P-Chip and P-Chip Bienzyme Electrodes Based on Recombinant Forms of Horseradish Peroxidase Immobilized on Gold Electrodes
E. E. Ferapontova1*, V. G. Grigorenko2, and A. M. Egorov2,3
1Faculty of Pharmacy, Department of Analytical Chemistry, University of Alcala, E-28871 Alcala de Henares, Madrid, Spain; E-mail: elena.ferapontova@uah.es2Department of Chemical Enzymology, School of Chemistry, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-2742; E-mail: aegorov@enz.chem.msu.ru
3State Scientific Center for Antibiotics, Moscow, Russia; fax: (095) 111-4238; E-mail: egorov@antibiotics.mtu-net.ru
* To whom correspondence should be addressed.
Received January 26, 2001; Revision received March 5, 2001
Adsorption and bioelectrocatalytic activity of native horseradish peroxidase (HRP) and its recombinant forms on polycrystalline gold electrodes were studied. Recombinant forms of HRP were produced by a genetic engineering approach using an E. coli expression system. According to direct mass measurements with a quartz crystal microbalance, all the forms of HRP formed monolayer coverage of the enzyme on the gold surface. However, only gold electrodes modified with the recombinant HRP forms (non-glycosylated) exhibited high and stable current response to H2O2 due to its bioelectrocatalytic reduction based on direct electron transfer (ET) between gold and the active site of the enzyme. Introduction of a six-His tag either at the C-terminus or at the N-terminus of the enzyme molecule additionally increased the strength of the enzyme binding with the gold surface and the efficiency of direct ET. Immobilization of recombinant forms of HRP containing histidine functional groups on the surface of the gold electrode was used both for the development of a P-chip, a biosensor for hydrogen peroxide determination based on direct ET, and for the development of a bienzyme biosensor electrode for the determination of L-lysine based on co-immobilized recombinant forms of HRP and L-lysine-alpha-oxidase.
KEY WORDS: recombinant forms of horseradish peroxidase, amperometric biosensor, P-chip, direct electron transfer
Abbreviations: LO) lysine oxidase; NHisrHRP) recombinant horseradish peroxidase of wild type containing a 6-histidine tag at the N-terminus of the enzyme; PZC) piezoelectric quartz crystal; HRP) horseradish peroxidase; ET) electron transfer; rHRP) recombinant horseradish peroxidase of wild type; CHisrHRP) recombinant horseradish peroxidase of wild type containing a 6-histidine tag at the C-terminus of the enzyme; PBS) phosphate buffer solution; EQCM) electrochemical quartz crystal microbalance.
Horseradish peroxidase (HRP) (oxidoreductase, EC 1.11.1.7) is a heme-
and Ca2+-containing glycoprotein, a member of the plant
peroxidase superfamily, capable of reducing peroxide compounds by
catalyzing one electron oxidation of a wide variety of organic and
inorganic substrates [1, 2].
The high catalytic activity of HRP in H2O2
reduction makes it possible to use HRP as a highly effective
bioelectrocatalyst participating in electron transfer (ET) between the
electrode, the active site of the enzyme, and the substrate. Efficient
ET from the electrode surface to the active site of the enzyme makes
possible the development of a P-chip, a biosensor system of microscopic
size for the detection of hydrogen peroxide both ex vivo and
in vivo (inside tissues and cells), based on direct ET.
The simplest biosensor for the detection of hydrogen peroxide consists of a monolayer of HRP molecules adsorbed on the electrode surface (Fig. 1a). In the presence of the substrate at a certain applied electrode potential, the measured reduction current is proportional to the H2O2 concentration and the following reactions are expected to proceed [3]:
Here, HRP(Fe3+) denotes the initial form of the enzyme immobilized on the electrode surface (oxidation state +3), and E1 represents oxidized HRP (conventional oxidation state +5) consisting of oxyferryl iron (Fe4+=O) and a porphyrin pi cation radical.
Reaction (1) involves a two-electron oxidation of the ferriheme prosthetic group of the peroxidase by hydrogen peroxide. The direct electrochemical reduction of the oxidized form of HRP (compound E1) has been shown to be kinetically slow on a majority of electrode materials [3-13] and is considered as direct ET. This mediatorless bioelectrocatalytic reaction of H2O2 reduction has been observed for HRP adsorbed on carbon and graphite [3-9], gold [4, 10-12], and platinum [13].Fig. 1. Schematic description of a P-chip (a) and a bienzyme biosensor electrode based on a P-chip and an appropriate oxidase (b).
