Molecular Mechanisms of Regulation of the Activity of Sarcoplasmic Reticulum Ca-Release Channels (Ryanodine Receptors), Muscle Fatigue, and Severin's Phenomenon
A. M. Rubtsov
Department of Biochemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3955; E-mail: am_rubtsov@mail.ru
Received April 29, 2001; Revision received June 8, 2001
In this short review of the literature and our own data the characteristics of structural organization of sarcoplasmic reticulum Ca-release channels (ryanodine receptors) in different types of muscles, the participation of other sarcoplasmic reticulum proteins in excitation-contraction coupling and Ca-release channel operation, and the regulation of the channel activity by endogenous low molecular weight compounds are analyzed. Special attention is given to changes that occur in muscle cells during exhausting work and to the role of sarcoplasmic reticulum Ca-release channels in the loss of muscle contractile activity during the development of fatigue. It is concluded that the protection of muscle fibers against fatigue in the presence of the histidine-containing dipeptide carnosine, called in the literature “Severin's phenomenon”, is primarily connected with modulation of sarcoplasmic reticulum Ca-release channel activity by carnosine.
KEY WORDS: sarcoplasmic reticulum, Ca-release channels (ryanodine receptors), excitation-contraction coupling, Ca-binding proteins, protein kinases, carnosine, muscle fatigue
Abbreviations: cADPR) cyclic ADP-ribose; CaM) calmodulin; DHPR) dihydropyridine receptor; FKBP12) 12 kD-protein which binds immunosuppressive drug FK506; G3PDH) glyceraldehyde-3-phosphate dehydrogenase; PK) protein kinase; RyR) ryanodine receptor; SR) sarcoplasmic reticulum.
At the Department of Biochemistry of Lomonosov Moscow State University,
under the supervision of Academician S. E. Severin, the biological role
of the histidine-containing dipeptide carnosine
(beta-alanyl-L-histidine) and related compounds has been
extensively studied for many years. One of the brightest and most
impressive physiological effects of this dipeptide is the so-called
Severin's phenomenon: addition of carnosine into solution bathing
isolated frog muscle preparations at the stage of deep fatigue enhances
very quickly and efficiently the force of muscle contraction and
increases significantly (several-fold) the amount of muscle work that
such preparations can produce [1]. It was shown
later that carnosine not only restores contractility of fatigued muscle
preparations but also prevents the development of fatigue of the
preparations under different modes of stimulation [2]; however, the mechanism of the action of carnosine
on muscle fibers was unknown for a long time. At the Department of
Biochemistry and in number of foreign laboratories it was demonstrated
that carnosine activates contractile proteins of skeletal muscles and
stabilizes energy metabolism in this tissue. The details of these
investigations are presented in a monograph by Prof. A. A. Boldyrev [3]; this book also contains full bibliography of
papers describing studies on the biological effects of carnosine.
However, it has been suggested that all effects of carnosine are
nonspecific and probably connected with the pH buffering ability of
carnosine: at physiological pH values carnosine and other
histidine-containing dipeptides have relatively good buffering capacity
[4].
It should be noted that when Severin's phenomenon was discovered very little was known about the details of excitation-contraction coupling in skeletal muscles, the molecular mechanisms of muscle contraction and the development of muscle fatigue. Therefore, it is not surprising that the molecular targets of the action of carnosine in muscle cells were a mystery for a long time. Now the sequence of molecular events in muscle contraction and relaxation is well established. Excitation-contraction coupling begins from the transmission of excitation from nerve terminals to muscle fibers in the synapse region and prolongation of a depolarization wave on the plasma membrane to the tubules of the T-system. Sarcolemma depolarization in this region induces the opening of sarcoplasmic reticulum (SR) Ca-release channels, the release of Ca2+ from SR into cytoplasm, activation of Ca-binding proteins of the contractile apparatus, and muscle contraction. The subsequent removal of Ca2+ by SR Ca-ATPase induces muscle relaxation [5]. It should remember that contractile proteins, actin and myosin, were well known in the middle of the last century but SR Ca-ATPase was discovered only in the early 60s and SR Ca-release channels in late 80s despite their presence in SR membranes being postulated in the middle of the 70s [6]. Practically simultaneously it was suggested that muscle fatigue is connected primarily with disturbances of Ca2+ release from SR [7]. This suggestion was later supported by numerous experimental data and now it is well documented that SR Ca-release channels are the “weak link of the chain”, the disturbance of whose operation results in muscle fatigue [5, 8].
An abundance of recently published reviews focus on different aspects of SR Ca-release channel organization and operation; they contain detailed bibliographies of original research papers [8-16]. Therefore, in this article we will describe only some aspects of SR Ca-release channel structure, their interaction with other SR proteins, and their regulation by low molecular weight endogenous substances with special attention to the effects of carnosine and other histidine-containing compounds on SR Ca-release channels.
STRUCTURE OF SARCOPLASMIC RETICULUM Ca-RELEASE CHANNELS
Sarcoplasmic reticulum Ca-release channel (ryanodine receptor, RyR) is a homotetramer consisting of four identical 560-kD subunits each, so, functionally active Ca-release channel has molecular mass >2*106 daltons [13]. This protein is characterized by the ability to bind with high affinity the plant alkaloid ryanodine with Kd values from 2 to 7 nM for different isoforms [15]. RyR binds ryanodine in stoichiometry 1 mole/mole of tetramer; ryanodine interacts only with open Ca-release channel and fixes it in this state. The C-terminal regions of RyR molecule are necessary for ryanodine binding and an interaction of the RyR protomers is necessary for the formation of this binding site: the monomeric form of RyR does not bind ryanodine [14, 17].
According to current opinion [18], each RyR polypeptide contains 12 transmembrane alpha-helixes that form a pore for Ca2+ release from the SR lumen, a large cytoplasmic domain consisting of the N-terminal part of the molecule, and loops connecting transmembrane alpha-helixes (Fig. 1). Four transmembrane helixes contain negatively charged amino acids that are conserved and are present in all known RyR isoforms. Replacement of only one of these amino acids (E3885A in transmembrane domain M2) leads to a 10,000-fold decrease in the sensitivity of the RyR3 isoform for Ca2+ [18]. It is suggested that this particular amino acid in each monomer of RyR tetrameric complex participates in the formation of the high affinity Ca-binding site.
The C-terminal part of the RyR molecule is also highly conserved. Deletion of three C-terminal amino acids significantly decreases the activity of the RyR1 isoform of Ca-release channel, and deletion of only 15 C-terminal amino acids leads to its total inactivation (this isoform contains 5037 amino acids); however, such molecules are able to form tetramers [19]. This part of the molecule is probably necessary for the formation of functionally active Ca-release channel. The single mutation (R615C) in pig skeletal muscle RyR isoform leads to the development of a disease known as malignant hyperthermia [20]. In humans this disease can be a result of 17 different mutations in two parts of the RyR molecule (amino acid substitutions in regions 36-615 and 2163-2458). This disease involves increased sensitivity of Ca-release channels for Ca2+, and stress (in pigs) or the use of particular local anesthetics (in humans) trigger a sharp increase in body temperature that can even lead to death [20, 21]. Therefore, these parts of the RyR molecule are also involved in the formation of Ca-binding sites. An interaction of skeletal muscle RyR isoform with DHPR necessary for normal transduction of conformational signal during excitation-contraction coupling is provided by a part of RyR molecule consisting of 37 amino acids (residues 1076-1112) [22]. Additionally, the RyR hydrophilic domain contains binding sites for nucleotides, caffeine, CaM, FKBP12, and other regulatory proteins and low molecular weight compounds [13-15].Fig. 1. Schematic diagram of proposed topology in the RyR monomer of hydrophobic alpha-helixes located in SR membrane lipid bilayer and forming the hydrophobic domain of the RyR molecule (shown as white cylinders) and hydrophilic segments (N- and C-terminal parts of the RyR molecule and cytoplasmic loops connecting hydrophobic alpha-helixes) forming its hydrophilic domain of the Ca-release channel [18]. Negatively charged amino acid residues which are present in conserved hydrophobic sequences of all known RyR isoforms of vertebrates and invertebrates are shown in single-letter amino acid code. Replacement of one of these amino acids (E3885A) in the M2 transmembrane domain of the RyR3 isoform results in the loss of sensitivity of the Ca-release channel to Ca2+.
