What Can Medicine Learn from the Human DNA Sequence?
E. Hofmann
University of Leipzig, Medical Faculty, Institute of Biochemistry, Liebigstr. 16, D-04103 Leipzig, Germany; E-mail: EberhardRenate.Hofmann@t-online.de
Received April 27, 2001
The cooperation of biochemistry with clinical medicine consists of two overlapping temporal phases. Phase 1 of the cooperation, which still is not finished, is characterized by joint work on the pathogenesis and diagnostics of systemic metabolic diseases, whereas in phase 2 the cooperation on tissue and cell specific as well as on molecular diseases is prevailing. In view of the conceptual revolution and shift in paradigm, which biochemistry and medicine are presently experiencing, the content of cooperation between the two disciplines will profoundly change. It will become deeply influenced by the results of the research into the human genome and human proteome. Biochemistry will strongly be occupied to relate the thousands of protein coding genes to the structure and function of the encoded proteins, and medicine will be concerned in finding new protein markers for diagnostics, to identify novel drug targets, and to investigate, for example, the proteomes of the variety of tumors to aid tumor classification, to mention only a few areas of interest which medicine will have in the progress of human genome research. The review summarizes the recent achievements in sequencing the human DNA as published in February 2001 by the International Human Genome Sequencing Consortium and Celera Genomics and discusses their significance in respect to the further development of molecular, in particular genetic, medicine as an interdisciplinary field of the modern clinical sciences. Only biochemistry can provide the conceptual and experimental basis for the causal understanding of biological mechanisms as encoded in the genome of an organism.
KEY WORDS: human genome, human DNA sequence, genomics, proteomics, protein-coding genes, non-protein-coding genes, repetitive DNA, molecular medicine
[Footnote: In commemoration of my highly esteemed colleague and good friend Sergei Evgenievich Severin on the occasion of his 100th Anniversary.]
CONTENT AND QUALITY OF COOPERATION OF BIOCHEMISTRY WITH
MEDICINE
The cooperation of biochemistry with medicine is traditionally excellent and has proved very productive for both partners. In the past, two overlapping phases in cooperation may be distinguished. In phase 1 cooperation in the pathogenesis and diagnostics of systemic diseases with metabolic, hormonal, intestinal, nutritional, and hereditary background was dominating, whereas in phase 2, which presently is prevailing, cooperation is focussed towards tissue and cell specific diseases, like tumor, heart and circulation, hepatic, immunological, neurodegenerative, protein folding, as well as hereditary diseases. No doubt, the cooperation exerts synergetic effects and stimulates clinical and biochemical research as well and gives rise not only to new concepts and insights into basic knowledge but also to significant improvements of clinical practice.
In view of the conceptual revolution and the shift in paradigm that biochemistry and medicine are experiencing presently, the content of cooperation between the two disciplines will change again and will attain a new quality. The most obvious paradigm shift was initiated when biochemists learned to experiment with the information carrying and transferring macromolecules DNA and RNA. The achievements caused biochemistry to become a causal-analytically oriented science and pushed open the door to many novel questions and to the birth of molecular medicine as a highly productive interdisciplinary way to new visions and capabilities. The new problems initiated the development of high throughput technologies, relying on technical improvements to enhance, to amend, and to automate traditional as well as novel methods. Although the new technological developments turned out to be to the benefit of biochemistry and medicine, the danger cannot be ignored that the automated methods governing more and more modern biological research might dissociate scientists from the traditional hypothesis generating experimental basis of scientific investigation, which for more than one century was recognized widely as typical for biochemistry and related sciences [1]. Oppressive thoughts like that do arise in view of the automations of the methods in sequencing the human genome and in investigating the human proteome. However, they will be mitigated when biochemists start to translate the information of thousands of hitherto unknown protein-coding genes accommodated in the human and in other genomes into the structure and function of the encoded proteins. It can be anticipated that the coming new stage in the cooperation of biochemistry with medicine will mainly be based on two fields, on the achievements of modern protein biochemistry and on the results of genome/proteome research. New activities in the large-scale purification of novel, hitherto unknown proteins, and in the investigation of their structure and function will be initiated. This will deepen our insights into cell biology and biological evolution and development. In respect to the collaboration with medicine, this will be the only way to identify hitherto unknown proteins and to investigate their function. These efforts will lead to new protein markers for medical diagnostics, to the identification of proteins as novel drug targets, and to investigate for example tumor proteomes to aid tumor classification and to improve tumor therapy, to mention only a few goals of the coming next phase of cooperation. The winners will be medicine and the welfare of human beings as well as basic science by gaining novel insights into fundamental knowledge.
The sequencing of the human genome as well as the structural and functional characterization of the great number of expressed and hitherto unknown proteins will open a new era in the collaboration between the basic and the clinical sciences. The message is that only biochemistry can provide the experimental basis for the causal understanding of biological mechanisms as encoded in the genomes of the great variety of biological species.
