Expression and Properties of Bacteriophage T4 Gene Product 11
L. P. Kurochkina1,2*, P. G. Leiman1,2, S. Yu. Venyaminov3, and V. V. Mesyanzhinov1
1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 336-6022; E-mail: vvm@ibch.siobc.ras.ru2Bach Institute of Biochemistry, Russian Academy of Sciences, Leninskii pr. 33, Moscow, 117071 Russia
3Department of Biochemistry and Molecular Biology, Mayo Foundation, 200 First Street SW, Rochester, Minnesota, 55905, USA
* To whom correspondence should be addressed.
Received September 13, 2000; Revision received November 5, 2000
A plasmid vector for expression of bacteriophage T4 gene product 11 (gp11) in E. coli cells has been constructed. Gp11 is a baseplate protein that connects short tail fibers providing irreversible adsorption of the virus on a cell. A method based on chromatography on hydroxyapatite has been developed for purification of recombinant gp11. The protein is active in an in vitro complementation assayand transforms defective phage particles lacking gp11 into infective ones. Gel filtration data suggest that the biologically active protein is a trimer. According to CD spectroscopy and sequence analysis data, the polypeptide chain of gp11 contains not less than 20% alpha-helical segments, about 30% beta-structure, and belongs to the class of alpha/beta structural proteins.
KEY WORDS: bacteriophage T4, baseplate, gene product 11, protein folding
The baseplate of Escherichia coli bacteriophage T4 is a complex molecular structure localized at the base of the distal end of the phage tail; it controls the process of host recognition and subsequent ejection of viral DNA into the cell. The baseplate is composed of six identical wedges (arms), each being a complex of seven gene products (gp): gp10, gp11, gp7, gp8, gp6, gp53, and gp25 [1]. Gene product 11 (gp11) is localized in angles of the bottom part of the baseplate and acts as a connector for short fibers encoded by gene 12, the latter being responsible for irreversible adsorption of phage particles on a cell.
The first intermediate in the assembly of a wedge is probably the complex of two proteins, gp10/11 [2]. According to published data [3, 4], this complex, formed in vivo as well as in vitro, contains two gp11 dimers that interact via their C-terminal domains with two dimers of gp10. However, according to gel-filtration data, gp11 isolated from an extract of 10--particles is a trimer [5].
Cells infected by amber mutants in gene 11 produce particles lacking gp11 (11--particles) that poorly adsorb on host cells and are not capable to infection even when adsorption does taken place [6]. Though these properties are mainly caused by the absence of short tail fibers (gp12), gp11 is apparently also necessary for contact to the cell surface. The gp11--particles can be transformed into infective particles by addition of extracts containing native gp11. Such extracts can be prepared by infection of cells by amber mutants in other genes of the baseplate (genes 9 and 10).
The polypeptide chain of gp11 contains 219 amino acids; the sequence of gene 11 was first determined in our laboratory [7, 8]. Gp11 is a convenient object for study of protein folding and oligomerization mechanisms. Many conditionally lethal (ts, temperature sensitive) mutations in gene 11 are known [9]. As in the case of the protein encoded by gene 9 of bacteriophage D22 [10], for gp11 it will probably be possible to select folding mutations (tsf, temperature sensitive folding) [11] and also suppressor mutations inside the gene to determine the role of different amino acids in polypeptide chain folding.
In this study, we have developed a method for expression and purification of recombinant gp11 that is biologically active in an in vitro complementation assay. The protein is a trimer; circular dichroism and amino acid analysis show that gp11 belongs to the class of alpha/beta structural proteins.
MATERIALS AND METHODS
Bacterial strains. The E. coli Top10 strain was used for the plasmid DNA preparation and transformation. The E. coli BL21(DE3) strain containing the RNA polymerase gene in the chromosome was used for expression of genes cloned into the vector under the control of the bacteriophage T7 promoter [12]. Be/1 strain (Su-, restrictive for amber mutants) and CR63 strain (Su+, permissive for amber mutants) of E. coli were used for preparation of defective extracts and titer determination of infective particles in the in vitro complementation assay.
