Antioxidant Systems in Tissues of Senescence Accelerated Mice
A. A. Boldyrev1,2*, M. O. Yuneva1, E. V. Sorokina1, G. G. Kramarenko2, T. N. Fedorova2, G. G. Konovalova3, and V. Z. Lankin3
1Department of Biochemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-1398; E-mail: aa_boldyrev@mail.ru2Institute of Neurology, Russian Academy of Medical Sciences, Volokolamskoe Shosse 80, Moscow, 123367 Russia; fax: (095) 490-2408
3Cardiology Research Center, 3-ya Cherepkovskaya ul. 15a, Moscow, 121552 Russia; fax: (095) 414-6727; E-mail: lankin@cardio.ru
* To whom correspondence should be addressed.
Received April 29, 2001
Significant decrease in the level of lipid antioxidants (measured from the kinetics of the induced chemiluminescence in brain homogenate) and of the hydrophilic antioxidant carnosine as well was observed in the brain of 14-16-month-old mice of SAMP1 line, which is characterized by accelerated accumulation of senile features, in comparison with the control line SAMR1. In the brain of SAMP1 animals the activity of cytosolic Cu/Zn-containing superoxide dismutase (SOD) was reduced, while the activity of membrane-bound Mn-SOD was at an extremely low level. The activity of glutathione-dependent enzymes (glutathione peroxidase, glutathione reductase, and glutathione transferase) did not differ in the brain of SAMP1 and SAMR1 animals, and catalase activity was similarly low in both cases. At the same time, excess concentration of excitotoxic compounds, significantly exceeding that for the control line, was determined in the brain and blood of SAMP1 animals. The activity of glutathione enzymes in liver and heart as well as the activity of cytosolic Cu/Zn-SOD in liver did not differ in the two studied lines, while the activity of erythrocyte glutathione peroxidase was slightly increased, and the activity of liver catalase and erythrocyte Cu/Zn-SOD was significantly decreased for SAMP1 compared with SAMR1. The results demonstrate that the accelerated ageing of SAMP1 animals is connected to a significant extent with the decreased efficiency of the systems utilizing reactive oxygen species (ROS) in tissues.
KEY WORDS: senescence accelerated mice, brain, antioxidant system
Senescence Accelerated Mice (line SAMP1, Prone) were selected by brother/sister mating of AKR/J mice and are characterized by rapid accumulation of senile features and shortened life span (1-1.5 years compared to 2-2.5 years in control animals) [1]. Until the age of 4 months, SAMP1 animals do not differ from control (SAMR1, Senescence Accelerated Mice, Resistant) either behaviorally or morphologically, but later they start rapidly accumulating senile changes. This results in disordering of their behavioral reactions and cognitive brain functions (orientation ability and learning capacity are particularly decreased) and appearance of age specific morphological changes (lordokyphosis, loss of hair, frequent eye cataract, and mucosal inflammation) [2].
Brain synaptosomes of 10-12-month-old animals of SAMP1 line are characterized by several times higher efficiency of NMDA binding by glutamate receptors, and mitochondrial fraction is characterized by twofold elevated monoamine oxidase B activity compared to the control line (SAMR1) [3]. The activation of these systems can cause increased generation of reactive oxygen species (ROS) in SAMP1 brain. At the same age, more intensive accumulation of end products of lipid peroxidation [4-6], including lipid hydroperoxides [7] in tissues as well as increased production of ROS accompanied by increased frequency of chromosomal aberrations in bone stem cells was noted for SAMP1 animals [8]. Increased oxidative damage of DNA in liver, brain, muscles, lungs, and mucosal brush border was also demonstrated [9].
The data indirectly suggest that disbalance between the rates of generation and utilization of ROS and organic free radicals can be one of the main reasons for accelerated aging of SAMP1 animals. Nevertheless, exhaustive analysis of the antioxidant defense system in SAMP1 tissues has not been done. Thus, we decided to comparatively study both the activity of antioxidant enzymes and the content of several natural nonenzymatic antioxidants in tissues of SAMP1 and SAMR1 animals.
MATERIALS AND METHODS
Animals. For the experiments, male mice of SAMP1 and SAMR1 lines were used at 14-16-months of age, i.e., at the age when distinct differences in behavior, exterior appearance, and some biochemical parameters are noted between the lines. Brain, liver, heart muscle, erythrocytes, and blood plasma were used for biochemical studies.
