transcription regulators and their expression [77]. Regulatory sites of most thrombin-induced factors contain the kappaB sequence for the interaction with NF-kappaB. NF-kappaB-binding sites also present in thrombin-regulated genes encoding E-selectin, MCP-1, PAI-1, and c-myc, and also cytokines (INF-gamma_and beta, IL-3, IL-6, IL-8, G-CSF), adhesion molecules (ICAM-1, VCAM-1) and vasoregulatory molecules (NOS) [78, 79]. The interaction of thrombin with PAR-1 of cultured vascular smooth muscle cells activated NF-kappaB and stimulated proliferation of these cells [80]. Treatment with anti-sense oligonucleotide to p65 NF-kappaB inhibited the thrombin-induced proliferation of the smooth muscle cells [81]. Stimulation of smooth muscle cells by TRAP and bFGF (basic fibroblast growth factor) induced degradation of IkappaBalpha_and activation of NF-kappaB [82]. MAP kinase inhibitor suppressed the transcriptional activity of NF-kappaB and proliferation of smooth muscle cells; this suggests the involvement of ERK1/2 activity. ERK1/2 may play an important role in the thrombin-induced activation of NF-kappaB.
The promoter of the PAR-1 gene contains regulatory sites for AP-2 and Sp1 [83]. The latter significantly reduces the activity of the PAR-1 promoter in vascular endothelial cell culture [84].
Besides the existence of NF-kappaB sites in thrombin-regulated genes, functional correspondence between thrombin effects and the expression of some genes has also been demonstrated. Thrombin-dependent activation of PAR-1 triggers the expression of ICAM-1 in endothelial cells; this mechanism involves the NF-kappaB site in the promoter and subsequent leukocyte adhesion [85]. Thrombin also induces the expression of P- and E-selectins, VCAM-1 and a chemokine secretion [86]. Thrombin-dependent activation of PAR-1 also stimulates expression of IL-8 in human monocyte cell culture and expression of IL-6 and G-CSF in synovial fibroblasts in rheumatoid arthritis [79, 87].
THROMBIN AS A REGULATOR OF REPARATIVE PROCESSES
Thrombin formed in a damaged site coordinates reparative processes in tissues; it stimulates cells involved in the main stages of wound healing. Thrombin activates adhesion and aggregation of platelets, activation of endothelial cells, release of growth factors from cells, and adhesion and recruitment of leukocytes (preferentially monocytes and T-lymphocytes); it stimulates proliferation of endothelial and epithelial cells, fibroblasts, neurons, and smooth muscle cells and interacts with mast cells [1-3, 5, 6, 9, 42, 58, 88-90].
Thrombin is considered an inflammatory transmitter because exposure to relatively high thrombin concentrations (>10 nM) is accompanied by rapid transient (during minutes) increase of endothelial permeability. Known inflammatory transmitters (cytokines) cause a long-term (days, hours) stable response [91]. Endothelial activation and the pro-inflammatory state are characterized by exposure of adhesion proteins and their receptors on the cell surface, release of platelet aggregation inducers and chemo-attractants responsible for monocyte activation, and their recruitment on the vascular surface.
Several factors are exposed on the surface of activated endothelium, providing leukocyte adhesion and rolling. These include adhesion molecules of the P (platelet) and E (endothelial) selectin families and molecules of the immunoglobulin family (ICAM-1 and VCAM-1) [92]. Thrombin also stimulates local secretion of endothelial chemo-attractants (MCP-1) and growth factors. Increase in cell migration and proliferation of fibroblasts and endothelial cells are characteristic features of the proliferative phase of wound healing. High concentrations of thrombin promote destabilization of intracellular junctions and impairment of focal contacts of the endothelial cell with the matrix [93]. This effect can also be attributed to local activation of prometalloproteases (MMP2) (gelatinases) by thrombin (these enzymes hydrolyze collagen and elastin and therefore accelerate cell migration and vascular remodeling) [94]. It was recently shown that proMMP2 activation by thrombin is a more complicated process than thought earlier [95]. Thrombin can cleave a propeptide from MMP2 activator (called MT1-MMP, or membrane type 1 MMP) that is localized on the endothelial cell membrane. This results in its binding with the N-terminal inhibitory domain of the inhibitor MMP2-TIMP2. At the same time, the C-terminal domain of TIMP2 binds MMP2; the latter can be activated by MT1-MMP on the cell surface provided that it is bound to the “receptor complex” TIMP2/MMP [95].
The difference in concentrations required for MMP hydrolysis (10-100 nM) and DNA synthesis during stimulation of endothelial proliferation (<0.1 nM) obviously reflects differences in mechanisms of thrombin action: receptor-independent cleavage of MMP-2 and PAR-1-dependent DNA synthesis and cell proliferation [96].
Some effects can stem from thrombin-mediated release of platelet activating factor (PAF), a potent inflammatory transmitter and vasodilatator increasing endothelial permeability and monocyte chemotaxis [91, 97].