Cloning of gene 11. A fragment containing full-length gene 11 was amplified by PCR using oligonucleotide primers containing point base substitutions to generate corresponding restriction sites. 5´-CCG CCA ACA TCT AGA ACT AAC primer was used as forward primer; 5´-CTA TCG GTA GGA TCC AAT TTT ACA was used as reverse primer (restriction sites are underlined). The reaction mixture contained (in 100 µl): ~1 ng of T4 DNA, 50 pmoles of each primer, 10 µl 10× PCR buffer (Promega, USA), 200 µmoles of each dNTP, and 2-5 units of Taq-polymerase (Qiagene, Germany). The reaction was started from preliminary DNA denaturation (97°C, 3 min); then 25 amplification cycles were run (94°C, 1 min; 52°C, 1 min; 72°C, 1 min) with subsequent incubation for 5 min at 72°C. After restriction, gene 11 was cloned into the XbaI-BamHI sites of pET-19b (Novagene, USA).
Expression of gp11 in E. coli BL21(DE3). Gp11 was expressed as described in [12] with insignificant modifications. Competent BL21(DE3) cells were transformed by the plasmid, plated on L-agar containing 1% glucose and ampicillin (100 µg/ml), and grown at 37°C to A600 ~ 0.6. IPTG (isopropylthio-beta-D-galactoside) was added to induce the expression to final concentration 1 mM, and the culture was incubated additionally for 3 h. The cells were centrifuged at 4000 rpm for 15 min (Megafuge 2.0 R, Heraeus Instruments, Germany). For preparative production of recombinant protein the volume of the cell culture was increased to 250-500 ml.
Electrophoretic analysis of the recombinant protein. A 100 µl sample of cell culture after expression was mixed with 30 µl of 4× sample buffer (0.25 M Tris-HCl, pH 6.8, 8% SDS, 40% (v/v) glycerol, 20% (v/v) 2-mercaptoethanol, 0.02% (w/v) bromphenol blue), heated for 3 min at 100°C, and then 10-30 µl were loaded on a gel.
SDS electrophoresis was run in 15% polyacrylamide gel according to the Laemmli method [13]. Gels were stained with 0.3% Coomassie R (Sigma, USA) in acetic acid-ethanol-water (1 : 3 : 6 v/v) solution and destained with 50% ethanol and 7% acetic acid solution. As markers, the standard LMW protein mixture (Pharmacia, Sweden) containing alpha-lactalbumin (14.4 kD), trypsin inhibitor (20.1 kD), carboanhydrase (30.0 kD), ovalbumin (43.0 kD), albumin (67.0 kD), and phosphorylase B (94 kD) was used.
Preparation of cell lysates. A 0.5-ml sample of cell culture was pelleted and resuspended in 100 µl of DMM (10-fold diluted M9 salt buffer containing 20 mM MgSO4) and incubated with lysozyme (0.5 mg/ml) and DNAse I (0.05 mg/ml) for 10 min at room temperature. The mixture then was subjected to three cycles of freezing in liquid nitrogen with subsequent thawing in a water bath at 30°C. The lysate was centrifuged at 12,000g for 2 min, and the freshly prepared supernatant was used in the in vitro complementation assay.
Preparation of concentrated 11--particle suspension. A plaque of CR63 phage lysis from top agar was extracted and transferred into 0.5 ml of physiological solution containing 20 mM MgSO4 and incubated for 1 h at room temperature; this solution was used for infecting 5 ml of liquid culture of E. coli CR63 in exponential growth phase with 2·108 cells per ml with following incubation for 16 h at 25°C. The cells were lysed by adding 2-3 drops of chloroform. The debris was removed by centrifuging at 15,000 rpm for 10 min. The titer of infective particles was ~5·1010 plaque forming units per ml.