Treatment of samples. To determine the activity of antioxidant enzymes, tissue homogenates were centrifuged at 10,000g for 15 min (4°C) to sediment mitochondria. The supernatant was re-centrifuged at 100,000g for 1 h. The resulting supernatant was used for measuring the activity of cytosolic enzymes. To measure SOD, erythrocytes were subjected to hypoosmotic treatment in cool 5 mM K,Na-phosphate buffer (pH 7.4), and heme-containing proteins were precipitated by extraction with chloroform-ethanol (3 : 5 v/v).
All spectrophotometric measurements were done using a Hitachi 557 (Japan) spectrophotometer equipped with an automatic recording unit. Enzyme activity was measured in kinetic regime determining the initial rate of enzyme reaction. The period corresponding to linear dependence of this value on the amount of protein added was determined preliminarily for each enzyme studied. The activities of the enzymes studied are expressed as units per mg protein determined by the Lowry procedure. Erythrocyte SOD is expressed as units per mg hemoglobin (Hb) measured in lysates by the hemocyanin method.
Glutathione peroxidase activity was determined at 340 nm in reaction medium containing 50 mM K,Na-phosphate buffer (pH 7.4 at 30°C), 1 mM EDTA, 0.12 mM NADPH, 0.85 mM glutathione reduced (GSH), 0.5 unit/ml yeast glutathione reductase, and 0.2 mM tert-butyl hydroperoxide as a substrate [10]. The amount of the enzyme converting 1 µmol GSH per min was taken as 1 activity unit.
Glutathione-S-transferase activity was measured at 340 nm in 100 mM K,Na-phosphate buffer (pH 7.0 at 30°C) in the presence of 1 mM EDTA, 1 mM GSH, and 1 mM 1-chloro-2,4-dinitrobenzene [11]. The amount of enzyme conjugating 1 µmol GSH per min was taken as 1 unit of activity.
Glutathione reductase activity was measured at 340 nm in medium containing 50 mM K,Na-phosphate buffer (pH 7.4 at 25°C), 1 mM EDTA, 0.16 mM NADPH, and 0.8 mM glutathione oxidized (GSSG) [12]. The amount of the enzyme reducing 1 µmol GSSG per min was taken as 1 activity unit.
Catalase activity was determined at 240 nm by measuring the rate of H2O2 utilization as described elsewhere [13] with the molar extinction coefficient for H2O2 being 43.6 M-1*cm-1. The amount of the enzyme utilizing 1 µmol H2O2 per min was taken as 1 activity unit.
Determination of superoxide dismutase activity. SOD activity was measured at 560 nm as the rate of suppression of reduction of nitrotetrazolium blue when superoxide anion radical was generated during oxidation of xanthine by xanthine oxidase. The reaction mixture contained 50 mM sodium carbonate dissolved in 50 mM K,Na-phosphate buffer (pH 7.8 at 25°C), 0.1 mM EDTA, 0.1 mM xanthine, 0.025 mM nitrotetrazolium blue. For 1 unit of activity, the amount of protein was taken which provided 50% inhibition of nitrotetrazolium blue reduction under standard conditions. Membrane-bound Mn-containing SOD was determined as the difference between total SOD activity and that of Cu/Zn-enzyme determined after treatment of the sample with 2% SDS [14]. Cu/Zn-SOD was measured in cytosolic fraction of brain and liver and in lysates obtained from erythrocytes.
Amount of non-protein SH-groups was analyzed in brain homogenates prepared in the presence of 20 mM EDTA (pH 4.7). After addition of 5% trichloroacetic acid to samples, protein was sedimented by centrifugation at 3000g for 10 min. Supernatant aliquots were mixed with 200 mM Tris-HCl (pH 8.9) in the ratio 1 : 3 and 4 volumes of methanol containing 0.125 mM 5,5´-dithiobis-2-nitrobenzoic acid. Sample absorbance was measured at 412 nm [15]. Under the conditions described, tissue glutathione (GSH) was generally determined, so the molar extinction coefficient for GSH of 13,700 M-1*cm-1 was used to calculate the SH-group content. The values obtained were ascribed to the amount of glutathione in the brain samples.