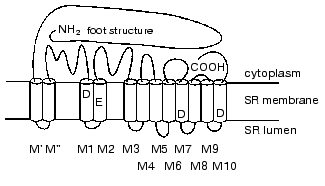
According to high-resolution cryo-electron microscopy data, the RyR tetrameric complex appears as a mushroom-like structure with a square shaped “cap” (27 × 27 × 11 nm) and a “stem” (12 × 12 × 6.5 nm) [23, 24]. The stem of the mushroom (the hydrophobic RyR domain which forms a transmembrane pore) anchors RyR in the SR membrane and the cap connects RyR with the plasma membrane and interacts with DHPR. Such molecules form in the SR membrane regular arrays in which RyR tetramers interact with each other by the “corners” of their hydrophilic domains, which is necessary, for example, for their cooperative interaction during Ca2+ binding [13, 15, 23, 24]. The opening of the Ca-release channel is accompanied by large-scale conformational changes that involve both the hydrophilic and the hydrophobic domains of the RyR molecule. These conformational changes increase the diameter of the central ion-conducting pore in a manner similar to the opening of the diaphragm of a camera or the iris of an eye [24, 25].
RYANODINE RECEPTOR ISOFORMS AND CHARACTERISTICS OF
EXCITATION-CONTRACTION COUPLING IN DIFFERENT TYPES OF MUSCLES
In mammalian tissues, RyR is present in three isoforms with high levels of homology (65-70%) that are encoded by genes located on different chromosomes in the human genome [10, 15]. The RyR1 isoform (5037 amino acid residues) is expressed predominantly in skeletal muscles and cerebellum, RyR2 (4976 amino acid residues) in cardiac muscle and brain, and RyR3 (4872 amino acid residues) in epithelial cells and specific regions of the brain (hippocampus, corpus striatum, and thalamus). Tetrameric complexes of all RyR isoforms consist of only identical polypeptides. They have similar topology, bind ryanodine with high affinity in stoichiometry 1 mole/mole tetramer, and are activated by low Ca2+ concentrations (EC50 is 1-4 µM for RyR1 and RyR2 and 10-14 µM for RyR3), adenine nucleotides, and caffeine. They are inhibited by millimolar Ca2+ concentrations, Mg2+, ruthenium red, and the local anesthetic procaine. They form pores in the membrane with conductance (in the presence of 500 mM KCl) 630, 550, and 743 pS for RyR1, RyR2, and RyR3, respectively [15].
In skeletal muscles of non-mammalian vertebrates (birds, reptiles, amphibians, and fish) two isoforms of RyR were found which are present in equal amounts--alpha-RyR with 80% homology to RyR1 of mammals and beta-RyR with 86% homology to RyR3 [10]. In addition, in heart of non-mammalian vertebrates a third RyR isoform homologous to the RyR2 isoform is present. As in the case with mammals, all these RyR isoforms are homotetramers consisting only from identical subunits [15].
It was recently found that in mammalian skeletal muscles not only RyR1 but also RyR3 is expressed; however, the functional role of the RyR3 isoform in excitation-contraction coupling still is not clear [15, 24]. The maximal amount of RyR3 was found in diaphragm and some slow muscles of several mammals but in other skeletal muscles this isoform appears before birth, then its content increases to a maximal level at 2-4 weeks, and then decreases to 1-2% of the content of RyR1 [15, 24]. The expression of RyR3 is also under control of muscle activity: in disuse atrophy of the soleus muscle the level of RyR1 expression is increased whereas the level of RyR3 expression is decreased [26]. It is interesting to note that the contractile ability of skeletal muscles of adult mice with ryr3 gene knockout is not affected, but in newborn mice this ability is strongly depressed [27]. In animals with knockout of the ryr1 gene or with knockout of both genes practically total degeneration of skeletal muscles, especially expressed in the latter case, is observed [28, 29].
It is now well documented that direct contact of RyR1 with DHPR of sarcolemma is responsible for the activation of the SR Ca-release channel [9-16]. The RyR1 molecules are localized in so-called junctional zones of SR membranes--areas of SR terminal cisterns which are in close contact with plasma membrane invaginations penetrating into muscle fibers--T-tubules. The so-called junctional feet (hydrophilic domains of RyR1 tetrameric complexes) are located between membranes of T-tubules and membranes of the junctional face of SR [9, 10, 13]. One half of RyR1 molecules (in muscles of birds and amphibians--only alpha-RyR) contacts with 4 molecules of DHPR (so-called tetrads) and every RyR monomer interacts with the alpha1-subunit of DHPR. The intracellular loop connecting transmembrane repeats II and III of the skeletal muscle isoform of DHPR alpha1-subunit and amino acid residues 1076-1112 in the RyR1 molecule are responsible for this interaction [15, 16, 22, 30]. During excitation-contraction coupling, DHPR plays the role of “voltage sensor”: following sarcolemma depolarization, its conformation is changed in such way that particular domains of the alpha1-subunit are moved in the direction normal to the membrane plane. This movement induces changes in the RyR1 molecule conformation that results in channel opening. The second half of the RyR1 molecules and, probably, RyR3 molecules (in muscles of birds and amphibians--beta-RyR) are not connected with DHPR. Nevertheless, these molecules of Ca-release channels are opened practically simultaneously with other channels. It is suggested that such synchronous operation of Ca-release channels is concerted by a tight interaction of RyR1 molecules. As mentioned above, RyR molecules form in SR membranes an ordered structures consisting of 2-3 arrays of molecules [13, 15, 30]. RyR tetramers interact with each other by the “corners” of their hydrophilic domains, and the presence of FKBP12 is necessary for this interaction [23, 24]. In the presence of FKBP12, molecules of Ca-release channels are opened synchronously even after their incorporation into artificial lipid bilayers, and removal of FKBP12 disrupts the normal interaction of RyR molecules [31]. Perhaps the local increase in concentration of Ca2+ released from SR during activation of Ca-release channels, which are in direct contact with DHPR, plays a particular role in activation of RyR1, RyR3, and beta-RyR molecules that are not connected with DHPR [9, 10, 15, 16]. This is suggested by the fact that in mutant mice lacking FKBP12 the normal functioning of skeletal muscles is preserved [32].
Excitation-contraction coupling in heart is provided by another mechanism--Ca-induced activation of SR Ca-release channels [10, 15, 16]. The system of T-tubules in heart is poorly developed in comparison with skeletal muscles and most RyR2 molecules are not in contact with sarcolemma. The Ca-influx into cytoplasm from extracellular medium through voltage-sensitive Ca-channels is necessary for activation of RyR2. The cardiac isoform of FKBP12, FKBP12.6, is probably strictly required for normal functioning of RyR2 because in mutant mice lacking this protein severe forms of cardiomyopathy develop [32]. The question of the participation of other intracellular messengers, especially cADPR, in the regulation of functional activity of RyR2 is still open. It was shown that the cADPR antagonist 8-amino-cADPR reduces contraction and intracellular level of Ca2+ in guinea pig cardiac myocytes during electrical stimulation [33]. It is suggested that cADPR increases the sensitivity of RyR2 for Ca2+ during Ca-induced activation of Ca-release.