Several attempts have been made to employ thrombin and its peptide fragments as growth factors for wound healing [2, 5, 98]. The use of thrombin was limited by its lability and the anti-inflammatory effect of high concentrations. However, fibrin glue is employed as a hemostatic and wound healing agent. A fibrin clot can be formed directly in the wound by treating the wound surface with a mixture of fibrinogen and thrombin solutions (in the presence of Ca2+, adhesion proteins, fibrin stabilizing factor XIII, and fibrinolysis inhibitors) [99, 100]. Fibrin glue preparations are quite expensive because they require careful purification. Thrombin has also been included in polymer composites formed during the thermal precipitation of polyvinyl caprolactam calcium alginate hydrogel. Study of the influence of hydrogels with encapsulated enzyme on wound healing in experimental wounds in rats and mice revealed significant acceleration of reparative processes caused by increased migration, cell proliferation, and angiogenesis (neovascularization) in the granular tissue [2, 101, 102]. A possible reason for thrombin-induced angiogenesis may be an effect (direct or indirect) of thrombin on the expression of VEGF receptors (VEGF is one of the main promoters of proliferation of endothelial cells and development of new vessels) [103].
The stimulatory effect on migration and proliferation of cells (including smooth muscle cells) underlies the involvement of thrombin in processes of wound healing and the development of atheromatous vascular damage. Increased expression of PAR-1 was noted in areas of vascular damage and in atheromas [42].
Thrombin can activate smooth muscle cells by stimulating STAT 3/SIF-A (signal transducers and activators of 3/sis-induced factor A transcription); it can also regulate the effect on aortal smooth muscle cells of such cytokines as IL-6, LIF, CNTF by inhibiting the STAT 3/SIF-A activity induced by these cytokines [104]. The latest studies by Libby's laboratory revealed that hirulog, a peptide inhibitor of thrombin, did not influence expression of inflammation markers (ICAM-1, TNF-alpha, IL-1) in the early response phase after balloon damage to rabbit aorta [105]. They concluded that thrombin formed during vascular damage is not a critical stimulus for the inflammatory response and smooth cell proliferation, and this probably explains the insufficient effectiveness of thrombin inhibitors in prevention of restenosis in patients [105]. Thrombin can not only stimulate but also regulate the activity of cells involved in inflammation and proliferation. The regulatory functions are mediated by PAR-1 and other PAR-family receptors (PAR-3 and PAR-4). It is possible that other (unknown) thrombin receptors are also involved. For example, the thrombin effect as a factor of myoblast survival is not related to PAR-1 activation, although thrombin participates in the reparation and development of muscle tissue by stimulating myoblast proliferation and inhibiting apoptosis of cultured cells [106].
However, PAR-1 activation by thrombin can result in either a classical pro-inflammatory or anti-inflammatory cell response [107, 108]. The latter is mediated by NO released from monocytes in response to the activation of endothelin (ET-1) receptor. The activation of endothelial PAR-1 by thrombin or PAR-1 agonist peptide can result in rapid release of endothelin, which binds to the monocyte receptor and stimulates NO release. TRAP can also release NO from monocytes, and this effect is blocked by pretreatment of cells with antagonist of the ET-1 receptor. NO inhibits monocyte adhesion to endothelium. However, formation of a large amount of thrombin (seen under such pathological conditions as diabetes and atherosclerosis) is accompanied by a sharp increase in endothelin release, and this causes desensitization of endothelin receptors on monocytes. This results in a decrease in NO release (related to normal functioning of endothelin receptor functioning) and promotes cell adhesion, the development of the inflammatory response, and the progression of the disease [107]. Thus, the activation of thrombin receptor (PAR-1) can inhibit cell adhesion to the endothelium; this effect is mediated by ET-1 receptor-dependent NO formation. So, thrombin can promote the manifestation of certain regulatory mechanisms of the cell response in sites of vascular lesions.
The anti-inflammatory effect of NO may also be attributed to the inhibition of expression of ICAM-1 and other adhesion molecules in endothelial cells by influencing transcriptional factors. NO stabilizes the inactive heterotrimer NF-kappaB/IkappaBalpha_in cytosol by stimulating gene expression of the IkappaBalpha_inhibitor [109].
Thrombin can release NO from the endothelium. Local NO formation by endothelial cells prevents expression of adhesion molecules usually inducible by various atherogenic stimuli (which can activate NF-kappaB). This mechanism operates in the intact vascular wall. However, in atheromatous regions this anti-inflammatory mechanism is less effective [109].
Chronic inhibition of endothelial NO release by L-NAME administration to rats caused the development of an inflammatory response: transmigration of monocytes into coronary vessels, activation of NF-kappaB, and expression of MCP-1. Administration of anti-sense oligonucleotides to NF-kappaB into rat hearts prevented the development of the inflammation induced by L-NAME administration [110]. This suggests the involvement of transcriptional factor NF-kappaB in the development of the early inflammatory response and contribution of endothelial NO (released by thrombin) to anti-inflammatory endothelial mechanisms.
However, thrombin can also inhibit the expression of inducible NOS (iNOS) and release of large quantities of NO, thus exhibiting cytotoxic effects in IL-1-activated cultured aortal smooth muscle cells [111].