THE GENERAL STRUCTURE OF THE HUMAN GENOME
Two qualitatively different and competitive approaches were started to sequence the human genome, the publicly funded International Human Genome Project, which includes 20 laboratories and hundreds of scientists around the world, the results of which being totally available worldwide, and a privately funded project performed by the company Celera Genomics, that hopes to sell the information. In February 2001 two drafts of the human genome sequence were published [2, 3]. Because both approaches are incomplete hitherto and still have many gaps and imperfections, the description of the human genome is not yet of high precision. Nevertheless, many valuable conclusions can be drawn from the published data and a series of interpretations performed. For comparison, the results of sequencing of four other eukaryotic genomes turned out to be of great value, that of the yeast Saccharomyces cerevisiae [4], the worm Caenorhabditis elegans [5], the fruit fly Drosophila melanogaster [6], and the plant Arabidopsis thaliana [7]. Now, the 3.1 giga base pairs (Gbp) of the haploid human genome are sequenced to about 85%. With respect to the weakly staining and gene rich euchromatic parts of the genome (2.95 Gbp) the human DNA sequence is about 90% complete [2, 3].
As known from previous achievements in the investigation of the genomes of a great number of eukaryotes, also the human DNA sequence is built up of several regions: protein-coding regions (possibly not more than approximately 2% of the total genome), non-protein-coding genes (“RNA only” coding genes) (estimated up to about 20% of the total genome), repeated sequences (more than 50% of the total genome); because cloning of these regions often is difficult or even impossible, numerous gaps in the sequence are left; fortunately, the repetitive regions of the genome are very poor in protein-coding genes.
Gaps in the draft genome sequence. In the published human genome sequence approximately 145,514 gaps remain. Three types of gaps must be distinguished: gaps within unfinished sequenced clones (“sequence gaps”); gaps between sequenced-clone contigs, but within fingerprint clone contigs; gaps between fingerprint clone contigs. The sequence gaps in the draft sequence cover about 80 mega base pairs (Mbp) (approximately 3% of the sequence) and the average size of a gap is 554 bp. About 9% of the genome resides in the two other types of gaps. From these numbers one can calculate that approximately 88% of the human genome is represented in the two published draft genome sequences.
The GC content and CpG islands in the human genome. Within the human genome long-range GC-rich and GC-poor regions are distinguishable. These regions differ in gene density, composition of repeat sequences, and in recombination rate. The distribution of the CpG islands across the genome is of particular interest because very often this dinucleotide is associated with the 5´ ends of genes [8]. Characteristically, CpG islands are underrepresented in human DNA. In the human DNA sequence altogether 50,267 CpG islands can be found and in the sequence masked to eliminate repeat sequences 28,890 CpG islands are distinguishable. The difference is due to the fact that the Alu element (300 bp) as major repeating sequence in the human genome is CG rich.
NON-PROTEIN-CODING GENES
By far the largest part of the human genome does not encode proteins. This part is composed of a great number of different portions of DNA-sequences, which may be subdivided roughly into “RNA only” transcribed DNA-segments and DNA-repeat regions. Thousands of human genes are transcribed only and produce several classes of non-protein-coding RNA-species (ncRNA).
Transfer-RNA genes: in the human genome 497 tRNA genes and one SeCys tRNA gene were identified. For comparison, in the worm 585, in the fly 285, and in yeast 274 tRNA genes (no SeCys tRNA gene) were found.
Ribosomal RNA: although previously published investigations revealed several hundred copies of the four different types of rRNA genes (18S rRNA, 5.8S rRNA, 28S rRNA and 5S rRNA) clustered on the short arms of the acrocentric chromosomes no true complete copies of rRNA repeats could really be identified yet in the human genome except four genes for the 5S rRNA. This puzzle will perhaps not be resolved before the human DNA sequence is completed. In the light of the results of high resolution X-ray crystallography of the large ribosomal subunit, which showed that peptide formation is catalyzed by rRNA, apparently not by protein, the disclosure of the rRNA genes is of particular interest [9, 10].
Small nuclear RNA (snRNA genes) as constituents of spliceosomes: approximately 80 genes encoding of spliceosomal RNAs were found.
Small nucleolar (sno) RNA genes: these ncRNAs have functions in rRNA processing and base modification; 97 known human snoRNA genes were collected in the human genome, 84 of them existing in one copy. One gene encoding the telomerase RNA and three genes encoding the 7SL RNA of the signal recognition particle, but 773 genes related to 7SL RNA (pseudogenes, fragments and paralogs) could be identified [2].
ncRNA genes and ncRNA-derived pseudogenes exhibit a remarkable proliferation. In the human DNA sequence a great number of sequences related to non-coding RNA genes could be identified [2]. Because ncRNA are small and do not have group specific structural characteristics, for example they are not polyadenylated, novel ncRNA genes cannot easily be found by computational gene-finding approaches. Possibly their number is much greater than recognized so far. It is estimated that approximately 20% of the human DNA might be composed of “RNA only” transcribable segments.