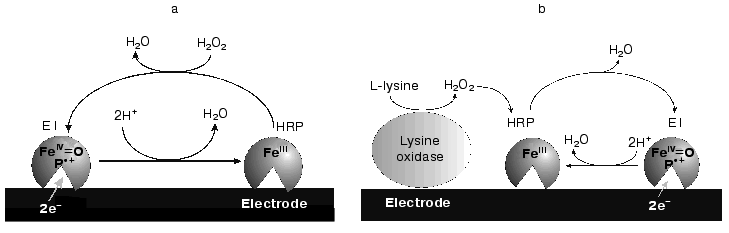
Peroxidase immobilized at the electrode surface and capable of highly efficient direct ET can be used to construct a mediatorless biosensor for the detection of peroxides and compounds (glucose, lysine, etc.) whose oxidation in the presence of the appropriate oxidases (glucose oxidase, lysine oxidase, etc.) yields hydrogen peroxide (Fig. 1b). However, the efficiency of direct electrochemical reduction of compound E1 on the majority of investigated electrode materials [3-13] was negligible, the rate constant of heterogeneous direct ET, ks, being generally below 1 electron per second [14]. The reasons for this are probably the long ET distance between the electrode surface and the active site of HRP, low surface concentration of the active enzyme, and/or unfavorable for direct ET orientation of the molecule at the electrode surface.
Thus, the key problem in the creation and further development of such enzyme electrodes based on direct ET is the optimization of the protein immobilization at the electrode surface and achievement of strong adsorption of the protein molecules onto the electrode surface. Unfortunately, all investigated methods for covalent immobilization through a special link increased the distance of the active site from the electrode surface, hindering direct ET. However, genetic engineering can be used to directly modify the surface of the HRP molecule by introduction of additional functional groups that can provide specific oriented immobilization of the enzyme on the conductive support, the choice of the electrode material playing an important role.
Gold electrodes for use as conductive supports for immobilization are of the clear interest in spite of the fact that the efficiency of direct ET in the gold electrode-native HRP system is extremely low [4, 10]. Gold is a chemically inert and highly hydrophobic material. Hence, it is favorable for direct ET after adsorption of recombinant (i.e., non-glycosylated) forms of HRP through hydrophobic interactions between the hydrophobic surface clusters of the recombinant HRP molecules and the hydrophobic gold surface. Moreover, because known strategies for immobilization of biomolecules onto gold surfaces are based on strong chemisorption of some functional groups [5], e.g., those of histidine and cysteine [15, 16], orientation and adsorption of the enzyme on the gold electrode surface in a way favoring direct ET might be achieved by genetic engineering design of protein molecule surface with similar groups.
The goal of the present work was to study and compare the adsorption and bioelectrocatalytic behavior of native HRP and its different recombinant forms on gold polycrystalline electrodes. The kinetic data were to be used for subsequent development of P-chips based on recombinant forms of HRP and for development of a P-chip bienzyme biosensor electrode for the detection of L-lysine (Fig. 1b).
MATERIALS AND METHODS
Reagents. Native HRP (isoenzyme C) and L-lysine-alpha-oxidase (LO, oxygen oxidoreductase, EC 1.4.3.14, from Trichoderma viride, activity 20 U/mg) from Sigma-Aldrich (USA) and substrates and buffer solution components from Merck (Germany) were used. All reagents were of analytical grade or of ultra-high purity and used as received. All solutions were prepared with de-ionized Milli-Q water (Millipore, France).
Production of recombinant forms of HRP. Recombinant forms of HRP (isoenzyme C), specifically, recombinant wild type HRP (rHRP), rHRP containing a 6-histidine tag at the C-terminus of the enzyme (CHisrHRP), and rHRP containing 6-histidine tag at the N-terminus of the enzyme (NHisrHRP) were produced by genetic engineering in E. coli strain BL21(DE3)pLysS transformed with the appropriate pET-based expression vectors (Novagen). The procedure of rHRP expression, refolding, and purification is given in detail elsewhere [17, 18].
To produce wild type rHRP, the HRP gene was amplified by PCR using the proofreading polymerase Pfu (Stratagene) where the forward and reverse primers were 5´-AACATATGCAGTTAACGCCGACTTTCTACG-3´ and 5´- TTCGGCCGTCATGAGTTCGAGTTTACGACTCGGCAGTTC-3´, respectively, with pETHRPhis [18] as the template. Bold italic letters indicate the introduced restriction sites (NdeI and XmaIII); the stop codon introduced just after Ser308 is underlined. The amplified PCR fragment was first ligated into pCRScript vector (Stratagene) and further recloned into pET20 (Novagen) cut with NdeI and XmaIII. The final construct pETHRP corresponding to wild type rHRP was confirmed by sequencing.