The role of thrombin as a regulator of NO release is also important during non-immune activation of mast cells.
EFFECT OF THROMBIN ON MAST CELLS
Mast cells are actively involved in wound healing. They release many potent transmitters in response to activation by immune (IgE and specific antigens) and non-immune liberators (components of the complement system, neuropeptides and regulatory peptides, lysosomal neutrophil proteins, insect venom, calcium ionophores, some drugs, cold stress, etc.). Among the released regulators are transmitters and modulators of inflammation, proliferation, and cell migration that are preformed and kept in cytoplasmic mast cell granules (histamine, neutral proteases, chymase and tryptase, acidic hydrolases, cathepsin G, carboxypeptidase, heparin, chondroitin sulfate, proteoglycans). Mast cells also release inflammatory transmitters synthesized after cell activation by PAF, PGD2, leukotriene C4, cytokines (IL-4, IL-5, IL-6, IL-8, IL-13, TNF-alpha), MIP-1alpha_(macrophage inflammatory protein 1alpha), and bFGF. Some cytokines (e.g., TNF-alpha) can be preformed in mast cells and also synthesized during the cell activation. Mast cells also release such regulators as NO, TGF-beta1, activator of fibrinolysis tPA (but not inhibitors of plasminogen activators: PAI-1 and PAI-2); they expose receptors of IgE, urokinase, growth factor (c-kit), etc. [14-16, 112-115]. The specific binding of FITC-labeled thrombin with rat peritoneal mast cells suggests the presence of thrombin receptor on these cells [116]. Evidence for the existence of mast cell PAR-1 was obtained by Strukova et al. [117]. They showed an increase in intracellular Ca2+ in response to the PAR-1 receptor agonist TRAP-6. Recently, PAR-1 mRNA was detected in peritoneal mast cells [57].
In many tissues, thrombin contacts mast cells that are localized along blood and lymphatic vessels at sites of tissue damage. Some evidence exists that thrombin can be formed by macrophages (where prothrombin mRNA was found) and nervous and developing muscular tissue [118, 119]. Little is known about the interaction of thrombin with mast cells. Mast cell deficient mice were characterized by increased incidences of thrombembolia after the provocation of thrombus formation [120]. Increased numbers of mast cells were found in the heart of patients with atrial thrombosis and in the adventitia of thrombosed profound veins [113, 115].
Thrombin can cause degranulation of cultured mast cells of bone marrow and skin. High concentrations of thrombin stimulate degranulation of rat peritoneal mast cells [1, 58, 121]. Heparin-dependent proteases secreted by mast cells can cleave exogenous thrombin [122]. Inflammation induced by chronic administration of bacterial lipopolysaccharide or thioglycocholic acid salts significantly reduced thrombin inactivation by cells [123].
The pro-inflammatory effect of thrombin in vivo may be mediated by mast cell PAR-1 receptor; administration of TRAP analog peptides increases vascular permeability and induces rat paw edema [11, 124]. Histochemical analysis revealed mast cell degranulation [11]. However, the conclusion that mast cell degranulation stems from a direct effect of TRAP on these cells requires better experimental documentation.
Patch-clamp potential studies and intracellular pH (pHin) measurements revealed that thrombin caused a dose-dependent increase in mast cell membrane conductivity and a biphasic change of pHin (initial decrease followed by subsequent increase of pHin mediated by PKC-dependent activation Na+/H+ exchange) [58, 125, 126]. High concentrations of thrombin can cause mast cell degranulation and histamine release, whereas low concentrations of thrombin increase cGMP in these cells and reduce spontaneous histamine secretion [58]. This is consistent with data on a biphasic effect of thrombin on the permeability of an epithelial cell monolayer (low concentrations reduce whereas high concentrations increase cell permeability) [127]. It is possible that--as in the case of platelets, fibroblasts, and endothelial cells--mast cell activation by thrombin involves reactions mediated by several receptors (including PAR-1). However, the mechanisms underlying the modulation of mast cell reactivity by thrombin remain unknown.
NO can be an endogenous regulator of mast cells: it activates guanylate cyclase [128, 129] and inhibits histamine and PAF secretion by cytokine-activated mast cells [130]. PAR-1-mediated modulation of mast cell reactivity was demonstrated during a study of the effect of TRAP-6 on NO release [131]: mast cell activation caused NO release, which blocked inducible platelet aggregation. This effect of TRAP-6 was blocked by L-NAME (inhibitor of NO formation) or calmidazolium (an inhibitor of calmodulin and constitutive NOS).
Calcium-ionophore-stimulated mast cells (a model of non-immune activation) released PAF, the potent inflammatory transmitter and platelet aggregation inducer [130, 131]. Pretreatment of mast cells with TRAP-6 (before calcium ionophore stimulation) caused significant (3-fold) reduction in PAF-induced increase of inducible platelet aggregation [131]. It is possible that thrombin acting at PAR-1 operates as a non-immune regulator of mast cell activity by stimulating NO formation and inhibiting secretion of PAF. These data support the hypothesis of a role of NO as a modulator mast cell activity and mast cell-induced recruitment of leukocytes in the inflammatory nidus [132]. NO donors inhibit histamine release by lipopolysaccharide-activated mast cells, mast-cell activation of leukocytes in vivo and in vitro, and histamine-induced increase in endothelial permeability [133-135].