REPETITIVE DNA
It is well-known that there is no correlation between the amount of DNA per cell and the complexity of an organism (C-value paradox, the C-value is the total amount of DNA in a haploid genome). For instance, the human genome is 200-fold larger than that of the S. cerevisiae, but 200-fold smaller than the genome of Amoeba dubia. The cause of this mystery was disclosed when it was recognized that the genome of eukaryotes consists to a great extent of repetitive DNA sequences being present in more or less large excess in relation to the protein-coding genes. It is estimated that in the human genome more than 50% of the DNA fall to the share of repetitive sequences, which often were considered uninteresting and designated as “junk”. The underestimation of the repetitive DNA led to many erroneous conclusions. F. Crick classified this DNA as parasitic and selfish [11]. By definition, selfish DNA spreads by making copies of itself within the genome and does not make a specific contribution to the phenotype. Most of that DNA arises from reverse transcription of RNA. Parts of the genome are like a sea of reverse-transcribed DNA with a small number of islands of coding genes [12]. The repeats can be subdivided in five classes. 1. Long and short interspersed repeats derivable from transposons; most of the human repeat sequences are apparently derived from transposable elements (about 45% of the human genome). 2. Inactive retroposed copies of cellular genes referred to as pseudogenes. 3. Simple sequence repeats (SSR) composed either of short repeating units (n = 1-13 bases, “microsatellites”) or of longer repeating units (n = 14-500 bases, “minisatellites”) of (A)n, (AC)n, (AT)n, (AAT)n or (AAC)n (3%). Because the SSR have a high degree of length polymorphism in the human population due to frequent slippage by DNA polymerase, they are very important in human genetic studies. 4. Duplications of larger DNA-segments (1-200 kb), copied from one region of the genome to another one, either inter- or intrachromosomally (5%). 5. Tandemly repeated sequences, for example near the chromosomal centromeres and in the telomeric regions of human chromosomes. In the telomeric regions often a structural polymorphism occurs. This can be observed in the presence or absence of G-protein coupled olfactory receptor segments. The olfactory receptors constitute a large family of about 1000 members occupying nearly 1% of the human genome.
Transposon derived repeats. In mammals the transposable elements can be categorized in four types: 1) long interspersed elements (LINEs); 2) short interspersed elements (SINEs); 3) retrotransposons, and 4) DNA transposons. In humans, LINEs are 5 to 8 kb long and appear in about 850,000 copies. They contain an internal RNA polymerase II promoter and two ORFs encoding a revertase and an endonuclease. These two enzymes combine with their LINE RNA in the cytoplasm and the resulting aggregates move into the nucleus, where the endonuclease makes a nick in the DNA and the revertase uses the nicked DNA for priming reverse transcription of the LINE RNA brought along with it. Because the revertase frequently fails to complete the transcription process many truncated, nonfunctional insertions in the DNA result. This machinery is probably responsible for most reverse transcriptions in the genome, also for the retrotransposition of the non-autonomous SINEs [13, 14]. The SINEs are short, contain 100-300 bp and have a copy number of 1.5*106. They encode no proteins but have an RNA polymerase III promotor in their sequence. One family of the SINEs is derived from the 7SL RNA of the signal recognition particle and includes Alu. This is the only active SINE in the human genome. The structure of Alu is characteristic for a processed pseudogene and is believed to have spread through an RNA mediated reverse transcription process. Alu has a length of 300 bp. It has a single AluI restriction site (named after the restriction enzyme AluI isolated from Arthrobacter lutens) and its around 1 million copies are distributed throughout the human genome. Alu covers at least 10% of the total DNA of a human cell. LINEs are mainly found in AT-rich DNA whereas SINEs, including Alu, occur mainly in GC-rich DNA. The LINEs constitute about 21% and the SINEs 13% of the human DNA. The autonomous retrotransposons (retrovirus-like elements), flanked by long terminal repeats, carry the gag, pol (encoding a protease as well as a revertase, RNase H, and integrase), and parts of the env genes. Their transposition occurs via the known retroviral mechanism with reverse transcription and primed by tRNA. Together with the non-autonomous retrovirus-like elements they form a fraction of 8% in the human genome. The DNA transposons (3% of the human genome) behave similar to bacterial transposons. They encode a transposase that mediates their mobility within the genome through a “cut and paste” mechanism.