To produce recombinant HRP with a 6-histidine tag at the N-terminus, pETHRP was digested with NdeI and PstI. Two fragments, NdeI-PstI and PstI-PstI (one PstI site in the HRP gene and one in the bla gene in the vector), were purified from agarose gel and ligated into pET15 (Novagen) cut with NdeI and PstI and transformed into E. coli DH5alpha cells. Several colonies (only those with correct orientation of the PstI-PstI fragment able to grow on ampicillin plates) were picked and analyzed by restriction with NdeI and PstI and further confirmed by sequencing.
Instrumentation. Data on the adsorption of peroxidase on gold were obtained with electrochemical quartz crystal microbalance (EQCM, Poland). Piezoelectric quartz crystals (PZC) coated with gold (OMIG, Poland) were used as the gold electrodes. The PZCs were AT-cut with a basic resonant frequency of 10 MHz; the gold coating diameter was 5 mm. The gold-coated PZC electrode was inserted into a three-electrode flow-injection cell as one of the cell walls, and it served as the working electrode. A silver ring, pressed into Teflon was used as the pseudo-reference electrode (the supporting electrolyte always contained 0.15 M NaCl), and a platinum ring of a larger diameter, also pressed into Teflon, served as the auxiliary electrode.
Amperometric measurements with polycrystalline gold disk electrodes (BAS, USA) of diameter 0.15 mm were performed in a standard three-electrode flow-injection cell [8]. The cell contained a silver-silver chloride (0.1 M KCl) reference electrode, the auxiliary electrode was a platinum wire, and a gold rod pressed into Teflon served as the working electrode. The flow of the solutions was maintained by a MINIPULS 2 peristaltic pump (Gilson, France).
Electrochemical measurements with gold electrodes were carried out with a µAUTOLAB three-electrode potentiostat (Eco Chemie, Netherlands) equipped with GPES 4.7 software (Eco Chemie). All experiments were performed at ambient temperature (22 ± 1°C).
Immobilization and measurements with the quartz crystal microbalance. The PZC electrodes used for HRP immobilization were pretreated as follows: 60 µl of a freshly prepared hot mixture of concentrated sulfuric acid and 30% hydrogen peroxide solution (1 : 1) were dropped onto the surface of the PZC electrode placed in the cell. After 2 min of activation, the electrode surface was rinsed with deionized water leaving a thin layer of H2O in the cell compartment to prevent any direct contact of the cleaned gold surface with air. Then 200 µl of 0.01 mg/ml HRP solution in 0.01 M phosphate buffer containing 0.15 M NaCl (PBS), pH 7.4, was pipetted into the cell compartment with the working PZC electrode. By immediately setting the frequency counter to zero, the EQCM measurement of the adsorption of peroxidase on gold was started. After the measurement of the adsorption process, i.e., when the EQCM frequency stabilized, a buffer solution was aspirated through the cell and the frequency change corresponding to the amount of HRP removed with the buffer solution flow was registered.
Immobilization and measurements in the flow-injection system. Prior to immobilization, the surface of the gold disk electrode (BAS) was polished to a mirror luster with alumina suspension (particle size 0.1 µl) in water, rinsed with deionized water, treated with a freshly prepared hot mixture of concentrated sulfuric acid and 30% hydrogen peroxide solution (1 : 1) for 2 min, and then rinsed again with water. Immediately after that, for adsorption of HRP, the electrodes were immersed in a 0.01 mg/ml HRP solution in 0.01 M PBS, pH 6.0, for 2 h. For co-adsorption of CHisrHRP and LO, the electrodes were kept for 2 h in PBS, pH 7.4, containing 0.01 mg/ml CHisrHRP and 0.06 mg/ml LO. Then the electrodes were rinsed in PBS and inserted into the electrochemical cell for subsequent amperometric measurements.
During the amperometric measurements, buffer solution was pumped through the cell at flow rate 900 µl/min, and a steady-state baseline current was registered at -50 mV. For the measurement of the amperometric response of the HRP-modified electrodes to H2O2, flow-carrier buffer solution containing different concentrations of hydrogen peroxide was used. Similar solution also containing L-lysine was used for measurement of the amperometric response of the bienzyme electrode modified with co-adsorbed CHisrHRP and LO. The signal difference between the background current and the steady-state current in the presence of the substrate was taken as the response signal corresponding to bioelectrocatalytic reduction of H2O2 on the modified gold electrodes.
The reproducibility of the data was verified by measurements with at least three equivalently prepared electrodes.