Differences in the effects of thrombin and TRAP analog peptides [136] suggest that the pro- and anti-inflammatory effects of thrombin can involve PAR-1-independent mechanisms. For example, thrombin-induced rat paw edema was almost completely abolished by preliminary mast cell exhaustion by a degranulator or cyclooxygenase inhibitor (indomethacin); this suggests the involvement of mast cells in the effect of thrombin. However, the peptide-induced edema was insensitive to indomethacin treatment and only slightly reduced in rats treated with s mast-cell degranulator. Co-administration of thrombin and peptides revealed an anti-inflammatory effect of thrombin. However, the control peptide FSLLRY, which did not activate PAR-1, caused the same edema as the experimental peptide.
PAR-2 expressed by numerous cells involved in inflammatory processes (leukocytes, endothelial cells, neurons) is not a thrombin receptor (table). It is activated by trypsin, mast cell tryptase, and PAR-2 agonist peptides; PAR-2 mediates acute inflammatory responses characterized by the development of edema and leukocyte infiltration [137-139]. Rat paw edema induced by PAR-2 agonist administration was not suppressed by pretreatment of the animals with a mast cell degranulator or NOS inhibitor; this suggests that the development of the inflammatory response occurs by a mechanism independent from mast cell activation and NO generation [139].
Thus, activation of blood clotting during damage to vascular endothelium is a part of the protective mechanism that coordinates cell and humoral responses accompanying inflammation and reparative processes in tissues. Thrombin is a key enzyme of blood clotting; it is actively involved in blood clotting and inflammation processes. Paradoxically, thrombin can operate as a procoagulant (converting fibrinogen into fibrin, activating blood clotting factors and blood cells) and as an anticoagulant (binding thrombomodulin and activating anticoagulant system of protein C). Allosteric changes in the thrombin molecule conformation can determine its pro- and anticoagulant properties. Moreover, thrombin can operate as an inflammatory transmitter activating endothelial cells, adhesion, monocyte recruitment, and increasing endothelial permeability. At the same time, low concentrations of thrombin regulate inflammation processes by blocking monocyte adhesion to endothelium, platelet aggregation, and by releasing NO from cells. The same (low) concentrations exhibit growth factor-like activity and accelerate proliferation of fibroblasts and endothelial and smooth muscle cells. Literally speaking, thrombin functions as a conductor of cell responses during inflammation and reparative processes in tissues. The effects of thrombin are mainly mediated by binding to and subsequent cleavage of PAR family receptors coupled to G-proteins. The mechanism of receptor activation by thrombin includes cleavage of an N-terminal receptor fragment followed by opening of a bound ligand that activates the receptor. Receptors of the PAR family (table) expressed by numerous cell types are involved in the mechanisms of inflammation, growth, development, and tissue repair. PAR activation by thrombin stimulates signal transduction mechanisms which may end in activation of transcription factors regulating the expression of the tissue factor, adhesion protein, growth factors, cytokines, other ligands involved in inflammation processes, and migration and proliferation of cells. PAR-1 agonist peptides simulate thrombin effects on cells but without receptor cleavage. The use of these agonist peptides and PAR antagonists might be useful for regulation of cells involved in blood clotting, inflammation, and tissue repair.
REFERENCES
1.Strukova, S. M., Kireeva, E. G., and Dugina, T. N.
(1997) Vestnik MGU, Biol. Ser., 16, 8-14.
2.Strukova, S. M., Dugina, T. N., Chistov, I. V.,
Markvicheva, E. A., Kuptsova, S. V., Kolokolchikova, E. G., Rumsh, L.
D., Zubov, V. P., and Glusa, E. (1998) Bioorg. Khim., 24,
288-292.
3.Henrikson, K. P., Salazar, S. L., Fenton, J. W.,
and Pentecost, B. T. (1999) Br. J. Cancer, 79,
401-406.
4.Glusa, E., Paintz, M., and Bretschneider, E. (1996)
Semin. Thromb. Hemost., 22, 261-266.
5.Carney, D. H., Redin, W., and McCroskey, L. (1992)
Semin. Thromb. Hemost., 18, 91-103.
6.Grand, R., Turnell, A., and Grabham, P. (1996)
Biochem. J., 313, 353-358.
7.Strukova, S. M., Sereiskaya, A. A., and Osadchuk,
T. V. (1989) Usp. Sovr. Biol., 107, 41-54.
8.Stubbs, M., and Bode, W. (1993) Thromb.
Res., 69, 1-58.
9.Fenton, J. W., II. (1995) Thromb. Haemost.,
74, 493-498.
10.Stubbs, M., and Bode, W. (1995) Trends
Biochem. Sci., 20, 23-28.