As selfish pieces of DNA, the primary force for the expansion of the transposons is not to provide a selective advantage for the host but to secure their ability to create progeny. However, selfish DNA may be responsible for valuable innovations in the host genome, for example by creating new regulatory elements and even generating new genes. In the human genome, 47 genes have been recognized as derivable from DNA transposons [2, 15].
PROTEIN-CODING GENES
On the basis of computational algorithms, which are based on the present knowledge on the general structure of genes and of protein domains but which obviously are far from being perfect, the number of protein-coding genes in the human genome have been calculated. The International Consortium counted 31,780 protein-coding genes and Celera Genomics found 39,114 (Table 1) [2, 3]. It should be emphasized that these numbers are rather preliminary and much lower than previous estimates, which amounted to 100,000-140,000 genes. On the other hand, the human proteome might contain up to 250,000 different proteins. Possibly, humans use genes in a more flexible way than other organisms and it will need years before a final answer for the exact number of protein-coding genes in the human genome can be given.
Table 1. Gene number and gene density in
five sequenced eukaryotic genomes
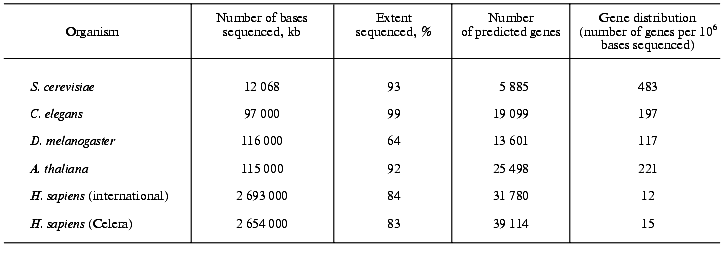
Comparison of the density of protein-coding genes in the genomes of eu- and prokaryotes. The genomes of the yeast S. cerevisiae, the plant Arabidopsis thaliana, the fruit fly Drosophila melanogaster and the worm Caenorhabditis elegans have a much higher percentage of protein-coding genes (up to 80%) than the human genome. The human genome shows only a moderate increase in the number of protein-coding genes compared with the other four eukaryotes (Table 1). In the yeast genome 483 genes are found per million of bases sequenced, in Arabidopsis thaliana this number amounts to 221, in the fly to 117, in the worm to 197 and in humans to only 12 (public project) or 15 (Celera project), respectively [16]. In comparison, in archaea, prokaryotes and viruses, in which practically the total DNA is coding and the same DNA sequence may encode more than one protein, 900 to 1200 genes can be found per million of bases sequenced.
General characteristics of the human genes. The typical human gene is composed of about 28,000 bases and has approximately eight exons. Its coding sequence consists in average of 1,340 bp and encodes a protein having 447 amino acid residues. The largest gene found in the human genome is that of the muscle protein dystrophin with 2.4*106 bp. The fibrillar protein titin as responsible for the passive elastic properties of the skeletal muscle is composed of 27,000 amino acids. Its gene contains, according to Celera, 234 exons [3]. This is the largest number of exons found in a human protein-coding gene.
The structure and the arrangement of the human genes are much more complicated than the genes of other sequenced eukaryotes. Very often they are interrupted by large introns, and some genes might be transcribed with different reading frames. According to rough and very preliminary estimations about 35% of the human genes might be read in different frames and 40% might be alternatively spliced. Hence, one DNA-sequence could probably produce more than one mRNA species.
The extent of genetic polymorphism. From the beginnings of the Human Genome Project the investigators were captured by the problem that no two individuals (except identical twins) in the human population are genetically the same [17, 18]. This view originates in the fact that the sequence variations between individuals in terms of single base exchanges, designated as single nucleotide polymorphisms (SNPs) serve as important markers of genes in genetic analysis. In the human DNA-sequence 1.42 million SNPs were found and their positions in the genome were precisely identified [18]. Their density is one SNP per 1.91 kb. Sixty thousand SNPs have been found within genes. This means that the SNP density should be higher in gene-containing regions than in the DNA repeats. When occurring in exons they are called “coding SNPs”. Nearly every human gene is marked by a sequence variation. The human sex chromosomes have the lowest rate in variation and X chromosomes have much less variations than Y chromosomes. Hence, sex chromosomes are less variable than non-sex chromosomes. There are genomic regions with lower and others with higher variations than the average. For example, the MHC-regions coding for proteins that present processed antigens to immune cells, show a high degree of variation.
The biological significance of genetic polymorphism is evident. Apo E4 polymorphism leads to an increase in the senile plaque density as being characteristic for Alzheimer's disease. A deletion of base pair 32 in the chemokine receptor gene CCR5 leads to resistance to HIV infection, because HIV does not only need the CD4-receptor for binding to the T4 lymphocytes but requires also the CCR5 as coreceptor for binding and uptake by the cells. This double signal is necessary for HIV infectivity.