RESULTS AND DISCUSSION
Immobilization of HRP on gold PZC electrodes. To compare the bioelectrocatalytic activity of the different forms of HRP immobilized on gold electrodes, it was necessary to compare the efficiency of binding of these HRP forms with the gold surface. Thus, the course of immobilization of the peroxidases was studied with the quartz crystal microbalance, giving direct data on the amount of the enzyme bound to the electrode surface [19]. Correct assessment of this amount makes it possible to estimate the effect of surface mutations of the protein molecule on the direct ET characteristics by separation of the effect of the amount of the enzyme on the electrode surface and the effect of orientation of the different forms of HRP immobilized at the electrode surface.
Native HRP and the recombinant forms: rHRP, CHisrHRP, and NHisrHRP were immobilized directly on the surface of gold PZC electrodes. All the recombinant forms of HRP were non-glycosylated, the last two forms additionally containing a histidine tag at either the C- or the N-terminus of the polypeptide chain.
Figure 2 shows the data on the resonant frequency shift with time due to the adsorption of NHisrHRP on the gold PZC electrodes. The general shape of the curves and frequency shifts for the studied forms, both recombinant and native, were quantitatively and qualitatively very close to that presented in Fig. 2 (see insert in Fig. 2). The decrease of the resonance frequency of the gold-quartz electrodes presented in Fig. 2 is due to the enzyme binding to the gold surface. The relationship between the change of the surface mass Deltam (grams) and the variation of the resonant frequency Deltaf (Hz) for AT-cut PZC electrodes is given by the Sauerbrey equation [20]:
where A is the surface area covered by adsorbed material (cm2), µq is the quartz shear modulus (µq = 2.947·1011 dyne/cm2), rhoq is the quartz density (rhoq = 2.648 g/cm3), and f0 is the fundamental resonant frequency of the unloaded crystal oscillator.
The results of the adsorption measurements obtained with the EQCM are tabulated in Table 1. From the data presented, it can be seen that the amount of the immobilized enzyme, calculated from the frequency shift of the gold PZC electrode due to immobilization of HRP, is virtually the same for the different recombinant forms to within the experimental error. The amount of native glycosylated HRP (in moles) adsorbed at the electrode surface is about 30% lower than that for the recombinant forms due to the higher molecular weight of the protein. From the data on the amount of the adsorbed enzyme, the geometrical sizes of the HRP molecules were calculated with the assumption that the adsorbed enzyme molecules are globular. As a result, the following values of the molecular diameters were obtained for the HRP molecules adsorbed at the electrode surface: 3 nm for the native form and 2.6-2.7 nm for the recombinant forms. This numbers are in good agreement with the crystallographic dimensions of HRP [21]. On subsequent concurrent mass and amperometric measurements with EQCM in a flow of 10-4 M H2O2 (polarization at -50 mV), native HRP completely desorbed from the electrode surface, in contrast to the recombinant form. This fact suggests that the binding of native HRP with the gold electrode surface is weaker than that of the recombinant forms.Fig. 2. Variation of the resonant frequency shift for the gold PZC electrodes during the immobilization of NHisrHRP from 0.01 mg/ml enzyme solution in PBS, pH 7.4. Insert: data for native HRP.
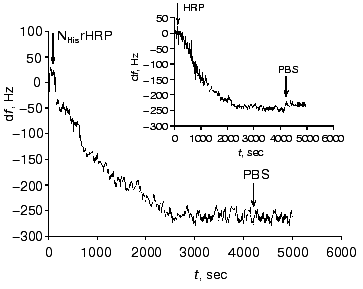
Table 1. Adsorption of native and
recombinant forms of HRP on the surface of gold PZC electrodes
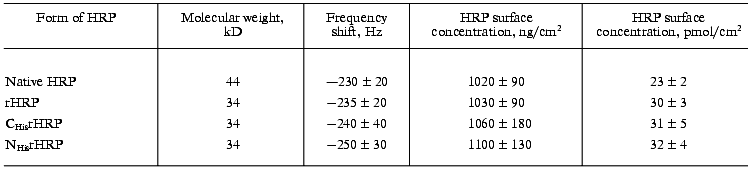
Thus, the microbalance measurements demonstrated that: a) the adsorption process results in monolayer coverage of the gold electrode surface with the HRP molecules; b) the adsorbed amounts of the enzyme are equal for the different recombinant forms of HRP; c) the strength of the enzyme binding with the gold electrode surface is much higher for the recombinant forms of HRP.