11.Cirino, G., Cicala, C., Bucci, M., Sorrentino,
L., Maraganore, J., and Stone, S. (1996) J. Exp. Med.,
183, 821-827.
12.Kranzhofer, R., Clinton, S., Ishii, K., Coughlin,
S., Fenton, J., and Libby, P. (1996) Circ. Res., 79,
286-294.
13.Morris, R., Winjard, P., Blake, D., and Morris,
C. (1994) Ann. Reum. Dis., 53, 72-79.
14.Valent, P., Sillaber, C., Baghestanian, M.,
Bankl, H. C., Kiener, H. P., Lechner, K., and Binder, B. R. (1998)
Int. Arch. Allerg. Immunol., 115, 2-8.
15.Bissonnette, E., Enciso, J., and Befus, A.
(1997) Am. J. Respir. Cell Mol. Biol., 16, 275-282.
16.Galli, S. J. (2000) Curr. Opin. Hematol.,
7, 32-39.
17.Taubman, M. B., Fallon, J., Schecter, A., Giesen,
P., Mendlowitz, M., Fyte, B., Marmur, J., and Nemerson, Y. (1997)
Thromb. Haemost., 78, 200-204.
18.Osterud, B. (1997) Thromb. Haemost.,
78, 755-758.
19.Libby, P., Egan, D., and Skarlatos, S. (1998)
Circulation, 96, 4095-4103.
20.Banner, D., D'Arcy, A., Chene, C., Winkler, F.,
Guha, A., Koningsberg, W., Nemerson, Y., and Kirchhofer, D. (1996)
Nature, 380, 41-46.
21.Ruf, W., and Mueller, B. (1999) Thromb.
Haemost., 82, 175-182.
22.Prydz, H., Camerer, E., Rottingen, J.-A., Wiiger,
M. T., and Gjernes, E. (1999) Thromb. Haemost., 82,
183-192.
23.Mann, K. (1999) Thromb. Haemost.,
82, 165-174.
24.Roberts, H. R., Monroe, D. M., Oliver, J. A.,
Chang, J. Y., and Hoffman, M. (1998) Haemophilia, 4,
331-334.
25.Mann, K., Nesheim, M., Church, W., Haley, P., and
Krishnaswamy, S. (1990) Blood, 76, 1-16.
26.Plescia, J., and Altieri, D. (1996) Biochem.
J., 319, 873-879.
27.Kalafatis, M., Rand, M., and Mann, K. G. (1994)
J. Biol. Chem., 269, 1869-1880.
28.Rosing, J., and Tans, G. (1997) Thromb.
Haemost., 78, 427-433.
29.Nesheim, M., Wang, W., Boffa, M., Nagashima, M.,
Moser, J., and Bajzar, L. (1997) Thromb. Haemost., 78,
386-391.
30.Coughlin, S. (1999) Proc. Natl. Acad. Sci.
USA, 96, 11023-11027.
31.Yang, Z., Arnet, U., Bauer, E., Segesser, L.,
Siebenmann, R., Turina, M., and Lusher, T. (1994)
Circulation, 89, 2266-2272.
32.Ogletree, M. L. (1996) in The Endothelial Cell
in Health and Disease (Vane, J., Born, G., and Welzel, D., eds.)
Schattauer. Stuttg., N. Y., pp. 85-96.
33.Bombeli, T., Mueller, M., and Haeberli, A. (1997)
Thromb. Haemost., 77, 408-423.
34.Bode, W., Brandstetter, H., Mather, T., and
Stubbs, M. (1997) Thromb. Haemost., 78, 501-511.
35.Wells, C. M., and di Cera, E. (1992)
Biochemistry, 31, 11721.
36.Di Cera, E., Guinto, E., Vindigni, A., Dang, Q.,
Ayala, Y., Wuyi, M., and Tulinsky, A. J. (1995) J. Biol. Chem.,
270, 22089-22092.
37.Zhang, E., and Tulinsky, A. (1997) Biophys.
Chem., 63, 185-200.
38.Strukova, S., Umarova, B., Kireeva, E., and
Kudryashov, B. (1978) Biokhimiya, 43, 708-716.
39.Orthner, C., and Kosow, D. (1980) Arch.
Biochem. Biophys., 202, 63-67.
40.Di Cera, E. (1998) Trends Card. Med.,
8, 340-350.
41.Guinto, A., and di Cera, E. (1997) Biophys.
Chem., 64,103-109.
42.Dery, O., Corvery, C. U., Steinhorff, M., and
Bunnett, N. W. (1998) Am. J. Physiol., 274,
C1429-C1452.
43.Altieri, D. (1995) J. Leukoc. Biol.,
58,120-127.
44.Cirino, G., Cicala, C., Bucci, M., Sorrentino,
A., Ambrosini, G., de Dominicis, G., and Altieri, D. (1997) J. Clin.
Invest., 99, 2446-2451.