The human SNP map covers the entire human genome and is essential for elucidating the contributions of individual genes to diseases with a complex and multigenic background. By comparing the patterns and the frequencies of SNPs in patients and healthy individuals, SNPs associated with a certain disease can be directly identified. Research into these relations will stimulate molecular medicine and will change profoundly many areas in clinical and theoretical medicine. It can be anticipated that in the future human genome research will be focussed towards the further exploration of human genetic polymorphism.
FROM GENOME TO PROTEOME
The term “proteome” was coined in 1994 as linguistic equivalent to the concept of the genome. It describes the complete set of proteins expressed by the genome in the lifetime of a cell. Proteomics may be considered the most important “post-genomic” approach to understand the functions of genes [19]. Indeed, the most practical applications of proteomics may be expected in medicine, in particular in the identification of protein markers of diverse diseases as new diagnostic tools and for the development of new drugs. The use of a genome-based technology raises new possibilities in the identification of the genomic effects of drugs and in the study of drug-genome interactions [2]. Recent activities give rise to the development of a new field of research and application called “pharmacogenomics”.
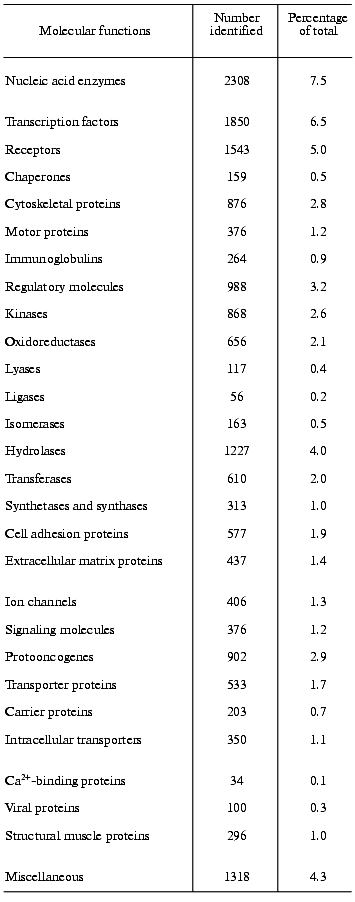
One of the main preconditions of the functional analysis of the human genome will be the knowledge of the complete human proteome. Frequently, valuable indication about the function of a gene comes from sequence similarity with a gene-product of known function in another organism. However, initial studies of that kind must necessarily be rather preliminary and must be completed by systematic analysis of the human proteome to determine the actual functions of the gene-products in humans. An example should explain the problem [2]. In yeast, 35 proteins are known to be involved in the vacuolar protein-sorting machinery. In the human DNA sequence, 34 genes can be found encoding homologs of these proteins. Nine genes of them clearly encode human orthologs (i.e., homologs in different species that have the same function). In twelve genes matches to a family of human paralogs occur (i.e., human homologs that diverge after speciation and have different functions), and in 13 cases matches to specific protein domains can be found. A comparison of the human proteome with the human genome must presently still be very preliminary, mainly because of two reasons, namely, because of the incompleteness of the human DNA sequence and because of our incomplete knowledge about the protein structure. The assignment of function from sequence information alone must be considered with great caution [20]. Nevertheless, from the research into the human DNA sequence many important insights can be gained into the mechanisms generating functional diversity, creating protein domains, enlarging protein and domain families, developing new protein architectures, and understanding horizontal transfer of genes. In an initial and provisional analysis based on protein families as listed in publicly available databases and on protein domains Celera functionally classified about 26,383 of the protein-coding genes, representing about 60% of the total amount of protein-coding genes found in the Celera Genome Project. However, approximately 40% of the genes and their protein products remained functionally unclassified by this initial analysis [3]. Approaches like these do not yield unequivocal results, because, as outlined above, it is presently still very difficult to coordinate sequence information to functional properties, despite the substantial knowledge we have about homologous, analogous, orthologous, and paralogous proteins. Table 2 presents the results of such a distribution analysis for the molecular functions of selected protein families [3].
Table 2. The functions of selected human
gene products. According to Celera Genomics the human genome harbors
39,192 protein-coding genes; out of these, the molecular functions of
12,809 (41.7%) is not known [3]
Did sequencing of the human genome reveal new oncogenes? Hitherto, 30 recessive oncogenes and more than 100 dominant oncogenes have been identified. By searching the human genome for paralogs no new oncogenes have been found [21]. The lack of novel paralogous oncogenes might mean that most of them have already been found by conventional analysis. This underlines the high medical significance of that gene family. However, presently it cannot be not excluded that additional paralogs are buried in hitherto unsequenced regions of the genome or that they remained undetected because of insufficiencies in the identification of the respective genes.