Extractable compounds in blood plasma and brain homogenates of the mice were determined by reversed phase hydrophobic high pressure chromatography using o-phthalic aldehyde to label the amino acids [16]. Measurements were done on an Altex 334 chromatograph (USA) with a Schoeffel GM970 fluorescent detecting unit (excitation wavelength 340 nm, fluorescence detection at 455 nm) using a 250 × 4.6 mm column (Serva, Germany) loaded with Silica S 1100-Polyol-RP8 as the sorbent. A mixture of 100 mM acetate buffer (pH 5.8) and methanol (70 : 30 v/v) was used as the mobile phase.
Oxidizability of brain membranes was characterized by the amplitude of initial fast flash and duration of lag-period of Fe2+-induced chemiluminescence as described earlier [17]. The measurements were done using an LKB 1251 chemiluminometer (Sweden).
Reagents. EDTA, NADPH, GSH, GSSG, nitrotetrazolium blue, 1-chloro-2,4-dinitrobenzene, 5,5´-dithiobis-2-nitrobenzoic acid, and glutathione reductase were from Sigma (USA); tert-butyl hydroperoxide and H2O2 were from Merck (Germany); xanthine oxidase was from Serva.
RESULTS AND DISCUSSION
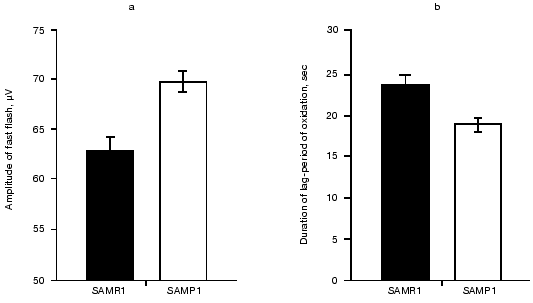
To estimate the resistance of cellular structures of brain toward oxidation, we measured the kinetics of Fe2+-induced chemiluminescence of brain homogenates from mice of the two lines. It was found (see figure) that addition of 5 mM FeSO4 to the samples induced significantly higher fast chemiluminescence flash in the case of SAMP1 brain compared to that of SAMR1. Also, the duration of the lag-period of the Fe2+-induced chemiluminescence was 27% less for SAMP1 than for SAMR1. Because the initial burst of chemiluminescence correlates with the content of alkoxy-radicals formed during hemolysis, this parameter is taken to be proportional to the initial amount of lipid hydroperoxides, whereas the duration of the lag-period of chemiluminescence characterizes the stability of the sample towards oxidation by generated hydroxyl radicals. The higher level of hydroperoxides in combination with a shorter lag-period of oxidation indicates significant decrease in the level of hydrophobic antioxidants in SAMP1 brain.
As Packer noted [18], decrease in the tissue level of hydrophobic antioxidants takes place at the latest step of oxidative stress and is preceded by prior decrease in hydrophilic antioxidant content. To estimate the participation of the latter in stabilization of the brain against oxidative stress we compared the amount of glutathione, taurine, and carnosine in brain of the studied animals. It is known that GSH participating in enzymatic reduction of membrane hydroperoxy-phospholipids prevents the formation of secondary alkoxy-radicals when organic peroxides are homolyzed [19], whereas two other hydrophilic antioxidants--carnosine and taurine--protect brain from ROS damage at the earliest (the former) and latest (the latter) steps of oxidative stress [20].Amplitude of the fast flash (a, µV) and duration of lag-period (b, sec) calculated after measurements of Fe2+-induced chemiluminescence of membranes prepared from brain of 14-month-old SAMR1 (black bars) and SAMP1 (white bars) animals. Significance of the difference between (a) and (b) corresponds to p < 0.01 and p < 0.05, respectively.
From Table 1 one can see that both GSH and taurine content in SAMP1 and SAMR1 brain did not differ significantly, whereas carnosine level in SAMP1 brain was more than twofold decreased. This may indicate both a specific role of carnosine in brain as an important antioxidant and disordering in the brain antioxidant system in SAMP1 animals.