45.Esmon, C., Xu, J., Gu, J.-M., Qu, D., Laszik, Z.,
Ferrell, G., Stearns-Kurosawa, D., Kurosawa, S., Taylor, F., and Esmon,
N. (1999) Thromb. Haemost., 82, 251-258.
46.Koshelnick, Y., Ehart, M., Stockinger, H., and
Binder, B. (1999) Thromb. Haemost., 82, 305-311.
47.Cupit, L., Schmidt, V., and Bahou, W. (1999)
Trends Cardiovasc. Med., 9, 42-48.
48.Vu, T.-K., Hung, D., Wheaton, V., and Coughlin,
S. (1991) Cell, 64, 1057-1068.
49.Rasmussen, U., Vouret-Craviari, V., Jallot, S.,
Schlesinger, Y., Pages, G., Parirani, A., Lecocq, J.-P., Pouyssegur,
J., and van Obberghen-Schilling, E. (1991) FEBS Lett.,
288, 123-128.
50.Brass, L. (1997) Coronary Artery Disease,
8, 49-58.
51.Molino, M., Barnathan, E., Numerof, M., Clark,
J., Dreyer, M., Cumashi, A., Hoxi, J., Schechter, N., Woolkalis, M.,
and Brass, L. (1997) J. Biol. Chem., 272, 4043-4049.
52.Ishihara, H., Connolly, A., Zeng, D., Kahan, M.,
Zheg, Y., Timmons, C., Tram, T., and Coughlin, S. (1997)
Nature, 386, 502-506.
53.D'Andrea, M., Derian, C., Leturcq, D., Baker, S.,
Brunmark, A., Ling, P., Darrow, A., Santulli, R. J., Brass, L. F., and
Andrade-Gordon, P. (1998) J. Histochem. Cytochem., 46,
157-164.
54.Scarborough, R., Naughton, M., Teng, W., Hung,
D., Pose, J., Vu, T., Wheaton, V., Turck, C., and Coughlin, S. (1992)
J. Biol. Chem., 267, 13146-13149.
55.Nanevicz, T., Wang, L., Chen, M., Ishii, M., and
Coughlin, S. (1996) J. Biol. Chem., 271, 702-706.
56.Schini-Kerth, V., Bassus, S., and Fissithaler, B.
(1997) Circulation, 96, 3888-3896.
57.Nishikawa, H., Kawabata, A., Kuroda, R., Nishida,
M., and Kawai, K. (2000) Jpn. J. Pharmacol., 82,
74-77.
58.Strukova, S. M., Dugina, T. N., Khlgatian, S. V.,
Redkozubov, A. E., Redkozubova, G. P., and Pinelis, V. G. (1996)
Semin. Thromb. Hemost., 22, 145-150.
59.Nakanishi-Matsui, M., Zheng, Y. W., Sulciner, D.
J., Weiss, E. J., Ludeman, M. J., and Coughlin, S. R. (2000)
Nature, 404, 609-613.
60.Trejo, J., and Couhlin, S. R. (1999) J. Biol.
Chem., 274, 2218-2223.
61.Brass, L. F., Manning, D., Cichowski, K., and
Abrams, C. S. (1997) Thromb. Haemost., 78, 581-589.
62.Shattil, S. (1999) Thromb. Haemost.,
82, 318-325.
63.Fox, J. (1999) Thromb. Haemost.,
82, 385-391.
64.Cichowski, K., Orsini, M. J., and Brass, L. F.
(1999) Biochim. Biophys. Acta, 1450, 265-276.
65.Narpenter, G. (1999) J. Cell Biol.,
146, 697-702.
66.Anderson, R. G. W. (1998) Annu. Rev.
Biochem., 67, 199-225.
67.Sternberg, P. W., and Alberola-Ila, J. (1998)
Cell, 95, 447-450.
68.Lu, C., Giordano, F. J., Bao, X., Morris, K. C.,
and Rothman, A. (1998) Circulation, 98, 596-603.
69.Chen, Y., Grall, D., Salcini, A., Pelicci, P.,
Pouyssegur, J., and van Obberghen-Schilling, E. (1996) EMBO J.,
15, 1037-1044.
70.Mackman, N. (1997) Thromb. Haemost.,
78, 747-754.
71.Khachigian, L. M., Lindner, V., Williams, A. J.,
and Collins, T. (1996) Science, 271, 1427-1431.
72.Bourcier, T., Sukhova, G., and Libby, P. (1997)
J. Biol. Chem., 272, 15817-15824.
73.Heissmeyer, V., Krappmann, D., Wulczyn, F. G.,
and Scheidereit, C. (1999) EMBO J., 18,4766-4778.
74.Chen, Z., Hagler, J., Palombella, V., Melandri,
F., Scherer, D., Ballard, D., and Maniatis, T. (1995) Genes
Dev., 9, 1586-1597.
75.Simeonidis, S., Stauber, D., Chen, G.,
Hendrickson, W., and Thanos, D. (1999) Proc. Natl. Acad. Sci.
USA, 96, 49-54.
76.Khachigian, L. M., and Collins, T. (1998) J.
Mol. Med., 76, 613-616.