Identification of human disease genes. An important precondition in the identification of human disease genes is the functional classification of these genes and their gene products. For classification a list of 923 disease genes, causing monogenic diseases and genes that increase the susceptibility for complex traits, was prepared [22]. Genes of the mitochondrial genome were not included. Each gene was categorized according to the function of the gene product in terms of pathology and clinical presentation (onset, mode of inheritance, frequency, severity, tissue involvement and association with malformations). The largest functional category comprises genes encoding enzymes (31% of the total), followed by modulators of protein function, which include protein activators and stabilizers, folding helpers and related proteins (14%). Each of another twelve categories (receptors, transcription factors, matrix proteins, transmembrane transporters, etc.) include less than 10% of the total set of disease genes. The correlation between the function of a gene product and the age of onset of its associated disease was analyzed. Diseases associated with genes encoding enzymes dominate at any stage of life, whereas those diseases associated with genes encoding transcription factors are over-represented in the group with onset in utero. This observation reflects the importance of transcription factors in tuning the processes of differentiation and development.
Human genes shared with yeast, worm, and fly. The human genome shares 61% of its genes with the genome of the fly, 43% with that of the worm, and 46% with that of yeast [2]. In the genomes of the latter three species 1,308 groups of proteins could be identified that contain at least one predicted ortholog of them in each species. Many were found to contain additional paralogs. These groups contain 3,129 human proteins, 1,445 fly proteins, 1,503 worm proteins, and 1,441 yeast proteins. One may consider these groups as representing a core of genes encoding enzymes and functional proteins that are mostly responsible for basic “housekeeping functions” of a cell, including basic metabolism, DNA replication and repair, as well as protein biosynthesis.
Horizontal gene transfer. In the human genome 223 genes have been found the products of which having significant similarities to proteins from bacteria, but without any similarity to proteins from yeast, worm, fly, and plant or to other non-vertebrate eukaryotes [2]. At least 113 of these genes are widespread among bacteria and are present only in vertebrates. In this group of bacterial and vertebrate genes orthologous enzymes and functional proteins are found like formiminotransferase, Na+/glucose cotransporter, aldose 1-epimerase, monoamine oxidase (MAO), ADP-ribosylglycohydrolase and thymidine phosphorylase/(platelet-derived growth factor (PDGF), acting as endothelium cell growth factor) (this is a “moonlighting protein” with more than one function). Principally, this finding may have two reasons. Either, the genes encoding these proteins were present in early prokaryotes and eukaryotes, but were lost in the lineages of yeast, worm, fly, and plant, possibly, also in other non-vertebrate eukaryote lineages or, more probable, these genes invaded the vertebrate lineage by horizontal transfer from bacteria. The introns, which can be found in many of these genes, were acquired presumably after the transfer event.
GENE EXPANSION IN THE HUMAN GENOME IN COMPARISON WITH THE OTHER
EUKARYOTIC GENOMES
Table 3 presents the functions of seven groups of genes expanded in the human genome in comparison with the genomes of Drosophila melanogaster and Caenorhabditis elegans [2, 3].
1. Defense and immunity. An important characteristic of the human genome is the existence of a great number of genes involved in acquired immunity as a significant achievement of vertebrates [3]. In the human genome, 22 MHC I and the same number of MHC II genes, 114 other immunoglobulin genes, and 59 genes of related immunoglobulin receptors could be classified. In addition, many cytokines and chemokines as well as components of their intracellular signal transduction pathways (STAT proteins as signal transducers, transcription activators, and suppressors of cytokine signaling) were found. In contrast, many protein domains, playing a role in the innate immune system, like Toll receptors, are not significantly expanded in the human genome in comparison with the two animals. A search in the human genome sequence for members of tumor necrosis factor alpha receptor family (TNFalphaR), which controls proliferation and apoptosis in lymphocytes identified 21 of 22 known TNFR-members. In addition, the genes of 21 proteins exhibiting homology to the B7 family of costimulatory proteins (CD80, CD86, ICOSL, B7H-1, SIRPs, etc.) and the genes of many hemopoietic cytokines were found [23].
Table 3. Gene expansion in the human genome
compared with the genomes of Drosophila melanogaster,
Caenorhabditis elegans, Arabidopsis thaliana, and
Saccharomyces cerevisiae [2, 3]

2. Development, structure, and function of the central nervous system; proteins involved in the cytoskeleton and in vesicle trafficking. A significant characteristic of the human genome is the expansion of protein families involved in neural development, such as neurotrophic factors, nerve growth factor, signaling molecules, myelin proteins, voltage-gated ion channels, and synaptic proteins. The cell adhesion mediating connexin domain-containing proteins forming intercellular channels can only be found in humans and not in the worm and the fly [3]. From the cytoskeletal systems of humans and higher vertebrates, six mammalian actins, 70 families of actin-binding proteins, six alpha- and beta-tubulins, about twelve microtubule-binding proteins, and more than 30 human intermediate filament proteins could be distinguished by traditional biochemical and genetic methods [24]. In its present state the human genome sequence confirmed most of these genes and disclosed at least fourteen new genes, seven divergent actin genes and seven new arp (“actin related proteins”) genes. Presently, it is unknown whether these genes are functional. Forty myosin genes and 40 kinesin genes are already known from conventional cloning. The genes of nearly all of these huge and multi-domain motor proteins have been found in the human DNA sequence together with several additional related genes, but many of them as gene fragments.