The Ca-induced Ca-release and cADPR play the main role in RyR3 isoform functioning in different types of tissues [15, 24, 34]. An increase in Ca2+ concentration in cytoplasm after stimulation of voltage-sensitive and receptor-operated Ca-channels of plasma membrane activates RyR3 in reticulum membranes. The same activation occurs after release of Ca2+ from intracellular stores through another type of Ca-release channels, the InsP3-receptors [15, 34]. It is interesting to note that cADPR activates RyR3 via interaction with FKBP12.6, at least in insulin-secreting cells of the pancreas [35]. It is suggested also that the stimulating action of NO on Ca2+ exchange in many cases is realized via changes in cADPR level [34].
INTERACTION OF RYANODINE RECEPTOR WITH PROTEINS OF THE
SARCOPLASMIC RETICULUM
It is now well documented that SR Ca-release channels interact with many soluble and intrinsic membrane proteins that provide fine regulation of their functioning (for review, see [11, 13-15]), and the list of these proteins is constantly increasing. The protein FKBP12 was mentioned above. This protein with molecular mass 12 kD belongs to the big family of immunophilins, which are responsible the effect of immunosuppressive drugs such as cyclosporin, rapamycin, and FK506 (the name of protein FKBP12 arose from its ability to bind the drug FK506). Proteins of this family possess cis-trans peptidyl-prolyl isomerase activity and are probably involved in post-translational folding of proteins [36]. Additionally, FKBP12 and FKBP12.6 form tight complexes with intracellular Ca-release channels, RyR and InsP3-receptors, which are not destroyed even during channel purification in the presence of detergents. The ability of these proteins to bind RyR, providing tight contacts between tetrameric complexes of Ca-release channels, is probably not connected in any way with their prolyl isomerase activity [14, 36].
RyR molecules also contain specific binding sites for another soluble protein, Ca-binding protein CaM [14, 23]. The effect of CaM on RyR activity probably depends on Ca2+ concentration: in the presence of nanomolar Ca2+ concentrations, CaM activates the RyR1 (but not RyR2) isoform increasing 3-fold the rate of Ca2+ release from SR and 6-fold the sensitivity of this isoform to activation by Ca2+ [37]. In contrast, in the presence of micromolar Ca2+ concentrations, CaM inhibits Ca-release channel activity [11, 14].
The best known intrinsic membrane protein of the junctional face of SR terminal cisterns is triadin, a protein with molecular mass 95 kD that interacts both with RyR1 and alpha1-subunit of DHPR [14]. It was suggested that triadin provides the contact of these proteins and can directly participate in the transduction of conformational signal from DHPR to RyR1 during excitation-contraction coupling in skeletal muscles. However, triadin has only one transmembrane alpha-helix and its cytoplasmic N-terminal domain consists of only 47 amino acid residues; this is probably not enough for direct contact with DHPR located in sarcolemma. However, the main SR Ca-binding protein calsequestrin is bound with the C-terminal domain of triadin that is enriched with positively charged amino acid residues. As a result of such interaction, calsequestrin is located in close proximity to RyR [11, 14]. Calsequestrin in particular and other lumenal SR Ca-binding proteins and their role in Ca-release channel regulation are now the subject of many studies.
Calsequestrin is a glycoprotein with molecular mass of the protein part of ~40 kD. It can bind up to 40-50 moles Ca2+/mole protein with low affinity (Kd ~ 1 mM) and can release Ca2+ during every cycle of contraction and relaxation [38]. It was found that during stimulation of Ca2+ release from SR by different RyR agonists a rapid increase of free Ca2+ concentration in SR lumen occurs that is followed by the release of this Ca2+ into the cytoplasm through Ca-channels. The removal of calsequestrin eliminates such changes of Ca2+ level in SR lumen and its superexpression in cardiac myocytes of mice increases 10-fold the amount of released Ca2+ [14]. It was suggested that triadin is necessary for locating calsequestrin near RyR. However, it was shown recently that at least RyR1 itself binds calsequestrin with high affinity (Kd ~ 100 nM) [39]. Calsequestrin has a few sites for phosphorylation by protein kinases and is present in SR in a phosphorylated state. It was believed that phosphorylation is necessary for normal post-translational modification (glycosylation) of this protein and its correct targeting to SR terminal cisterns but is not important for its interaction with RyR. However, mutant forms of calsequestrin lacking phosphorylation sites are also found in SR terminal cisterns [40]. Moreover, it was found that only the dephosphorylated form of calsequestrin activates RyR, and dephosphorylation of only 1% of calsequestrin present near the contact zone of SR membranes is enough for half-maximal RyR activation [39, 41]. Therefore, phosphorylation-dephosphorylation of calsequestrin plays an important role in the regulation of SR Ca-release channel functional activity.
It is interesting to note that in the heart of hibernating animals spending winter time in a state of torpor a specific form of calsequestrin was found that is different from all of its known isoforms in electrophoretic mobility [42]. Since the hearts of hibernators retain the ability to contract at low body temperatures (about 0°C) whereas the heart of most mammals stops when body temperature is decreased, it was suggested that the specific calsequestrin isoform provides SR Ca-release channel operation at low temperatures. In addition, the RyR2 itself in the heart of hibernating ground squirrels is characterized by low sensitivity to activating Ca2+ concentrations [42]. The isoform of calsequestrin which is present in skeletal muscles of hibernators is not different in electrophoretic mobility from other known isoforms present in mammalian skeletal muscles [42, 43]. However, we have shown in our investigations that the content of calsequestrin in skeletal muscles of a typical hibernator, the ground squirrel Spermophilus undulatus, is decreased 4-5-fold during the winter period [44, 45].
In addition to calsequestrin, two other high-molecular-weight Ca-binding glycoproteins, sarcalumenin and histidine-rich Ca-binding protein, are present in the SR lumen [14]. Phosphorylation of these proteins by endogenous PK inhibits RyR1 activity [46]. We have found that in skeletal muscle SR of the ground squirrel S. undulatus specific isoforms of these proteins are present that are different in apparent molecular masses from isoforms present in skeletal muscle SR of rats and rabbits [43]. In ground squirrel SR these proteins are also phosphorylated by endogenous PK [44] and during winter period their content is decreased 2.5-3-fold similarly to the content of calsequestrin [45].
In addition to the phosphorylation of Ca-binding proteins, endogenous and exogenous PK phosphorylate triadin [14] and a 30-kD calsequestrin-binding protein [44], but the functional significance of this phosphorylation is still not clear. Protein kinases A, G, and Ca/CaM-dependent PK phosphorylate RyR itself, probably, at the same site (S2483 in rabbit RyR1) with stoichiometry ~1 mole Pi/mole of monomer that results in Ca-release channel activation [11, 14]. Protein kinase C phosphorylates both RyR1 and RyR2 with stoichiometry of ~1 mole Pi/mole of tetramer that also activates the Ca-release channel, but data about the physiological significance of RyR phosphorylation by different PK are rather contradictory [14].
Some proteins of the glycolytic complex, in particular, glycogen phosphorylase, G3PDH, and aldolase, are bound with junctional face of SR membranes. Moreover, the latter two proteins interact directly with RyR1 [11, 14]. It is suggested that glycolysis in this compartment of muscle cell can occur relatively independently from the glycolysis in other parts of the cytoplasm, in particular because significant amounts of glycogen are bound with this part of SR membranes [8].
Despite severe degenerative changes in muscle fibers of mutant mice with knockout of ryr1 and ryr3 genes, the electron microscopy reveals in such fibers triads lacking junctional feet [28, 29]. This shows that in addition to RyR some other proteins are involved in formation of tight connection of membranes of the T-system and SR contact zone. One of these proteins is probably a yet unnamed 90-kD protein that interacts directly with RyR1 and probably participates in stabilization of triad structure [47]. Proteins with molecular mass 72 kD, named junctophilins, three isoforms of which were found in skeletal muscles and heart of mice, also play an important role in such stabilization [48]. The hydrophobic C-terminal region of these proteins spans the SR membrane, and a large cytoplasmic domain interacts specifically with sarcolemma. Mutant mice lacking cardiac junctophilin isoform die as embryos despite normal expression of RyR2 and DHPR [48]. Therefore, even brief analysis shows that the direct or indirect interaction of RyR with a number of soluble and intrinsic membrane proteins is necessary for the normal functioning of SR Ca-release channels.