Amperometric detection of H2O2 (direct ET). The bioelectrocatalytic activity of the different forms of HRP immobilized on gold was determined amperometrically by measuring the concentration dependence of H2O2 bioelectrocatalytic reduction current at -50 mV. Calibration curves, i.e., the dependence of the measured current response on the concentration of H2O2 for the electrodes modified with the native and recombinant forms of HRP are presented in Fig. 3. Since under the conditions of the electrochemical reaction the electrode serves as the electron donor (a proton is transferred from the solvent molecules or from any other proton donor present in the system), then the data shown in Fig. 3 virtually correspond to the bioelectrocatalytic activity of the studied forms of HRP on the gold electrodes. As seen, CHisrHRP and NHisrHRP gave the highest current response due to direct ET, the signal being lower for rHRP. However, these values are 8-11 times higher than that for native HRP (Table 2).
Table 2. Activity of peroxidases and characteristics of peroxidase-modified electrodesFig. 3. Dependence of the steady-state current of electroreduction on the H2O2 concentration determined with gold disk electrodes modified with: 1) native HRP; 2) rHRP; 3) CHisrHRP; and 4) NHisrHRP. The electrodes were placed in a flow-injection cell. The flow rate of the substrate in PBS, pH 7.4, was 900 µl/min. The applied potential was -50 mV vs. Ag/AgCl in 0.1 M KCl.
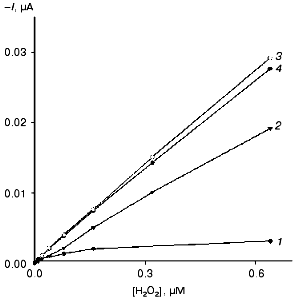

The electrodes modified with the recombinant HRPs exhibit high sensitivity for H2O2 as determined from the initial slopes of the calibration curves (Fig. 3). The calculated values of the sensitivity for H2O2 for the electrodes modified with recombinant HRPs are presented in Table 2. It can be seen that the sensitivity values are close to 1.4 A·M-1·cm-2. The sensitivity for the gold electrodes modified with the recombinant forms of HRP is 8-9 times higher than that for native HRP and is the best among existing HRP-modified electrodes based on direct ET [22, 23]. The rates of direct ET between the recombinant forms of HRP and gold are so high that the current response for H2O2 is determined only by the mass transfer of H2O2 to the electrode surface. The introduction of a mediator (catechol) into the system does not increase the sensitivity, in contrast to that for gold electrodes modified with native HRP (Table 2).
Since the measured surface concentrations of the recombinant forms under study are similar (Table 1) and only 30% higher than that for the native form. Thus, the higher bioelectrocatalytic activity of the recombinant forms (and as a result the sensitivity of the electrodes for H2O2) in the reaction of H2O2 reduction is due to more favorable arrangement of the enzyme molecules on the gold surface for establishing direct ET, and, hence, higher direct ET rate in the enzyme-electrode system. Thus, the data (Fig. 3 and Table 1) indicate that in the case of non-glycosylated forms of the enzyme the efficiency of direct ET in the recombinant HRP form-gold electrode system is much higher than that in the case of the glycosylated native HRP.
The electrodes modified with the recombinant forms of HRP exhibited good stability operated daily for 2 h and stored in buffer at 4°C between measurements. For CHisrHRP- and NHisrHRP-modified electrodes, stable current signal was observed during the first 24 h of storage, the half-life on the gold electrodes for both histidine HRPs being about 90 h (Fig. 4). For rHRP this parameter was lower (50 h) due to the weaker binding of rHRP with the gold surface. For H2O2 concentrations lower than 1 µM, 100% stability of the signal was retained for the entire test period of the biosensor operation (from 70 to 100 h). Electrodes modified with native HRP demonstrated constantly decreasing sensitivity in direct and mediated ET due to enzyme desorption from the electrode surface.
Effect of genetic-engineering modification of the rHRP molecule surface on the efficiency of direct ET. The data obtained amperometrically and with the quartz crystal microbalance allow us to estimate the effect of surface modifications of the rHRP molecule on the efficiency of direct ET in the HRP-gold electrode system. Since the rHRP-modified electrodes exhibit bioelectrochemical activity 25% lower than that for the histidine forms, the surface concentration of the HRPs being practically the same (Tables 1 and 2), the adsorption of CHisrHRP and NHisrHRP apparent occurs not only due to hydrophobic interactions between gold and the HRP molecules, but also through specific interaction of the histidine functional groups with the electrode surface. Histidine adsorbs irreversibly onto gold [15, 16], and this was specially used for the modification of the rHRP molecule surface to improve the adsorption process. The adsorption of the histidine forms of HRP presumably through the nitrogen atom of the imidazole ring [15] results, on one hand, in increasing the strength of binding of these forms of HRP with the gold surface, and on the other hand probably in more favorable orientation of the HRP molecules on the gold electrode surface for direct ET.Fig. 4. Stability of the amperometric response (data normalized to the initial current response) with time for gold electrodes modified with CHisrHRP. The concentration of H2O2: 1) 0.1; 2) 1; 3) 10; 4) 20; 5) 40; and 6) 100 µM. Curve 7 shows the data for native HRP in the concentration range of H2O2 from 10-5 to 4·10-5 M. Other conditions as in Fig. 3.