Furthermore, the genes encoding proteins involved in vesicle trafficking were compared between S. cerevisiae, D. melanogaster, C. elegans, and Homo sapiens [25]. In the human genome 53 genes encoding the coat multi-subunit protein complexes were identified as well as 60 genes encoding Rab proteins, which belong to the superfamily of small GTPases. This is twice as many as in the genomes of the fly and of the worm and five times more than in the yeast genome. Thirty-five genes encoding SNARE-proteins (also more than in the genomes of the other three species; SNARE abbreviated from “soluble N-ethylmaleinimide sensitive fusion protein attachment protein”) and seven genes encoding Sec1-proteins (Sec abbreviated from secretion, the Sec proteins belong to the Rab family) were found in the human genome. The Rab-family and to a lesser extent also the SNARE-family expand from yeast to man. This implies that multicellular organisms possibly exert more regulation over vesicle-trafficking pathways than unicellular ones.
3. Inter- and intracellular signaling pathways in development and homeostasis. In relation to invertebrates many protein families expanded in humans that are involved in the processes of development and differentiation. The expanded gene families include growth factors (TGFbeta, fibroblast growth factor, nerve growth factor, PDGF), hormones, receptors, intracellular signaling molecules, and transcription factors [3].
The human genome sequence also teaches something about drug addiction. The human genome was searched for genes with functions in the desensitization of receptors and involved in mediation of abusive drugs. Depending on the algorithms applied several hundred genes encoding G-protein receptor kinases, approximately five genes encoding arrestins, and 20 to 30 genes attributable to RGS-proteins could be identified [26].
4. Transcription and translation. In respect to the transcription factors and functional proteins involved in pre-mRNA splicing and polyadenylation, considerable but selective differences could be found between the human genome and that of Drosophila and Caenorhabditis [3]. In the group of nearly 2000 human genes for transcription factors and transcription activators, 900 members of the C2H2 zinc finger protein family were identified. This is more than twice as many as in the genome of the fly and approximately ten to 50 times more than in the genomes of the worm and of yeast. The human genome contains more than 200 homeo box genes, twice as many as the fly and three to ten times more than the worm and yeast, respectively [27]. In terms of translation, 28 different ribosomal subunits have been identified. In comparison with the worm and the fly, the genes of the ribosomal proteins are expanded in the human genome by approximately one order of magnitude [3]. A few of them have been shown to exert “moonlighting functions”, for example, by having actions in inducing apoptosis [28]. In the human genome the eEF1A family is expanded to 56 genes. Many of them are pseudogenes and/or intronless paralogs possibly generated by retrotransposition [29].
5. Hemostasis. In the human genome many groups of genes expanded that are known to regulate the coagulation pathway as well as the interactions between the vascular epithelium and blood platelets [3]. A significant expansion of extracellular adhesion domains in comparison to the fly and the worm can be observed mediating the surface interactions between hematopoietic cells and the vascular matrix. Interestingly, there is apparently no significant increase in the serine protease genes, but a significant expansion in that of matrix metalloproteinases.
6. Apoptosis. In comparison to the worm and the fly, the human genome exhibits an increase in the adapter and effector protein domains implicated in the apoptotic pathway as well as an expansion in the caspase and calpain families producing the apoptotic protease cascade [3].
7. Expansion of the glyceraldehyde-3-phosphate dehydrogenase. In the human genome not less than 46 genes for glyceraldehyde-3-phosphate dehydrogenase can be identified (though some of them might be retrotransposed pseudogenes) [3]. This is in contrast to the fly and the worm, in which only 3 or 4 genes, respectively, of that enzyme are found. In addition to its function in basic metabolism, the enzyme has previously been shown to be a “moonlighting protein”, i.e., to have also other functions than catalyzing the well-known oxidative formation of 1,3-bisphosphoglycerate from glyceraldehyde 3-phosphate in glycolysis. It has a second enzymatic function by acting as a uracil DNA glycosylase [30]. In addition, the protein operates as a cell cycle regulator [31] and is in involved in apoptosis [32].