REGULATION OF SR Ca-RELEASE CHANNELS BY ENDOGENOUS
COMPOUNDS
Among low-molecular-weight compounds and substances which can activate RyR, Ca2+ (at micromolar concentrations), adenine nucleotides, caffeine, ryanodine (at nanomolar concentrations), doxorubicine, as well as heavy metals ions, GSSG, hydrogen peroxide, and other SH-group modifiers are the best known. Ca2+ and Mg2+ (at millimolar concentrations), ruthenium red, ryanodine (at micromolar concentrations), lactic acid, a number of local anesthetics, and compounds reducing disulfide bonds in proteins are inhibitors of SR Ca-release channels [8, 11, 12, 15].
The dependence of SR Ca-release channel activity on Ca2+ concentration is bell-shaped with a maximum at ~100 µM Ca2+. In the presence of caffeine the sensitivity of RyR to the activating effect of Ca2+ is increased about 10-fold, but caffeine has practically no effect on inhibition of Ca-release channels by high Ca2+ concentrations [15]. During the study of caffeine-induced Ca2+ release from preparations of terminal cisterns isolated from rabbit skeletal muscles, we found that the amount of Ca2+ released depends hyperbolically on caffeine concentration with K0.5 ~ 0.3 mM [49]. Caffeine activates isolated SR Ca-release channels incorporated into artificial lipid bilayers [8]; this suggests that the RyR molecule contains a saturating binding site for this compound. Some other caffeine-binding proteins are also present in SR preparations. Using affinity chromatography on Reactive Red 120-agarose, we have shown that caffeine elutes from the column a 170-kD histidine-rich Ca-binding protein. High caffeine concentrations (10-20 mM) are necessary for the elution [49, 50]. It is interesting to note that some parameters of SR Ca-release channel operation, in particular the maximal activity, is simulated by caffeine only at concentrations >10 mM [15]. As mentioned above, histidine-rich Ca-binding protein can participate in the regulation of Ca-release channel activity [46]. This suggests that effect of caffeine on Ca-release channel functioning is provided by its interaction not only with RyR but also with other proteins of the SR contact zone [49].
The rate of Ca2+ release from SR and the amount of Ca2+ released are increased nonlinearly with increase in both total Ca2+ content and concentration of free Ca2+ inside the SR lumen [12, 51]. It is interesting to note that strict coordination of SR Ca-release channel and Ca-pump (Ca-ATPase) operation was recently found: in parallel with the increase of the rate of Ca2+ release from SR at moderate loading of SR by calcium (100-150 nmol Ca2+/mg of protein) the rate of its reaccumulation by Ca-ATPase is also increased [52]. Most likely the Ca2+ concentration gradient across the SR membrane plays the main role in the regulation of the operation of both Ca-release channels and Ca-ATPase, but it is also possible that the proteins of SR membrane and/or lumen participate in coordination of the operation of these systems [8, 14, 15, 52]. Since during operation of Ca-release channels significant amounts of Ca2+ are released from SR, the flow of these ions across the membrane is compensated by flow in the opposite direction of other cations, especially K+, Mg2+, and H+, and by release of Cl- from SR [53]. Because of this, the electric potential on the SR membrane is rapidly compensated. It should be noted that the activity of Ca-release channels is practically independent of the pH in the SR lumen in the range 6.0-7.5, but pH decrease to 5.0-5.5 results in their total inactivation, and subsequent increase in pH restores activity of only ~50% of Ca-channels [54]. Ca-release channels are more sensitive to pH value of the cytoplasm: decrease of pH from 8.0 to 5.0 (at constant pH value inside the SR lumen) results in total channel inhibition, at least for the RyR1 isoform, and half-maximal inhibition occurs at pH 6.5. Subsequent pH increase totally restores the activity of Ca-release channels during 1-60 sec (depending of the duration of SR exposure to acidic medium) [54].
An inhibiting effect of Mg2+ on SR Ca-release channels is provided by its interaction with two types of Ca-binding sites on the RyR molecule: magnesium is a competitive Ca2+ antagonist in the sites with high affinity (activating sites) and agonist in the sites with low affinity (inhibitory sites) [15]. Therefore, Mg2+ shifts the curve of the dependence of Ca-release channel activity on Ca2+ concentration to the right and decreases the channel maximal activity. Among RyR isoforms, RyR2 has the minimal sensitivity to Mg2+; because of this, in the heart the Ca-induced Ca2+ release from SR occurs during excitation-contraction coupling in the presence of relatively high physiological Mg2+ concentrations [10, 15, 16].
It is believed that adenine nucleotides, in contrast to caffeine and Mg2+, do not affect the affinity of Ca-release channels for Ca2+ but significantly increase the rate of Ca2+ release from SR over the whole range of calcium concentrations [15]. However, our results raise some doubts about this situation [50, 55, 56]. In our experiments, we actively loaded SR vesicles isolated from rabbit skeletal muscles with Ca2+ using the operation of the Ca-pump. Since Ca-ATPase can utilize not only ATP but also other nucleoside triphosphates as substrates, we loaded the SR vesicles with Ca2+ in medium containing ATP, GTP, CTP, ITP, or UTP. In ATP-containing medium after accumulation of Ca2+ into SR vesicles, addition of micromolar Ca2+ concentrations or millimolar caffeine concentrations into the medium induced fast ruthenium red-sensitive Ca2+ release from SR by activating Ca-release channels. However, in medium containing non-adenine nucleotides neither Ca-induced nor caffeine-induced Ca-release was registered [55, 56]. Therefore, in the absence of ATP, Ca2+ and caffeine either do not interact with the corresponding binding sites or this interaction does not activate Ca-release channels. However, addition of ATP or other adenine nucleotides to SR vesicles loaded with Ca2+ in the medium with non-adenine nucleotides induces fast ruthenium red-sensitive Ca2+ release through Ca-release channels [50, 55, 56]. This shows that after binding of adenine nucleotides at corresponding binding sites, the affinity of activating Ca-binding sites of RyR1 is sharply increased, this resulting in activation of Ca-release channels. The ability of adenine nucleotides to activate Ca-release channels in our experiments was decreased in the order: ATP ~ beta,gamma-CH2-ATP > ADP > AMP, in good agreement with data from the literature [15]. Recently the change of sensitivity of Ca-release channels to Ca2+ depending on ATP concentration was demonstrated also for isoforms RyR2 [57] and RyR3 [58].
Since the amount of Ca2+ released from SR vesicles after Ca-release channel activation by adenine nucleotides was dependent on the concentrations of nucleotides used, we estimated the affinity of nucleotide-binding sites of RyR for beta,gamma-CH2-ATP and AMP [56]. It was found that the affinity of Ca-release channels to adenine nucleotides depends on the non-adenine nucleotide that was present in the medium (the K0.5 values were different 2-3-fold in the presence of CTP or UTP), in other words, adenine and non-adenine nucleotides compete for the same binding sites on the RyR1 molecule. The affinity of nucleotide-binding sites to non-adenine nucleotides decreased in the order: CTP > ITP > UTP [56]. Therefore, both adenine and non-adenine nucleotides can interact with nucleotide-binding sites of RyR, but the binding of latter with RyR not only does not activate Ca-release channels, but probably even decreases their sensitivity to Ca2+.