Table 1. Content of low molecular weight
hydrophilic antioxidants in brain of studied mice

Brain carnosine deficiency may play some role in the syndrome of antioxidant system deficit accompanying the accumulation of senile features in SAMP1 mice and serve as one of the causes of non-compensated action of ROS resulting in accelerated aging. This suggestion is supported by independent data from the literature. Recently, in vitro experiments showed that incubation of neurons with carnosine decreased the production of intracellular ROS when the neurons were exposed to excitotoxic compounds [21] as well as removed the toxic effect of glutamate on the brain neurons [22]. Administration of carnosine in vivo also protected animal brain from ischemic injury [23]. Moreover, using carnosine as food additive for SAMP1 mice was found to increase average life span of the animals and to prevent accumulation of senile features [24].
Activities of glutathione peroxidase and glutathione-S-transferase, both glutathione-dependent enzymes responsible for utilization of H2O2 and lipid hydroperoxides did not statistically differ in SAMP1 and SAMR1 brain (Table 2), which correlates well with the absence of difference in GSH content in their brains (Table 1). Similarly, in SAMP1 and SAMR1 brain no reliable difference in activity of the key enzyme of GSH bioregeneration, glutathione reductases, was found (Table 2). While GSH-dependent enzymes play an important role in preventing cellular damage induced by alkoxyl and hydroxyl radicals [19], the data obtained suggested that these enzymes are not directly involved in oxidative stress of aged SAMP1 brain.
Table 2. Activity of antioxidant enzyme
system (units/mg protein) in brain of SAMR1 and SAMP1 mice
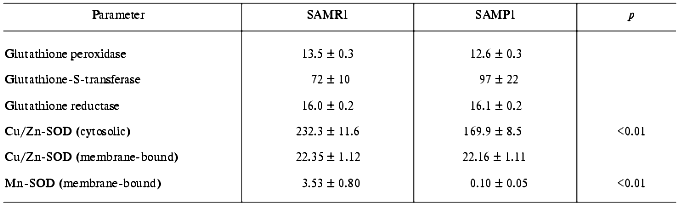
Brain is well known to contain little catalase [25], which we also confirmed for the brain of both SAMP1 and SAMR1 (the data are not presented because of the low level of the enzyme activity). Under such circumstances, thorough study of activity of several isoforms of SOD was of special interest--this enzyme is responsible for superoxide anion detoxication in several cell compartments. We did measure activity of Cu/Zn-SOD located in cell cytosol (soluble isoform) and in peroxisomes (membrane-bound isoform), as well as activity of Mn-SOD belonging to mitochondria. In comparing SAMP1 and SAMR1, we found that only membrane-bound Cu/Zn-SOD showed no significant difference in activity, while both cytosolic (Cu,Zn-containing) and membrane-bound (Mn-containing) SOD isoforms in brain of mice with accelerated aging were significantly lower than in the control. Actually, the Mn-isoform of SOD in SAMP1 brain was nearly absent (Table 2). The results suggest that the deficit in these two SOD isoforms can be the cause of ROS accumulation in SAMP1 brain being responsible for brain dysfunction in aged SAMP1 animals.
Deficit in SOD activity in brain and liver of SAMP1 mice was noted earlier. However, the authors did measure total SOD activity [4]. Similar data on SOD activity in rats are also known [26]. The separate analysis of different isoforms of the enzyme that was made in our study showed that decrease in total activity is explained by suppression not of the peroxisomal membrane-bound SOD but that of cytosolic and mitochondrial isoforms perhaps being the most vulnerable for ROS. Actually, SOD is an enzyme that is significantly damaged during hyper-production of ROS. Recently, in in vitro experiments continuous generation of hydroxyl radicals was found to result in oxidative modification of SOD, its fragmentation, and consequent loss of activity [27]. It is important to note that in these experiments carnosine was found to be among other factors preventing both fragmentation and decrease enzyme activity. Thus, the deficit of SOD accompanied by twofold decrease in carnosine content (Table 1) in SAMP1 brain could be related.
It is important to note that decrease in efficiency of the antioxidant system in SAMP1 brain appeared simultaneously with increased amount of excitotoxic amino acids--aspartate, glutamate, asparagine, and gamma-aminobutyrate (GABA) (Table 3). These mediators performing both excitation (glutamate, aspartate) and suppression (GABA) functions, under over-loading conditions, can stimulate ROS production [28, 29], inducing conditions for oxidative stress in neurons (thus, they are usually named excitotoxic mediators). Oxidative stress, in turn, can result in increased activity of NMDA receptors and activation of such ROS producing enzymes as monoamine oxidase B, which, in fact, was characteristic of aged brain of SAMP1 animals [4, 5].