77.Edmead, C., Kanthou, C., and Benzakour, O. (1999)
Anal. Biochem., 275, 180-186.
78.Nakajima, T., Kitajima, I., Shin, H., Takasaki,
I., Sitigeta, K., Abeyana, K., Yamashita, Y., Tokioka, T., Soejima, Y.,
and Maruyama, I. (1994) Biochem. Biophys. Res. Commun.,
204, 950-955.
79.Shin, H., Kitajima, I., Nakajiama, T., Shao, Q.,
Tokioka, T., Takasaki, I., Hanyu, N., Kubo, T., and Maruyama, I. (1999)
Ann. Rheum. Dis., 58, 55-60.
80.Maryama, I., Shigeta, K., Miyahara, H., Nakajima,
T., Shin, H., Ide, S., and Kitajima, I. (1997) Ann. N. Y. Acad.
Sci., 811, 429-436.
81.Autieri, M. V., Yue, T. L., Ferstein, G. Z., and
Ohlstein, E. (1995) Biochem. Biophys. Res. Commun., 213,
827-836.
82.Hoshi, S., Goto, M., Koyama, N., Nomoto, K., and
Tanaka, H. (2000) J. Biol. Chem., 275, 883-889.
83.Schmidt, V. A., Vitale, E., and Bahou, W. F.
(1996) J. Biol. Chem., 271, 9307-9312.
84.Wu, Y., Ruef, J., Rao, G., Patterson, C., and
Runge, M. S. (1998) Biochem. J., 330, 1469-1474.
85.Rahman, A., Anwar, K. N., True, A. L., and Malik,
A. B. (1999) J. Immunol., 162, 5466-5476.
86.Kaplanski, G., Marin, V., Fabrigoule, M., Boulay,
V., Benoliel, A. M., Bongrand, P., Kaplanski, S., and Farnarier, C.
(1998) Blood, 92, 1259-1267.
87.Suk, K., and Cha, S. (1999) Immunol Lett.,
67, 223-227.
88.Coughlin, S. R. (2000) Nature, 407,
258-264.
89.Umarova, B. A., Khlgatian, S. V., and Strukova,
S. M. (1989) Byul. Eksp. Biol. Med., 107, 131-133.
90.Strukova, S. M., and Khlgatian, S. V. (1991)
Thromb. Res., 64, 795-796.
91.Dejana, E., and Mashio, A. D. (1995) Thromb.
Haemost., 74, 309-312.
92.Alpin, A. E., Howe, A., Alahari, S. K., and
Juliano, R. L. (1998) Pharmacol Rev., 50, 197-263.
93.Lim, H., and Malik, A. B. (1996) Can. J.
Physiol. Pharmacol., 74, 787-800.
94.Werb, Z. (1997) Cell, 91,
439-442.
95.Silletti, S., and Cheresh, D. A. (1999)
Fibrinol. Proteolysis, 13,226-238.
96.Zucker, S., Mirza, H., and Conner, C. (1998)
Int. J. Cancer, 75, 780-786.
97.Strukova, S. M. (1995) in Inflammation
(Serov, V. V., and Paukov, V. S., eds.) [in Russian], Meditsina,
Moscow, pp. 52 -81.
98.Stiernberg, J., Redin, W., Warner, W., and
Carney, D. (1993) Thromb. Haemost., 70, 158-162.
99.Dunn, C., and Goa, K. L. (1999) Drug,
58, 863-886.
100.Spotnitz, W. (1995) Thromb. Haemost.,
74, 482-485.
101.Strukova, S., Dugina, T., Umarova, B., Chistov,
I., Markvicheva, E., Kuptsova, S., Kolokolchikova, E., Rumsh, L.,
Pinelis, V., and Glusa, E. (1998) Cardiovasc. Pathol., 7,
346.
102.Markvicheva, E., Dugina, T., Kuptsova, S.,
Chistov, I., Lange, M., Zubov, V., and Strukova, S. (1999) in INTAS
Symp. “Microbiol and Cellular System for Pharmacology,
Biotechnology, Medicine and Environment”, Dialogue-MSU,
Moscow, pp. 43-44.
103.Tsopanoglou, N. E., and Maragoudakis, M. E.
(1999) J. Biol. Chem., 274,23969-23976.
104.Bhat, G. J., Hunt, R. A., and Baker, K. M.
(1998) Arch. Biochem. Biophys., 350, 307-314.
105.Kranzhofer, R., Maragonore, J. M., Baciu, R.,
and Libby, P. (1999) Cardiovasc. Drugs Ther., 13,
429-434.
106.Chinn, C., de Niese, M. R., Tew, D. J.,
Jenkins, A. L., Bottomley, S. P., and Mackie, E. J. (1999) J. Biol.
Chem., 274, 9169-9174.
107.King, J., Srivastava, K. D., Stefano, G.,
Bilfinger, T., Bahou, W., and Magazine, H. I. (1997) J.
Immunol., 158, 880-886.
108.Srivastava, K. D., and Magazine, H. I. (1998)
J. Immunol., 161, 5039-5044.
109.Libby, P., Sukhova, G., Lee, R., and Liao, J.