The activity of SR Ca-release channels can be regulated also by the interaction of transmembrane RyR domain with hydrophobic compounds [59, 60]. Using a large number of 1,4-dihydropyridine derivatives (agonists, antagonists, and compounds which do not affect DHPR activity), we have demonstrated that these compounds can inhibit Ca-release channels in rabbit skeletal muscle SR [59]. Despite a tight interaction of DHPR and RyR1, the effect of these compounds on the RyR channel activity was not connected with their ability to activate or inhibit DHPR but correlated very well with their hydrophobicity. Hydrophilic compounds did not affect SR Ca-release channels, and the inhibitory action of hydrophobic compounds was in good agreement with their ability to be incorporated into the membrane lipid bilayer and to interact with other intrinsic membrane proteins, in particular with SR Ca-ATPase [59]. The stable hydrophobic verdazil free radicals, which are used for studying the properties of biological membranes, also affected the activity of SR Ca-release channels. Depending of their structure, the verdazil radicals either inhibited or activated Ca-induced Ca2+ release and increased the amount of Ca2+ released from SR vesicles by caffeine [60].
As mentioned above, compounds which oxidize RyR SH-groups--heavy metals ions, GSSG, reactive oxygen species, NO, and many others--are potent activators of SR Ca-release channels [15, 61]. Oxidizing agent-induced Ca2+ release from SR is activated by RyR stimulators such as adenine nucleotides, caffeine, and low Ca2+ concentrations and is inhibited by ruthenium red, tetracaine, and reducing agents. It is believed that oxidation of SH-groups sharply increases the sensitivity of RyR to the activating action of Ca2+, and though it is unlikely that reversible formation and reduction of disulfide bonds play an important role in a single cycle of Ca-release channel opening and closing, the general redox status of the cell can significantly affect their functional properties.
Since mainly the pair GSH/GSSG supports redox potential in muscle cell and in many other tissues, the role of these compounds in the regulation of Ca-release channels is now intensively studied. It is known that under normal conditions the ratio GSH/GSSG in cytoplasm is supported at the level of 30 : 1 that provides redox potential -220 mV, whereas in the SR lumen this ratio is between 3 : 1 and 1 : 1 (redox potential -180 mV) [62, 63]. Among 50 SH-groups of the RyR1 monomer, particular groups have enhanced reactivity (so-called hyperreactive sulfhydryls) and the redox potential of these groups (about -165 mV in the presence of 10-50 µM Ca2+) is changed in the presence of inhibitors and activators of Ca-release channels that results in their opening or closing. Thus, low Ca2+ concentrations and caffeine decrease the potential of hyperreactive SH-groups that facilitate their oxidation and activates Ca-release channels, but Mg2+ and high Ca2+ concentrations, in contrast, increase the potential that provides reduction of disulfides (formation of free SH-groups) and Ca-release channel closing [62]. The rate of ryanodine binding with RyR1 is increased ~10-fold following the increase of redox potential of the medium from -240 to -80 mV. After covalent modification of hyperreactive SH-groups the binding of ryanodine with RyR1 became insensitive to the changes of redox potential, i.e., it is these groups that play the role of redox “sensor” of the cell.
On the other hand, recent data suggest that not redox potential of the cytoplasm itself but the gradient of redox potential on the SR membrane plays the key role in the regulation of Ca-release channel activity [63]. Thus, the open state probability of SR Ca-release channels was minimal when the gradient of redox potential on the SR membrane (created with the use of different ratios of GSH/GSSG) was 40 mV (-180 mV inside SR vesicles and -220 mV outside, which corresponds to normal physiological values) and graduate decrease of gradient value to 0 mV progressively and reversibly increased the Ca-release channel activity [63]. It is interesting to note that the sensitivity of Ca-release channels to redox gradient on SR membrane was abolished by covalent modification of hyperreactive SH-groups. Since SR membranes contain a specific GSH transporter colocalized with RyR1 in the same zone of SR membrane, it is suggested that the local changes of GSH/GSSG ratio on the two sides of the SR membrane are probably part of an important physiological mechanism that regulates the functional activity of Ca-release channels [63].
In addition to formation of disulfide bonds, the effect of another potent physiological regulator of Ca-release channel activity, NO, may also be dependent on redox potential of particular SH-groups in the RyR molecule [64, 65]. Thus, RyR1 and RyR2 isoforms are isolated from skeletal muscles and cardiac tissue, respectively, in S-nitrosylated form. Moreover, S-nitrosylation of 12 out of 84 free SH-groups/tetramer of RyR2 in vitro results in reversible activation of the Ca-release channel and the level of activation correlates very well with the number of modified groups. However, oxidation of 20-24 SH-groups/tetramer of RyR2 has practically no effect on Ca-release channel activity, but further oxidation leads to irreversible and uncontrolled channel activation [64].
It should be noted that most investigations concerning Ca-release channel regulation, in particular, analysis of the action of oxidizing and reducing agents on their activity, were carried out under ambient oxygen tension (~150 mm Hg). This is probably correct for the study of cardiac RyR2 isoform, whereas in skeletal muscles under normal conditions this tension is significantly lower (10-20 mm Hg). Thus, the studies in which the conditions of experiment were close to physiological are of the most interest. It was found that under ambient oxygen tension 24-32 SH-groups/tetramer of RyR1 are in the oxidized state; under these conditions NO does not affect RyR1 activity [65]. Under physiological values of pO2 these SH-groups convert into the reduced state and S-nitrosylation of a single SH-group/monomer of RyR1 in the presence of nanomolar NO concentrations activates the Ca-release channel. S-nitrosylation of this group probably eliminates the inhibiting effect of CaM on the Ca-channel [65]. Therefore, the functional activity of RyR is under fine control of the oxidation and reduction of particular SH-groups and both GSH/GSSG ratio and pO2 in cells and tissues play an important role in this process. The molecular mechanisms responsible for the effect of cell redox status on Ca-release channel activity are still not fully understood, however, it can be said with confidence that S-nitrosylation and reversible formation of disulfide bonds in the RyR molecule are very important in this process.
POSSIBLE MECHANISMS OF DYSFUNCTION OF Ca-RELEASE CHANNEL
OPERATION DURING MUSCLE FATIGUE
As mentioned above, among the reasons for the sharp decrease of skeletal muscle contractile activity during intensive work the changes in the properties of Ca-release channels that result in the disturbances of normal Ca2+ exchange play a main, but not the sole, role. Since many publications have been devoted to this question, we will focus here only on the main factors that affect Ca-release channel operation (for review, see [5, 8, 66]). First, during intensive muscle work the level of free Ca2+ in cytoplasm is increased significantly and high free Ca2+ concentrations inhibit Ca-release channels [8, 15]. Additionally, the decrease of the Ca2+ gradient across the SR membrane can also contribute to disturbances of Ca-release channel operation [12, 51, 52]. Since under particular stimulation protocols the disturbances in muscle fibers became irreversible, it is suggested that the elevation of free Ca2+ level in cytoplasm activates Ca-dependent proteases, especially calpain, which can use RyR and other SR proteins as substrates [67]. Specific pH and temperature dependences of fatigue development indicate that probably other enzymes, in particular Ca-dependent protein kinases and protein phosphatases, are involved in this process [8].
Intensive muscle work is also accompanied by significant (according to some data, 2-fold) increase of free Mg2+ concentration in the cytoplasm. This can be connected both with the decrease of ATP level, in complex with which significant amounts of this cation are present, and with replacement of Mg2+ by Ca2+ in cation-binding sites of Ca-binding proteins when free Ca2+ concentration in cytoplasm is increased [8, 66]. In any case, since Mg2+ is an inhibitor of SR Ca-release channels even small changes of its concentration can disturb normal channel operation.