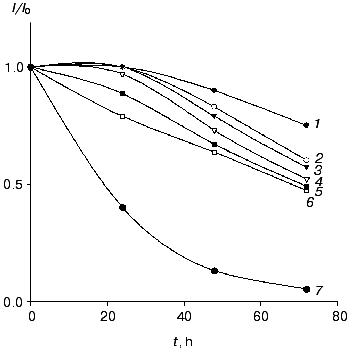
The supposition of the adsorption of CHisrHRP and NHisrHRP through the histidine groups introduced in different positions of the rHRP polypeptide chain allows evaluation of the effect of the rHRP molecule orientation on the gold electrode surface on the efficiency of direct ET as well. Both CHisrHRP and NHisrHRP, when immobilized on gold, gave the highest current response due to direct ET. But comparing the enzymatic activity of the samples with their amperometric responses (Table 2), it can be seen that, despite a 40% decrease in an enzymatic activity, NHisrHRP exhibits the same bioelectrochemical activity as CHisrHRP, and this fact can be considered as evidence of orientation of the NHisrHRP molecules on the surface of the gold electrode more favorable for direct ET. Thus, the genetic engineering design of the rHRP molecule surface with histidine sequences resulted in oriented immobilization of the recombinant forms of HRP on the surface of the gold electrode.
P-Chip development. The lower detection limit of the biosensors based on the studied forms of HRP was determined to be 10-8 M H2O2. The detection limit was determined as the minimal value of the H2O2 concentration that gave a signal in the region of the linear dependence of calibration plots (Fig. 3). In the case of HRP-biosensors based on chemiluminescence methods of H2O2 determination, the detection limit is not better than 1 µM H2O2 [24, 25]. Among biosensor electrodes, the most sensitive are graphite electrodes modified with HRP and a redox mediator, the detection limit of these electrodes for H2O2 being .·10-9 M [26].
High sensitivity (1.4 A·M-1·cm-2), low detection limit for H2O2, and stability of the electrodes modified with the recombinant forms of HRP allow the development of a P-chip, a miniaturized biosensing device capable of determining low concentrations of hydrogen peroxide in vivo. Fast and reliable control of the content both of H2O2 and of compounds yielding H2O2 in the course of their transformations in small-volume samples, specifically in living cells and tissues, is restricted at present by the availability of corresponding miniature mediatorless biosensing devices. Existing equipment can measure currents of the order of 1 pA or even 0.1 pA. Thus, for an intracellular concentration of H2O2 of 10-8 M (the detection limit for the biosensors based on the studied forms of HRP) and the sensitivity of 1.4 A·M-1·cm-2, the surface area of the working electrode can be reduced to 7·10-6 cm2. This size corresponds to a disk electrode with diameter of 30 µm (or to a probe 5 µm in diameter and 44.2 µm long) and represents itself a P-chip--a microbiosensor that can be manufactured with currently existing thin film technologies [27] and can be used for effective and reliable measurements of hydrogen peroxide content in vivo.
Development of bienzyme biosensor electrodes. Co-immobilization on the gold electrode surface of HRP, that possesses high catalytic activity in the reaction of H2O2 reduction, and a number of oxidases capable of oxidizing their corresponding substrates yielding hydrogen peroxide, makes it possible to develop mediatorless bienzyme biosensors for the detection of these substrates. For example, co-adsorption of HRP and LO can be used for amperometric determination L-lysine, an amino acid essential for balanced nutrition of human beings and animals and which is a basic component in histone synthesis. Then the concentration of L-lysine will be proportional to the concentration of hydrogen peroxide that is released during the enzymatic oxidation of L-lysine according to Eq. (4):
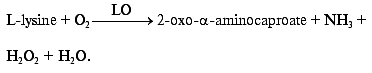 (4)
(4)Existing electrochemical methods for the detection of products of enzymatic degradation of L-lysine or oxygen consumption in the course of the reaction (4) have certain drawbacks. Amperometric detection of oxygen consumption requires strict control of air saturation and temperature [28]. Direct electrochemical oxidation of hydrogen peroxide is accompanied by oxidation of other components present in the sample [29]. Potentiometric detection of ammonia is affected by the presence of endogenous ammonia in a sample and is disturbed by other cationic species such as potassium ions[30]. A bienzyme biosensor based on a P-chip and LO will work at moderate potentials (from 0 to -50 mV) and will by highly specific in the determination of hydrogen peroxide and L-lysine if direct ET between the electrode surface and the active site of HRP can be established.