(1997) Int. J. Cardiol., 62, S23-S29.
110.Kitamoto, S., Egashira, K., Kataoka, C.,
Koyanadi, M., Katoh, M., Shimokawa, H., Morishita, R., Kaneda, Y.,
Sueshi, K., and Takeshita, A. (2000) Circulation, 102,
806-812.
111.Schini, V. B., Catovsky, S., and Durante, W.
(1993) Amer. J. Physiol., 264, 611-616.
112.Gottwald, T., Coerper, S., Schaffer, M.,
Koveker, G., and Stead, R. H. (1998) Wound Repair Regen.,
6, 8-20.
113.Bankl, H. C., Grosschmidt, K., Pikula, B.,
Bankl, H., Lechner, K., and Valent, P. (1999) Hum. Pathol.,
30, 188-194.
114.Metcalfe, D. D., Baram, D., and Mekori, Y.
(1997) Physiol. Rev., 77,1033-1079.
115.Baghestanian, M., Hofbauer, R., and Kress, H.
(1997) Thromb. Haemost., 77, 577-584.
116.Strukova, S. M., Khlgatian, S. V., and Zaitsev,
S. V. (1991) Byul. Eksp. Biol. Med., 111, 385-387.
117.Strukova, S. M., Khlgatian, S. V., Dugina, T.
N., and Pinelis, V. G. (1995) Thromb. Haemost., 73,
932.
118.Lindahl, U., Peiler, G., Bozgwald, J., and
Seljelid, R. (1989) Arch. Biochem. Biophys., 273,
180-188.
119.Zoubine, M. N., Ma, J. Y., Smirnova, I. V.,
Citron, B. A., and Festoff, B. W. (1996) Develop. Biol.,
179, 447-457.
120.Kitamura, Y., Taguchi, T., and Yokoyama, M.
(1986) Am. J. Pathol., 122, 469-480.
121.Marx, G. (1986) Ann. N. Y. Acad. Sci.,
485, 425-431.
122.Pejler, G., and Sadler, J. E. (1999)
Biochemistry, 38, 12187-12195.
123.Peijler, G. (1999) Inflamm. Res.,
48, 344-350.
124.Kawabata, A., Kuroda, R., Nishikawa, H., Asai,
T., Kataoka, K., and Taneda, M. (1999) Br. J. Pharmacol.,
126, 1856-1862.
125.Strukova, S. M., Khlgatian, S. V., Pinelis, V.
G., Dugina, T. N., Kudinov, Yu. V., Markov, Kh. M., and Vrzhets, V. M.
(1992) Biokhimiya, 57, 927-936.
126.Redkozubov, A. E., Redkozubova, G. A.,
Strukova, S. M., and Dugina, T. N. (1992) Biol. Membr. (Moscow),
9, 1242-1247.
127.DeMichele, M., and Minnear, F. (1992) Semin.
Thromb. Hemost., 18, 287-295.
128.Scsmidt, H. H. H. W., Hofmann, H., and Ogilvie,
P. (1995) The Role of Nitric Oxide in Physiology and
Pathophysiology. B., Springer, Heidelberg.
129.Iikira, M., Takaishi, T., Hirai, K., Yamada,
H., Iida, M., Koshino, T., and Morita, Y. (1998) Int. Arch. Allergy
Immunol., 115, 129-136.
130.Hogaboam, C., Befus, A., and Wallace, J. (1993)
J. Immunol., 151, 3767-3774.
131.Strukova, S. M., Chistov, I. V., Umarova, B.
A., Dugina, T. N., Storozhevykh, T. P., Pinelis, V. G., and Glusa, E.
(1999) Biochemistry (Moscow), 64, 658-664.
132.Kubes, P., and Granger, D. N. (1996)
Cardiovasc. Res., 32, 699-708.
133.Salvemini, D., Masini, E., Pistlli, A.,
Mannaioni, P. F., and Vane, J. (1991) Cardiovasc. Pharmacol.,
17, S258.
134.Gaboury, J. P., Niu, X., and Kubes, P. (1996)
Circulation, 93, 318-324.
135.Al-Naemi, H., and Baldwin, A. L. (1999) Am.
J. Physiol., 277, H2010-H2016.
136.Vergnolle, N., Hollenberg, M., and Wallace, J.
R. (1999) Br. J. Pharmacol., 126, 1262-1268.
137.Steinhoff, M., Vergnolle, N., Young, S. H.,
Tognetto, M., Amadesi, S., Ennes, H. S., Trevisani, M., Hollenberg, M.
D., Wallace, J. L., Caughey, G. H., Mitchell, S. E., Williams, L. M.,
Geppetti, P., Mayer, E. A., and Bunnett, N. W. (2000) Nat. Med.,
6, 151-158.
138.Vergnolle, N. (1999) J. Immunol.,
163, 5064-5069.
139.Vergnolle, N., Hollenberg, M. D., Sharkey, K.
A., and Wallace, J. L. (1999) Br. J. Pharmacol., 127,
1083-1090.