Though the total ATP level in muscle fibers even during deep disturbances of contractile activity is changed not very significantly, the question of the content of this macroergic compound in different microcompartments of the cell is still open. Thus, it was shown that ATP that is synthesized in the triad region is not in equilibrium with the total pool of this nucleotide in muscle fiber [68]. Since both glycogen and glycolytic enzymes are bound with SR membranes, this cell compartment can operate relatively independently from other compartments; however, during exhaustive muscle work the amount of glycogen in the SR contact zone is probably not enough for the normal functioning of SR [8, 66]. The decrease of ATP level will disturb the operation of both Ca-release channels, which are activated by this nucleotide, and Ca-ATPase, which uses Mg-ATP complex as a substrate [57]. Since the affinity of ADP to Mg2+ is significantly lower than that of ATP, the decrease of the ATP/ADP ratio will increase the Mg2+ concentration that, as mentioned above, also will lead to the inhibition of Ca-release channels.
Exhaustive muscle work is accompanied by significant acidification of cytoplasm [8, 66] that can also disturb Ca-release channel operation because RyR is extremely sensitive to pH value on the cytoplasmic side of the SR membrane [54]. Moreover, Ca-release channels are inhibited by a glycolysis product, lactic acid, which is accumulated in cells during intensive muscle work. An inhibiting effect of lactic acid on Ca-release channels occurs even at neutral pH values [8].
Finally, since the normal operation of SR Ca-release channels depends specifically on redox status of RyR [62-65], reactive oxygen species, the level of which is sharply increased during intensive muscle work, can play a significant role in disturbances of Ca-release channel operation [8, 61]. Probably the mitochondrial respiratory chain is the main source of reactive oxygen species, especial superoxide radical [8]. At conditions of rest, ~2-4% of oxygen can leave mitochondria as free radicals. These are rapidly neutralized by superoxide dismutase, catalase, and GSH. However, during exhaustive muscle work, the consumption of oxygen by mitochondria is increased 20-fold and the amount of free radicals formed can exceed the possibilities of the antioxidative defense mechanisms of the cell [8]. In any case, an increase of muscle oxygen supply will increase pO2 value in muscle cells and, probably, will change the ratio GSH/GSSG that in turn will affect parameters of operation of SR Ca-release channels [62, 63, 65].
Therefore, intensive muscle work is accompanied by significant changes of the content in the cells of a number of compounds that are endogenous regulators of RyR functional activity. Due to these changes, the operation of SR Ca-release channels will be disturbed.
MODULATION OF SR Ca-RELEASE CHANNEL ACTIVITY BY CARNOSINE AND
SEVERIN'S PHENOMENON
Since carnosine effectively protects muscle preparations against fatigue [1, 2] and the development of muscle fatigue is connected in a great extent with the disturbances of SR Ca-release channels operation [5, 8, 66], we have studied the effects of this dipeptide and related compounds on functional parameters of Ca-release channels. In the first stages of investigation we demonstrated experimentally that carnosine is an effective scavenger of free radicals [69, 70]. In experiments using spin traps we have shown that carnosine interacts with OH-radical formed in the Fenton reaction or after illumination of H2O2 solution by UV-light [69]. Anserine, acetylcarnosine, and homocarnosine have the same ability [70]. Later it was demonstrated that carnosine and related compounds can quench not only OH-radical but also a wide variety of other reactive oxygen species (a detailed analysis of the antioxidant properties of histidine-containing dipeptides is presented in [3]). Therefore, carnosine can eliminate one of the factors that accompany the development of muscle fatigue. However, this ability of carnosine is not specifically directed toward the protection of SR Ca-release channels against damage by reactive oxygen species. Rather, the presence of high (millimolar) carnosine concentrations in muscle cell cytoplasm contributes into the overall antioxidant defense system that protects the cell against different types of oxidative stress.
The study of direct carnosine effects on parameters of Ca-release channel operation in heavy fraction of isolated SR preparations, from our point of view, was much more interesting. We have shown that increasing carnosine concentrations progressively increase the time of ATP-dependent accumulation of Ca2+ by SR vesicles [71]. Since it was demonstrated previously that this dipeptide not only does not inhibit the Ca-pump but even enhances efficiency of pump operation [72], we suggested that the decrease of the rate of Ca2+ accumulation by SR vesicles in the presence of carnosine is connected with an increase of Ca2+ leakage from vesicles, i.e., with the activation of Ca-release channels providing this leakage. In fact, carnosine induced fast ruthenium red-sensitive Ca2+ release from SR via activation of Ca-release channels [71, 73]. As in the case with caffeine-induced Ca2+ release [49], the amount of Ca2+ released depended hyperbolically on carnosine concentration, i.e., SR Ca-release channels probably have a saturable binding site for this dipeptide. The affinity of Ca-release channels to carnosine was significantly lower than that to caffeine (K0.5 values ~8.7 and ~0.3 mM, respectively), but the maximal amount of Ca2+ released after carnosine addition is ~1.5-fold higher than that from caffeine-induced Ca2+ release [71, 73]. This may suggest that carnosine interacts with Ca-release channels which are insensitive to caffeine (for example, not only with RyR1 but also with RyR3 isoform) or in the presence of this dipeptide Ca-release channels convert into close state more slowly. In addition, carnosine probably interacts not only with RyR but also with other SR proteins involved in operation of Ca-release channels, for example, with calsequestrin that will increase the amount of Ca2+ released in the SR lumen during activation of the channels.
It should be noted that not only carnosine but also a number of its derivatives activated SR Ca-release channels [74]. Thus anserine, like carnosine, induced Ca2+ release from SR vesicles; the affinity of Ca-release channels to anserine was higher than that to carnosine (K0.5 ~ 2.7 mM) and the maximal amount of Ca2+ released was 1.4-fold higher than that during carnosine-induced Ca2+ release. The ability of carnosine and related compounds to activate SR Ca-release channels decreased in the order: carcinine ~ anserine > carnosine ~ homocarnosine > 1-methyl-L-histidine ~ acethylcarnosine > ophidine. However, components of the carnosine molecule, L-histidine and beta-alanine, added either separately or together in equimolar concentrations do not activate Ca-release channels. Acetylhistidine and 3-methyl-L-histidine also do not induce Ca2+ release [74]. Therefore, particular spatial structure of the dipeptide molecule is necessary for its interaction with the RyR molecule. Introduction of different side groups into the imidazole ring of histidine (as in the case of methylated derivatives, anserine and ophidine) and in other parts of the carnosine molecule (acethylcarnosine) or its decarboxylation (carcinine) result in significant changes in the ability of carnosine derivatives to activate Ca-release channels [73, 74]. It was also shown that carnosine and other histidine-containing dipeptides activate not only the RyR1 isoform of Ca-release channel in SR preparations from rabbit skeletal muscles but also the RyR2 isoform from rat heart [75] and the cardiac RyR isoform from duck heart [74]. It is interesting to note that carnosine activated the RyR1 isoform more efficiently than acetylcarnosine, but for the duck cardiac RyR isoform the opposite dependence was found. Since in heart, in contrast with skeletal muscles, the presence of acetylated forms of histidine-containing dipeptides is typical [3], this may suggest that the interaction of different RyR isoforms with these compounds has particular tissue specificity [74].
In contrast to caffeine, carnosine is an endogenous component of muscle tissue and in skeletal muscles of different animal species its concentration can be 20 mM and even higher [3]. Thus, it was interesting to clarify the possible influence of carnosine on the interaction with SR Ca-release channels with other endogenous regulators of their activity. During the study of AMP-induced Ca2+ release from SR vesicles after their loading with Ca2+ using GTP as the energy source for the Ca-pump, it was found that carnosine (here and further the concentration used was 30 mM) increases ~2.5-fold the affinity of Ca-release channels to this nucleotide. The affinity of Ca-release channels to caffeine was also increased ~1.7-fold in the presence of carnosine [71, 73].