CONCLUDING REMARKS AND OUTLOOK
The DNA sequence of the human genome points to an increase in complexity in the evolutionary process from yeast to humans. However, the human genome shows only a moderate increase in the number of protein-coding genes compared with other eukaryotes. The human genome encodes five times more genes than Saccharomyces cerevisiae and approximately twice that of the worm Caenorhabditis elegans, the fruit fly Drosophila melanogaster, and the plant Arabidopsis thaliana, respectively. The higher complexity of the human proteome compared with the other eukaryotes is due to large-scale protein innovation and not simply because of the enlargement of the number of protein-coding genes. H. sapiens did not primarily invent new genes, but made use of existing structural domains by composing novel proteins equipped with novel functions. The human genome encodes more paralogs and multidomain proteins with a greater variety in function and domain architecture than the other eukaryotes. The explanation for the greater phenotypic complexity of vertebrates, particular of human beings, in comparison with non-vertebrates is still rather theoretical and speculative. It must find biological background and convincing biochemical mechanisms. It may be hypothesized that the explanation might lie in the combinatorial amplification of the moderate differences by a complexity of mechanisms that include alternative splicing, post-translational modification, and non-linear cellular regulatory networks.
REFERENCES
1.Brenner, S. (2000) Trends Biochem. Sci.,
25, 584.
2.International Human Genome Sequencing
Consortium: Initial Sequencing and Analysis of the Human Genome
(2001) Nature, 409, 860-921.
3.Venter, J. C., et al. (2001) Science,
291, 1304-1351.
4.Goffeau, A., et al. (1996) Science,
274, 546-567.
5.The C. elegans Sequencing Consortium: Genome
Sequence of the Nematode C. elegans: A Platform for Investigating
Biology (1998) Science, 282, 2012-2022.
6.Adams, M. D., et al. (2000) Science,
287, 2185-2195.
7.The Arabidopsis Genome Initiative: Analysis of
the Genome Sequence of the Flowering Plant Arabidopsis thaliana
(2000) Nature, 408, 796-815.
8.Bird, A. P. (1987) Trends Genet., 3,
342-347.
9.Ban, N., Nissen, P., Hansen, J., Moore, P. B., and
Steitz, T. A. (2000) Science, 289, 905-920.
10.Nissen, P., Hansen, J., Ban, N., Moore, P. B.,
and Steitz, T. A. (2000) Science, 289, 920-930.
11.Orgel, L. E., and Crick, F. H. C. (1980)
Nature, 284, 604-607.
12.Baltimore, D. (2001) Nature, 409,
814-816.
13.Okada, N., Hamada, M., Ogiwara, I., and Ohshima,
K. (1997) Gene, 205, 229-243.
14.Esnault, C., Maestre, J., and Heidmann, T. (2000)
Nature Genet., 24, 363-367.
15.Jurka, J., and Kapitonov, V. V. (1999)
Genetica, 107, 239-248.
16.Bork, P., and Copley, R. (2001) Nature,
409, 818-820.
17.Chakravarti, A. (2001) Nature, 409,
822-823.
18.The International SNP Map Working Group
(2001) Nature, 409, 928-933.
19.Abbott, A. (1999) Nature, 402,
715-720.
20.Gerlt, J. A., and Babitt, P. C. (2000)
http://genomebiology.com/2000/I/5/reviews/0005.I
21.Futreal, P. A., Kasprzyk, A., Birney, E.,
Mullikin, J. C., Wooster, R., and Stratton, M. R. (2001) Nature,
409, 850-852.
22.Jimenez-Sanchez, G., Childs, B., and Valle, D.
(2001) Nature, 409, 853-855.
23.Fahrer, A. M., Bazan, J. F., Papathanasiou, P.,
Nelms, K. A., and Goodnow, C. (2001) Nature, 409,
836-838.
24.Pollard, T. D. (2001) Nature, 409,
842-843.
25.Bock, J. B., Matern, H. T., Peden, A. A., and
Scheller, R. H. (2001) Nature, 409, 839-841.
26.Nestler, E. J., and Landsman, D. (2001)
Nature, 409, 834-835.
27.Tupler, R., Perini, G., and Green, M. R. (2001)
Nature, 409, 832-833.
28.Chen, F. W., and Ioannou, Y. A. (1999) Int.
Rev. Immunol., 18, 429-448.
29.Madsen, H. O., Poulsen, K., Dahl, O., Clark, B.
F., and Hjorth, J. P. (1990) Nucleic Acids Res., 18,
1513-1516.
30.Meyer-Siegler, K., et al. (1991) Proc. Natl.
Acad. Sci. USA, 88, 8460-8464.
31.Mansur, N. R., Meyer-Siegler, K., Wurzer, J. C.,
and Sirover, M. A. (1993) Nucleic Acids Res., 21,
993-998.
32.Tatton, N. A. (2000) Exp. Neurol.,
166, 29-43.