Table 3. Content of excitotoxic compounds in
brain and blood plasma of SAM animals
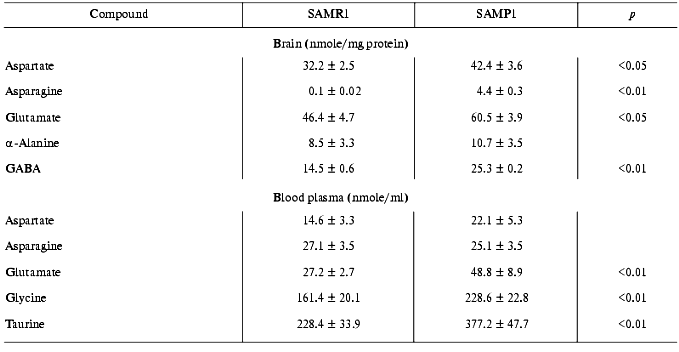
Increased basal level of several excitotoxic compounds was detected also in blood plasma of SAMP1 animals (Table 3). It is unlikely that this is a cause of their release from brain into the blood stream because of both the existence of the blood-brain barrier for these compounds and different ratios for them in brain and blood. It is more probable to suggest that increased level of aspartate, glutamate, glycine, and taurine in SAMP1 blood reflects the specificity of their tissue metabolism being among multiple reasons characterizing specificity of metabolic pathway of mice with accelerated aging under intensifying ROS production.
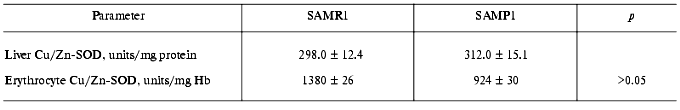
In subsequent experiments the activity of several antioxidant defense enzymes was measured in liver, heart, and erythrocytes. In these tissues similar features of antioxidant deficit were demonstrated. Thus, in SAMP1 liver significantly (nearly twofold) lower catalase activity was found--88.4 ± 4.4 versus 137.7 ± 6.9 units/mg protein, while activity of all the three GSH dependent enzymes (Table 4) and cytosolic Cu/Zn-SOD (Table 5) did not change. No change in the activities of GSH-dependent enzymes in heart was found between SAMP1 and SAMR1 (Table 4). In SAMP1 erythrocytes, however, glutathione peroxidase was somewhat higher than that in SAMR1, whereas glutathione-S-transferase and glutathione reductase did not differ. In contrast, erythrocyte Cu/Zn-SOD was significantly suppressed in SAMP1 versus SAMR1. Thus, disordering of the functioning of the antioxidant defense system in other tissues of SAMP1 is again indicated in the presence of serious changes in oxidative metabolism on the whole similar to that found in brain. However, the molecular mechanisms of the development of oxidative stress can be different in different tissues. In neuronal tissue, this process can be related to metabolism of excitotoxic compounds regulating ROS production within the neurons as well as with the shift in the functional activity of a number of proteins (for example, with activation of NMDA or monoamine oxidase B) and decreased defense provided by enzymatic and nonenzymatic antioxidants. The reasons for dysfunction of the antioxidant defense in other tissues are studied much less well. From the common point of view, it seems that primary changes in antioxidant enzymes activity are programmed genetically or induced by ROS attack [30]. Actually, deficit of mitochondrial Cu/Zn-SOD [31] appears on the base of pronounced mitochondrial dysfunction conceivably induced by increased level of intracellular ROS, though this parameter has not been measured until now [4]. Under these conditions, however, decrease in SOD activity was found to be related rather to processing of the protein resulting in its destabilization, than to decrease in the level of its mRNA [31, 32].