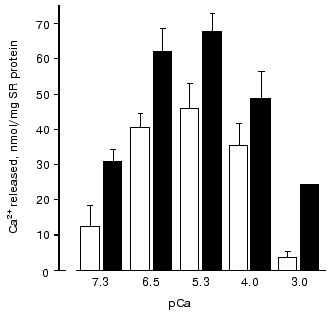
During study of Ca-induced Ca2+ release from SR vesicles after their passive loading with 45Ca2+ [73, 74], it was found that the amount of Ca2+ released during the first 15 sec of registration (i.e., the rate of Ca2+ release from SR) both in absence or in the presence of carnosine has a bell-shape dependence on Ca2+ concentration in the medium (Fig. 2). However, carnosine increased the amount of released Ca2+ at all Ca2+ concentrations used. It is necessary to note that the most significant effect of carnosine was found at low (sub-activating) and high (inhibitory) Ca2+ concentrations. Therefore, in the presence of carnosine the affinity of RyR activating Ca-binding sites to Ca2+ is increased and affinity of inhibitory Ca-binding sites is decreased. Carnosine also totally abolished the inhibitory effect on Ca-release channels of Mg2+ at 50 µM concentration, which corresponds approximately to the Ki value for this inhibitor, but did not prevent inhibition of Ca-release channels by high Mg2+ concentrations (1 mM) or by ruthenium red [73, 74].
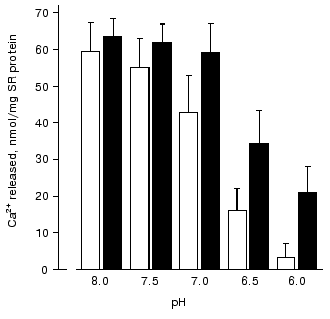
An analysis of the influence of pH on Ca2+ release from SR vesicles has shown [74] that in the absence of carnosine acidification of the medium to pH 6.0 totally inhibits Ca-release channels (Fig. 3). However, in the presence of carnosine at this pH value Ca-release channels retained ~30% of their activity. The protonated carnosine form is probably a more efficient activator of Ca-release channels because at pH 8.0 carnosine had practically no effect on Ca2+ release from SR (Fig. 3).Fig. 2. Dependence of the amount of Ca2+ released from rabbit skeletal muscle SR vesicles in 15 sec on the concentration of free Ca2+ in the medium in the absence (unfilled rectangles) and in the presence of 30 mM carnosine (filled rectangles) [73].
Thus, our investigation of the effects of carnosine and related compounds on the parameters of SR Ca-release channel operation has shown that these compounds are efficient activators of RyR that can induce under particular conditions Ca2+ release from SR. Moreover, Ca-release channels probably have saturable binding sites for these compounds [71, 73]. Since relatively high concentrations of histidine-containing dipeptides are always present in muscle cells, it is unlikely that the dipeptide-induced Ca2+ release occurs in vivo. It is much more interesting that carnosine modulates the interaction of Ca-release channels with other endogenous regulators of their activity: carnosine increases the affinity of Ca-release channels to activating Ca2+ concentrations and adenine nucleotides and prevents the inhibiting effect of high Ca2+ concentrations and, partially, of Mg2+ [71, 73]. In addition, carnosine prevents inhibition of Ca-release channels by acidification of the medium [74] and efficiently interacts with reactive oxygen species, in particular, with free radicals [3, 69, 70]. This suggests that carnosine can stabilize the operation of Ca-release channels during intensive muscle work, protecting them from inhibition by elevated Ca2+ and Mg2+ concentrations, free radicals, and low pH values of the cytoplasm and increasing the affinity of Ca-release channels to adenine nucleotides. Therefore, Severin's phenomenon--the carnosine-mediated protection of muscle fibers against fatigue--is mainly connected with the modulation of Ca-release channel activity by this dipeptide.Fig. 3. Dependence of the amount of Ca2+ released from SR vesicles of rabbit skeletal muscles during 15 sec on pH of incubation medium in the absence (unfilled rectangles) and in the presence of 30 mM carnosine (filled rectangles) [74].
The study of SR Ca-release channels at the Department of Biochemistry was made possible in part by the International Science Foundation and the Russian Government (grant JFF100) and the Russian Foundation for Basic Research (grants 95-04-11918, 98-04-49184, and 01-04-48237).
REFERENCES
1.Severin, S. E., Kirzon, M. V., and Kaftanova, T. M.
(1953) Dokl. Akad. Nauk SSSR, 91, 691-694.
2.Dupin, A. M., and Stvolinskii, S. L. (1986)
Biokhimiya, 51, 160-164.
3.Boldyrev, A. A. (1998) Carnosine. Biological
Significance and Possibilities of Medical Applications [in
Russian],MGU, Moscow.
4.Skulachev, V. P. (1992) Biokhimiya,
57, 1311-1316.
5.Stephenson, D. G., Lamb, G. D., and Stephenson, G.
M. M. (1998) Acta Physiol. Scand., 162, 229-245.
6.Ogawa, Y., and Ebashi, S. (1976) Biochem.
J., 80, 1149-1157.
7.Eberstein, A., and Sandow, A. (1976) in The
Effects of Use and Disuse in Neuromuscular Function (Guttman, E.,
and Hnik, P., eds.) Elsevier, Amsterdam, pp. 515-526.
8.Favero, G. F. (1999) J. Appl. Physiol.,
87(2), 471-483.
9.Meissner, G., and Lu, X. (1995) Biosci.
Rep., 15, 399-408.
10.Sutko, J. L., and Airey, J. A. (1996) Physiol.
Rev., 76, 1027-1071.
11.Rubtsov, A. M., and Batrukova, M. A. (1997)
Biochemistry (Moscow), 62, 933-945.
12.Sitsapesan, R., and Williams, A. J. (1997) J.
Membr. Biol., 159, 179-185.
13.Franzini-Armstrong, C., and Protasi, F. (1997)
Physiol. Rev., 77, 699-729.
14.Mackrill, J. J. (1999) Biochem. J.,
337, 345-361.
15.Ogawa, Y., Kurebayashi, N., and Murayama, T.
(1999) Adv. Biophys., 36, 27-64.
16.Lamb, G. D. (2000) Clin. Exp. Pharmacol.
Physiol., 27, 216-224.
17.Callaway, C., Seryshev, A., Wang, J. P., Slawik,
K. J., Needleman, D. H., Cantu, C. R., Wu, Y., Jayaraman, T., Marks, A.
R., and Hamilton, S. L. (1994) J. Biol. Chem., 269,
15876-15884.
18.Chen, S. R. W., Ebisawa, K., Li, X., and Zhang,
L. (1998) J. Biol. Chem., 273, 14675-14678.
19.Gao, L., Tripathy, A., and Meissner, G. (1997)
FEBS Lett., 412, 223-226.
20.Denborough, M. (1998) Lancet, 352,
1131-1136.
21.Tong, J., Oyamada, H., Demaurex, N., Grinstein,
S., McCarthy, T. V., and MacLennan, D. H. (1997) J. Biol. Chem.,
272, 26332-26339.
22.Leong, P., and MacLennan, D. H. (1998) J.
Biol. Chem., 273, 7791-7794.
23.Wagenknecht, T., Rademacher, M., Grassucci, R.,
Berkowitz, J., Xin, H.-B., and Fleisher, S. (1997) J. Biol.
Chem., 272, 32463-32471.
24.Sorrentino, V., and Reggiani, C. (1999) Trends
Cardiovasc. Med., 9, 54-61.
25.Serysheva, I. I., Schatz, M., van Heel, M., Chiu,
W., and Hamilton, S. L. (1999) Biophys. J., 77,
1936-1944.
26.Bastide, B., Conti, A., Sorrentino, V., and
Mounier, Y. (2000) Biochem. Biophys. Res. Commun., 270,
442-447.