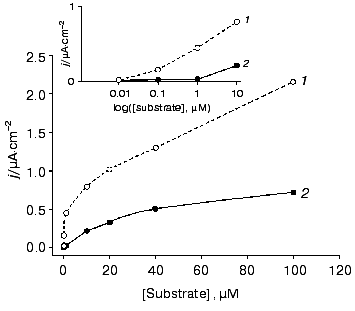
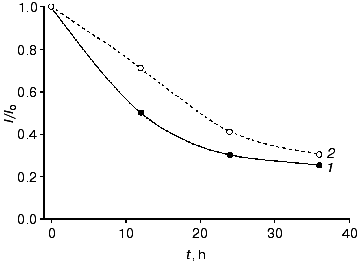
We directly co-immobilized LO and CHisrHRP on the surface of polycrystalline gold electrodes. Efficient direct ET between the gold electrode surface and the immobilized recombinant HRP made possible amperometric detection of hydrogen peroxide released during the enzymatic degradation of L-lysine (Figs. 1b and 5). During the amperometric detection of L-lysine in a flow of PBS, pH 7.4, a minimal signal corresponding to 1 µM L-lysine was also obtained for higher L-lysine concentrations due to the removal of formed H2O2 with the flow of the buffer solution. Decreasing the flow rate of the L-lysine-containing PBS to zero enhanced the current response due to the accumulation of H2O2 released during the enzymatic oxidation of L-lysine near the electrode and its further diffusion to the active sites of HRP (Fig. 1b). The time corresponding to the maximal current response varied from 2 to 18 min depending on the L-lysine concentration. The detection limit for L-lysine was 1 µM, the sensitivity of the detection being 0.03 A·cm-2·M-1 at -50 mV. Electrodes modified with co-adsorbed LO and CHisrHRP demonstrated constantly decreasing sensitivity in direct and mediated ET, probably due to desorption of the enzyme from the electrode surface (Fig. 6). The stability of the developed system was thus significantly lower than that of CHisrHRP alone immobilized on the gold electrode surface (compare Figs. 4 and 6).
Fig. 5. Dependence of the steady-state current density on (1) H2O2 and (2) L-lysine concentrations determined with the gold disk electrodes modified with CHisrHRP co-adsorbed with LO. The electrodes were placed into a flow-injection cell. The flow rate of the substrate in PBS, pH 7.4, was 900 µl/min. The applied potential was -50 mV vs. Ag/AgCl in 0.1 M KCl. Curve 1 was obtained in a stopped-flow regime achieved by decreasing the flow rate to zero.
Thus, the rate and efficiency of L-lysine detection is determined by its mass transfer at concentrations lower than 1 µM and by diffusion of the released H2O2 to the active centers of HRP at higher concentrations of L-lysine. The developed bienzyme system is promising for the detection of L-lysine at the concentrations above 1 µM: simplicity of manufacturing and minimization of the number and amounts of system components makes it attractive for further biosensor development, at least for a one-time determinations. However, further work is necessary for the improvement of the stability and for optimization of the operative conditions of the sensor.Fig. 6. Stability of the amperometric response (data normalized to the initial current response) with time for the gold electrodes modified with co-adsorbed CHisrHRP and LO with respect to (1) H2O2 and (2) L-lysine in the concentration range of substrates from 10 to 100 µM.
In conclusion, it has been shown that immobilization of recombinant forms of HRP on a gold electrode surface significantly increases the heterogeneous ET rate compared to electrodes modified with native HRP due to a decrease in the distance between the electrode surface and the active site of the enzyme. The introduction of histidine sequences at either the C- or N-termini of the enzyme molecule additionally increases the strength of the enzyme binding with the gold surface and the efficiency of direct ET. The efficient direct ET between the gold electrodes and the immobilized recombinant forms of HRP containing histidine groups enabled us to develop a P-chip, a biosensor for the detection of hydrogen peroxide based on direct ET, as well as a bienzyme biosensor electrode for the detection of L-lysine based on co-immobilized CHisrHRP and L-lysine-alpha-oxidase.
This work was supported by the European Commission (grant No. ERB IC15-CT96-1002) and by grant No. 4-59 of the Russian State Scientific and Technological Program “Advanced Methods of Bioengineering”. We thank Dr. M. Rubtsova for helpful discussions when preparing this manuscript.
REFERENCES
1.Dunford, H. B. (1982) Adv. Inorg. Biochem.,
4, 41-68.