Preparation of defective extract of 11--particles. Cell extract containing T4 phage 11--particles were prepared as described in [14]. E. coli Be/1 cells were grown in 50 ml of 2×TY medium containing 5 mM MgSO4 at 37°Cto a density of 2·108 cells/ml and infected by the amber mutant N128 with multiplicity of 5. After incubation for 15 min at 30°C, the concentration of MgSO4 was increased to 20 mMand the culture was additionally incubated for 20 min. The cells were pelleted at 4000 rpm for 20 min and resuspended in 2 ml of DMM containing 0.05 mg/ml DNAse I. Aliquots were stored at -70°C.
Complementation assay in vitro. A 20-µl sample of defective extract was mixed with 20 µl of cell lysate containing the expressed protein. The mixture was incubated for 2 h at 30°C. During incubation, 5 µl aliquots were diluted 1000-fold in physiological solution and titrated using E. coli CR63 cells.
Isolation and purification of the recombinant protein. Frozen cells were resuspended in 4-10 ml of 20 mM Tris-HCl buffer, pH 7.8, and lysed for 20 min at room temperature with lysozyme (0.1 mg/ml). The cell suspension was sonicated for 2 min, and cellular debris was removed by centrifuging for 10 min at 12,000g. Nucleic acids and accompanying proteins were removed by adding 30% (w/v) streptomycin sulfate solution to final concentration 3% and incubation for 15 min at 4°C. The precipitate was centrifuged under the same conditions. Saturated ammonium sulfate was slowly added to the supernatant to final concentration 25-30%, and the mixture was incubated for 1 h on ice. After centrifugation, the pellet containing protein was resuspended in 10 mM Tris-HCl buffer, pH 7.8, and dialyzed against the same buffer overnight. The protein solution was loaded onto a column (1 × 5 cm) packed by hydroxyapatite (HAP) (Bio-Rad, USA) equilibrated with 10 mM sodium phosphate buffer, pH 7.8. The protein was eluted using the same buffer, and the fractions were analyzed by electrophoresis.
UV spectroscopy. Absorption spectra were recorded at 25°C using a Cary 219 (Varian, USA) spectrophotometer over the 240-400 nm wavelength range with 1 nm resolution. All spectra were corrected for turbidity [15]. The absorption coefficient was obtained by a nitrogen micro method [16] assuming the nitrogen content of 16.6%. The measurements were recorded in 0.1 M sodium phosphate buffer, pH 7.2.
CD spectroscopy. CD spectra were recorded using a J-600 (JASCO, Japan) apparatus. For calculation of the molar ellipticity per amino acid residue, the value of the mean residue weight was taken as 107.5 daltons. The secondary structure was calculated using published methods [17, 18].
Gel filtration. Analytical gel filtration of purified gp11 was carried out on a column (1.4 × 30 cm) packed with Superose 12 (FPLC, Pharmacia Biotech, Sweden) in 50 mM Tris-HCl buffer, pH 8.0, containing 1 mM EDTA with flow rate 0.2 ml/min. The column was calibrated with albumin (67.0 kD) and chymotrypsinogen A (25 kD).
RESULTS AND DISCUSSION
The nucleotide sequence of gene 11 [7, 8] and the derived amino acid sequence of gp11 are shown in Fig. 1. The polypeptide chain of the protein contains 219 amino acids and has molecular weight 23.7 kD.
The DNA fragment containing full-length gene 11 was cloned into the XbaI-BamHI sites of pET-19b under the control of the bacteriophage T7 promoter. The designed construction was used for production of recombinant protein by the BL21(DE3) E. coli strain. The electrophoretic pattern of proteins from cell lysates before and after induction with IPTG is shown in Fig. 2. Comparison of lanes 2 and 3 shows the high level expression of gp11 in E. coli BL21(DE3)cells.Fig. 1. Nucleotide and derived amino acid sequences of gp11 with predicted secondary structure elements. Secondary structure elements: H, alpha-helix, high probability; h, alpha-helix, low probability; B, beta-structure; S, beta-strand; T, bend.

The biological activity of the recombinant protein was studied in an in vitro complementation assay. The cell lysate containing recombinant protein was added to the extract of 11--particles and the number of complemented infective phage particles was determined. The titer of the phage increases about two orders of magnitude compared to the control, this indicating functional activity of the recombinant protein and its proper folding and oligomerization.Fig. 2. Electrophoretic analysis in 15% SDS-polyacrylamide gel of the recombinant gp11 after expression (3) and purification (4). 1) Marker proteins; 2) E. coli cell lysate before induction.