27.Bertocchini, F., Ovitt, C. E., Conti, A., Barone,
V., Scholer, H. R., Bottinelli, R., Reggiani, C., and Sorrentino, V.
(1997) EMBO J., 16, 6956-6963.
28.Ikemoto, N., Komazaki, S., Takeshima, H., Nishi,
M., Noda, T., Iino, M., and Endo, M. (1997) J. Physiol.,
501, 305-312.
29.Barone, V., Bertocchini, F., Bottinelli, R.,
Protasi, F., Allen, P. D., Franzini-Armstrong, C., Reggiani, C., and
Sorrentino, V. (1998) FEBS Lett., 422, 160-164.
30.Franzini-Armstrong, C., Protasi, F., and Ramesh,
V. (1999) Biophys. J., 77, 1528-1539.
31.Marx, S. O., Ondrias, K., and Marks, A. R. (1998)
Science, 281, 818-821.
32.Shou, W., Aghdasi, B., Armstrong, D. L., Guo, Q.,
Bao, S., Charng, M. J., Mathews, L. M., Schneider, M. D., Hamilton, S.
L., and Matzuk, M. M. (1998) Nature, 391, 489-492.
33.Rakovic, S., Galione, A., Ashamu, G. A., Potter,
B. V., and Terrar, D. A. (1996) Curr. Biol., 6,
989-996.
34.Lee, H. C. (1997) Physiol. Rev.,
77, 1133-1164.
35.Noguchi, N., Takasawa, S., Nata, K., Tohgo, A.,
Kato, I., Ikehata, F., Yonekura, H., and Okamoto, H. (1997) J. Biol.
Chem., 272, 3133-3136.
36.Marks, A. R. (1996) Physiol. Rev.,
76, 631-649.
37.Fruen, B. R., Bardy, J. M., Byrem, T. M.,
Strasburg, G. M., and Louis, C. F. (2000) Am. J. Physiol. Cell.
Physiol., 279, C724-C733.
38.Yano, K., and Zarain-Herzberg, A. (1994) Mol.
Cell. Biochem., 135, 61-70.
39.Herzog, A., Szegedi, C., Jona, I., Herberg, F.
W., and Varsanyi, M. (2000) FEBS Lett., 472, 73-77.
40.Nori, A., Furlan, S., Patri, F., Cantini, M., and
Volpe, P. (2000) Exp. Cell Res., 260, 40-49.
41.Szegedi, C., Sarcozi, S., Jona, I., and Varsanyi,
M. (1999) Biochem. J., 337, 19-22.
42.Milner, R. E., Michalak, M., and Wang, L. C. H.
(1991) Biochim. Biophys. Acta, 1063, 120-128.
43.Shutova, A. N., Storey, K. B., Lopina, O. D., and
Rubtsov, A. M. (1999) Biochemistry (Moscow), 64,
1250-1257.
44.Rubtsov, A. M. (2001) in Cell and Molecular
Responces to Stress (Storey, K. B., and Storey, J. M., eds.) Vol.
2, Elsevier Science, Amsterdam, pp. 57-71.
45.Malysheva, A. N., Storey, K. B., Ziganshin, R.
Kh., Lopina, O. D., and Rubtsov, A. M. (2001) Biochemistry
(Moscow), 66, 918-925.
46.Shoshan-Barmatz, V., Orr, I., Well, S., Meyer,
H., Varsanyi, M., and Heilmeyer, L. M. (1996) Biochim. Biophys.
Acta, 1283, 89-100.
47.Froemming, G. R., Pette, D., and Ohlendieck, K.
(1999) Biochem. Biophys. Res. Commun., 261, 603-609.
48.Takeshima, H., Komazaki, S., Nishi, M., Iino, M.,
and Kangawa, K. (2000) Mol. Cell, 6, 11-22.
49.Rubtsov, A. M., and Murphy, A. J. (1988)
Biochem. Biophys. Res. Commun., 154, 462-468.
50.Smirnova, M. B., Rubtsov, A. M., and Boldyrev, A.
A. (1989) Ukr. Biokhim. Zh., 61, 57-64.
51.Shannon, T. R., Ginsburg, K. S., and Bers, D. M.
(2000) Biophys. J., 78, 334-343.
52.Saiki, Y., and Ikemoto, N. (1999)
Biochemistry, 38, 3112-3119.
53.Fink, R. H. A., and Veigel, C. (1996) Acta
Physiol. Scand., 156, 387-396.
54.Laver, D. R., Eager, K. R., Taoube, L., and Lamb,
G. D. (2000) Biophys. J., 78, 1835-1851.
55.Rubtsov, A. M., Quinn, P. J., and Boldyrev, A. A.
(1988) FEBS Lett., 238, 240-244.
56.Rubtsov, A. M., Smirnova, M. B., and Boldyrev, A.
A. (1988) Biochem. Int., 17, 629-636.
57.Yang, Z., and Steele, D. K. (2000) J.
Physiol., 523, 29-44.
58.Manunta, M., Rossi, D., Simeoni, I., Butelli, E.,
Romanin, C., Sorrentino, V., and Schindler, H. (2000) FEBS
Lett., 471, 256-260.
59.Rubtsov, A. M., Barkalaya, N. Z., Boldyrev, A.
A., Uldrikis, J. R., Kastron, V. V., Skrastins, I. P., Bisenieks, E.
A., Tirzitis, G. D., and Duburs, G. J. (1989) Biol. Membr.
(Moscow), 6, 18-27.
60.Uspanova, Zh. K., Rubtsov, A. M., Vigalok, I. V.,
and Boldyrev, A. A. (1988) Ukr. Biokhim. Zh., 60,
62-67.
61.Kourie, J. I. (1998) Am. J. Physiol. Cell.
Physiol., 275, C1-C24.
62.Xia, R., Stangler, T., and Abramson, J. J. (2000)
J. Biol. Chem., 275, 36556-36561.
63.Feng, W., Liu, G., Allen, P. D., and Pessah, I.
N. (2000) J. Biol. Chem., 275, 35902-35907.
64.Xu, L., Eu, J. P., Meissner, G., and Stamler, J.
S. (1998) Science, 279, 234-237.
65.Eu, J. P., Sun, J., Xu, L., Stalmer, J. S., and
Meissner, G. (2000) Cell, 102, 499-509.
66.Allen, D. G., Lannergren, J., and Westerblad, H.
(1995) Exp. Physiol., 80, 497-527.
67.Belcastro, A. N. (1993) J. Appl. Physiol.,
74, 1381-1386.
68.Han, J., Thieleczek, R., Varsanyi, M., and
Heilmeyer, L. M. J., Jr. (1992) Biochemistry, 31,
377-384.
69.Rubtsov, A. M., Schara, M., Sentjurc, M., and
Boldyrev, A. A. (1991) Acta Pharm. Jugosl., 41,
401-407.
70.Boldyrev, A. A., Kurella, E. G., Rubtsov, A. M.,
Tyulina, O. V., Schara, M., and Shentjurc, M. (1992) Biokhimiya,
57, 1360-1365.
71.Batrukova, M. A., Rubtsov, A. M., and Boldyrev,
A. A. (1992) Biokhimiya, 57, 904-910.
72.Lopina, O. D., and Boldyrev, A. A. (1975)
Dokl. Akad. Nauk SSSR, 220, 1218-1221.
73.Batrukova, M. A., and Rubtsov, A. M. (1997)
Biochim. Biophys. Acta, 1324, 142-150.
74.Batrukova, M. A. (1997) Effect of
Histidine-Containing Dipeptides on the Functional Characteristics of
Sarcoplasmic Reticulum Ca-Release Channels [in Russian], Ph. D.
Thesis, MGU, Moscow.
75.Zaloga, G. P., Roberts, P. R., and Nelson, T. E.
(1996) New Horiz., 4, 26-35.