2.Welinder, K. G. (1979) Eur. J. Biochem.,
96, 483-502.
3.Ruzgas, T., Gorton, L., Emneus, J., and
Marco-Varga, G. (1995) J. Electroanalyt. Chem., 391,
41-49.
4.Yaropolov, A. I., Tarasevich, M. R., and
Varfolomeev, S. D. (1978) Bioelectrochem. Bioenerg., 5,
18-24.
5.Bogdanovskaya, V. A. (1993) Elektrokhimiya,
29, 441-447.
6.Razumas, V., Jasaitis, J., and Kulys, J. (1984)
Bioelectrochem. Bioenerg., 12, 297-302.
7.Bogdanovskaya, V. A., Tarasevich, M. R., Hintsche,
R., and Sheller, F. (1988) Bioelectrochem. Bioenerg., 19,
581-584.
8.Lindgren, A., Munteanu, F., Gazaryan, I., Ruzgas,
T., and Gorton, L. (1998) J. Electroanalyt. Chem., 458,
113-120.
9.Lindgren, A., Tanaka, M., Ruzgas, T., Gorton, L.,
Gazaryan, I., Ishimori, K., and Morishima, I. (1999) Electrochem.
Commun., 1, 171-175.
10.Zhao, J., Henkens, R. W., Stonehuerner, J.,
O'Daly, J. P., and Crumbliss, A. L. (1992) J. Electroanalyt.
Chem., 32, 109-119.
11.Razumas, V. J., Gudavicius, A. V., and Kulys, J.
J. (1986) J. Electroanalyt. Chem., 198, 81-87.
12.Presnova, G., Grigorenko, V., Egorov, A., Ruzgas,
T., Lindgren, A., Gorton, L., and Borchers, T. (2000) Faraday
Discuss., 116, 281-289.
13.Durliat, H., Courteix, A., and Comtat, M. (1989)
Bioelectrochem. Bioenerg., 22, 197-209.
14.Ruzgas, T., Gorton, L., Emneus, J., and
Marco-Varga, G. (1996) Anal. Chim. Acta,330, 123-138.
15.Khudyakova, R. V., Soloshko, S. V., and Safronov,
A. Yu. (1997) Elektrokhimiya, 33, 1165-1171.
16.Safronov, A. Yu., Tarasevich, M. R.,
Bogdanovskaya, V. A., and Chernyak, A. S. (1983) Elektrokhimiya,
19, 421-424.
17.Egorov, A. M., Gazaryan, I. G., Kim, B. B.,
Doseeva, V. V., Kapeliuch, J. L., Veryovkin, A. N., and Fechina, V. A.
(1994) Annals NY Acad. Sci., 721, 73-81.
18.Grigorenko, V., Chubar, T., Kapeliuch, Yu.,
Burchers, T., Spener, F., and Egorov, A. (1999) Biocatalysis and
Biotransformations, 17(5), 359-379.
19.Ward, M. D., and Buttry, D. A. (1990)
Science, 249, 1000-1007.
20.Sauerbrey, G. Z. (1959) Z. Physik.,
155, 206-210.
21.Gajhede, M., Schuller, D. J., Henriksen, A.,
Smith, A. T., and Poulos, T. L. (1997) Nat. Struct. Biol.,
4(12), 1032-1038.
22.Csoregi, E., Jönsson-Pettersson, G., and
Gorton, L. (1993) J. Biotechnol., 30, 315-337.
23.Kulys, J., and Schmid, R. D. (1990)
Bioelectrochem. Bioenerg., 24, 305-311.
24.Eremin, S. A., Vlasenko, S. B., Osipov, A. P.,
Eremina, I. D., and Egorov, A. M. (1989) Analyt. Lett.,
22(9), 2037-2050.
25.Rubtsova, M., Kovba, G., and Egorov, A. (1998)
Biosens. Bioelectron., 13, 75-85.
26.Wendzinski, F., Grundig, B., Renneberg, R., and
Spener, F. (1997) Biosens. Bioelectron., 12, 43-52.
27.Yu, P., and Wilson, G. S. (2000) Faraday
Discuss., 116, 305-317.
28.Vrbova, E., Marek, M., and Ralys, E. (1992)
Analyt. Chim. Acta, 270, 131-136.
29.Curulli, A., Kelly, S., O'Sullivan, C.,
Guilbault, G. G., and Palleschi, G. (1998) Biosens.
Bioelectron., 13, 1245-1250.
30.Saurina, J., Hernandez-Cassou, S., Alegret, S.,
and Fabregas, E. (1999) Biosens. Bioelectron., 14,
211-220.