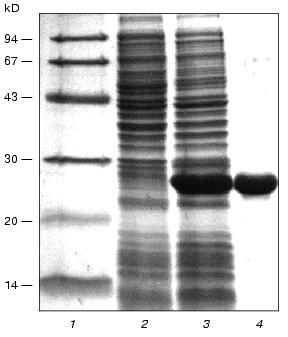
For further investigations, gp11 was isolated in preparative amounts and purified by chromatography on hydroxyapatite (see “Materials and Methods”). Electrophoretically pure gp11 was eluted from the column with 10 mM sodium phosphate buffer, pH 7.8 (Fig. 2, lane 4), this being evidence of low hydrophobicity of the protein. The molar absorption coefficient for purified gp11 at 282.5 nm is 27,000 Ì-1·cm-1.
To estimate the oligomeric organization of gp11, its resistance to detergent (SDS) at concentrations used for electrophoresis in polyacrylamide gel under denaturing conditions (2%) was investigated. As established earlier, some phage T4 structural proteins (fibritin, gp9, and also number of its deletion fragments) are stable trimers that are resistant to SDS treatment. In the presence of 2% SDS in polyacrylamide gel electrophoresis without preliminary heating of samples for 5 min at 95°C, all these proteins migrate with apparent molecular weights corresponding to their trimeric forms [19, 20]. The analysis of heated and unheated samples of gp11 in an SDS-polyacrylamide gel showed that both samples migrate identically with an apparent molecular weight corresponding to the monomeric form of the protein (data not shown), this indicating labile oligomeric structure with respect to SDS.
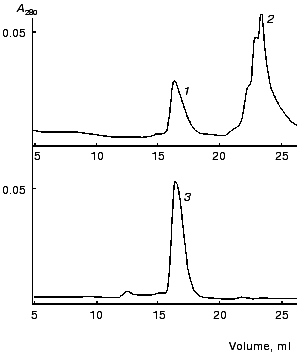
To estimate the oligomeric organization of the recombinant protein, gel filtration of purified gp11 was carried out. Two proteins (chymotrypsinogen A and albumin) having molecular weights close to those for the monomeric and trimeric forms of gp11, respectively, were used as markers for calibration of a column. The gel-filtration data are shown in Fig. 3. Gp11 (peak 3) was eluted at a volume appropriate to the elution volume of albumin (peak 1), this indicating that the recombinant protein is a trimer. Our results are in agreement with earlier obtained data showing trimeric organization of native gp11 isolated from an extract of (10-) phage mutant [5].
The shares of various types of structure in gp11 were calculated using data from circular dichroism spectroscopy. CD spectra for the recombinant protein in the far (a) and near (b) UV ranges are presented in Fig. 4. The predicted secondary structure elements of gp11 calculated by two different methods are shown in the table. The polypeptide chain of gp11 contains 21-23% of alpha-helical segments and 20-34% of beta-structure; thus, the protein can be assigned to the class of alpha/beta structural proteins.Fig. 3. Gel filtration on Superose 12: mixture of albumin (1) and chymotrypsinogen A (2); gp11 (3). Flow rate is 0.2 ml/min.
Calculation of the secondary structure of gp11Fig. 4. Circular dichroism spectra of gp11 in far (a) and near (b) UV ranges.
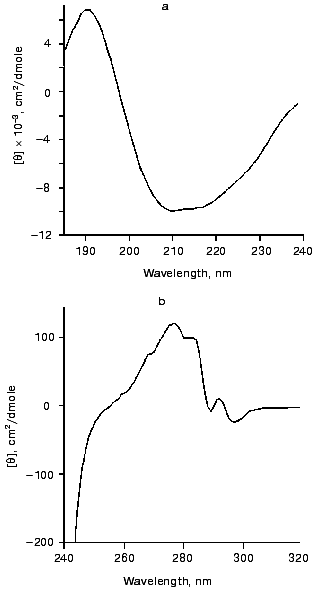

The secondary structure of the protein based on the derived gp11 amino acid sequence was predicted using the ALB program [21]. The distribution of the predicted secondary structure elements in the gp11 molecule is shown in Fig. 1. The protein contains approximately identical amount of alpha-helices and beta-structure (20 and 22%, respectively), this being in good agreement with the results of the CD spectroscopy.
Thus, a plasmid vector has been constructed and a system for expression and purification of biologically active recombinant gp11 has been developed. It was estimated that the biologically active protein is a trimer. CD spectroscopy data and secondary structure prediction indicate that gp11 is an alpha/beta structural protein. We recently obtained crystals of gp11, and now we are finishing the determination of its spatial structure using X-ray crystallographic analysis.
This study was supported by grants from the Howard Hughes Medical Institute (No. 75195-520803), the Russian Foundation for Basic Research (No. 99-04-48430), and the “Universities of Russia” program (No. 2608-415/98).
Note added in proof: The X-ray structure of gp11 has been solved recently (Leiman, P. G., Kostyuchenko, V. A., Shneider, M. M., Kurochkina, L. P., Mesyanzhinov, V. V., and Rossmann, M. G. (2000) J. Mol. Biol., 30, 975-985).
REFERENCES
1.Berget, P. B., and King, J. (1983) in
Bacteriophage T4 (Mathews, C. K., Kutter, E. M., Mosig, G., and
Berget, P. B., eds.) American Society for Microbiology, Washington, D.
C., pp. 246-258.
2.Berget, P. B., and King, J. (1978) J. Mol.
Biol., 124, 469-486.
3.Plishker, M. F., and Berget, P. B. (1984) J.
Mol. Biol., 178, 699-709.
4.Plishker, M. F., Chidambaram, M., and Berget, P. B.
(1983) J. Mol. Biol., 170, 119-135.
5.Terzaghi, E., and Terzaghi, B. E. (1974) J.
Biol. Chem., 249, 5119-5125.
6.Simon, L. D., Swan, J. G., and Flatgaard, J. E.
(1970) Virology, 41, 77-90.
7.Prilipov, A. G., Selivanov, N. A., Efimov, V. P.,
Marusich, E. I., and Mesyanzhinov, V. V. (1989) Nucleic Acids
Res., 17, 3303.
8.Barrett, B. K., and Berget, P. B. (1989)
DNA, 8, 287-295.
9.Smith, D. H., and King, J. (1981) J. Mol.
Biol., 145, 653-676.
10.Betts, S., and King, J. (1999) Structure Fold.
Des., 7, 131-139.
11.Goldberg, D., and King, J. (1981) J. Mol.
Biol., 145, 633-651.
12.Studier, F. W., Rosenberg, A. N., Dunn, A. H.,
and Dubendorff, J. W. (1990) Meth. Enzymol., 185,
60-89.
13.Laemmli, U. K. (1970) Nature, 227,
680-685.
14.Edgar, R. S., and Wood, W. B. (1966) Proc.
Natl. Acad. Sci. USA, 55, 498-505.
15.Venyaminov, S. Y., and Gogia, Z. V. (1982)
Eur. J. Biochem., 126, 299-309.
16.Jaenicke, L. (1974) Anal. Biochem.,
61, 623-627.
17.Chang, C. T., Wu, C.-S. C., and Yang, J. T.
(1978) Anal. Biochem., 91, 13-31.
18.Provencher, S. W., and Glockner, J. J. (1981)
Biochemistry, 20, 33-37.
19.Londer, Yu.Ya., and Mesyanzhinov, V. V. (1999)
Bioorg. Khim., 25, 257-263.
20.Navruzbekov, G. A., Kurochkina, L. P.,
Kostyuchenko, V. A., Zurabishvili, T. G., Venyaminov, S. Y., and
Mesyanzhinov, V. V. (1999) Biochemistry (Moscow), 64,
1266-1272.
21.Finkelstein, A. V., Badretdinov, A. Ya., and
Ptitsyn, O. B. (1991) Proteins, 10, 287-299.