Table 4. Activity of glutathione-dependent
enzymes in tissues of mice studied (in the case of liver and heart
activity is expressed as units/mg protein, in the case of blood as
units/mg Hb)
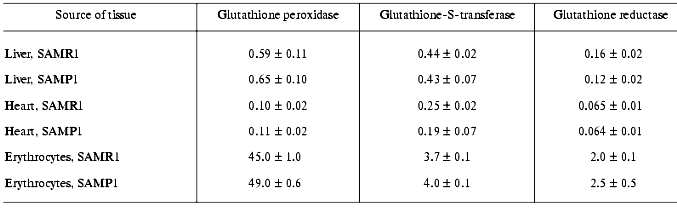
Table 5. Activity of cytosolic Cu/Zn-SOD in
tissues of SAM studied
In all the cases, the most important enzyme responsible for cell tolerance toward oxidative damage is SOD and disordering of its synthesis or processing in a cell can be an immediate reason for very early changes in ROS balance resulting in systemic shift in SAMP1 tissues and in accelerated accumulation of senescent features. Nevertheless, it is important to stress that this process is induced by many reasons. Among them decreased level of carnosine, accumulation of excitotoxic mediators, and oxidative modification of a number of cellular proteins are especially important for excitable tissues.
This work was supported by the Russian Foundation for Basic Research (grants No. 99-04-49420 and No. 00-04-48767).
REFERENCES
1.Takeda, T. (ed.) (1994) The SAM Model of
Senescence, Excerpta Medica, Amsterdam.
2.Hosokawa, M., Abe, T., Higuchi, K., Shimakawa, K.,
Omori, Y., Matsushita, T., Kogishi, K., Deguchi, E., Kishimoto, Y.,
Yasuokama, K., and Takeda, T. (1997) Exp. Gerontol., 32,
111-117.
3.Bulygina, E., Gallant, S., Kramarenko, G.,
Stvolinsky, S., Yuneva, M., and Boldyrev, A. (1999) J. Anti-Aging
Med., 2, 45-49.
4.Mori, A., Utsumi, K., Liu, J., and Hosokawa, M.
(1998) Ann. N. Y. Acad. Sci., 854, 239-250.
5.Yuneva, M. O., Bulygina, E. R., Stvolinsky, S. L.,
Kramarenko, G. G., Gallant, S., and Boldyrev, A. A. (1998)
Neirokhimiya, 15, 276-279.
6.Nakahara, H., Kanno, T., Inai, Y., Utsumi, K.,
Hiramatsu, M., Mori, A., and Packer, L. (1998) Free Rad. Biol.
Med., 24, 85-92.
7.Matsugo, S., Kitagawa, T., Minami, S., Esashi, Y.,
Oomura, Y., Tokumari, S., Kojo, S., Matsushima, K., and Sasaki, K.
(2000) Neurosci. Lett., 278, 105-108.
8.Yuneva, M. O., Guseva, N. V., and Boldyrev, A. A.
(2000) Uspekhi Gerontol., 4, 147-152.
9.Hosokawa, M., Fujisawa, H., Ax, S., Zahn-Daimler,
G., and Zahn, R. K. (2000) Mech. Ageing Dev., 118,
61-70.
10.Lankin, V. Z., Tikhase, A. K., Kovalevskaya, A.
L., Lemeshko, V. V., and Vikhert, A. M. (1981) Dokl. AN SSSR,
261, 1467-1470.
11.Keen, J. H., and Yakoby, W. B. (1978) J. Biol.
Chem., 253, 5654-5657.
12.Beutler, E., and Yeh, M. K. H. (1963)
Blood, 21, 573.
13.Aebi, H. (1984) Meth. Enzymol.,
105, 121-126.
14.Misra, H., and Fridovich, I. (1972) J.
Biochem., 247, 3170-3175.
15.Sedlak, J., and Lindsay, R. (1968) Analyt.
Biochem., 25, 192-205.
16.Roth, M. (1971) Analyt. Chem., 43,
880.
17.Fedorova, T. N., Boldyrev, A. A., and
Gannushkina, I. V. (1999) Biochemistry (Moscow), 64,
75-79.
18.Packer, L. (1995) in Oxidative Stress and
Aging (Kutler, R. G., Packer, L., Mori, A., and Bertram, J., eds.)
Birkhauser Verlag, Basel-Boston-Berlin, pp. 1-14.
19.Lankin, V. Z., Tikhase, A. K., and Belenkov, Yu.
N. (2000) Kardiologiya, 40, 48-61.
20.Boldyrev, A., Abe, H., Stvolinsky, S., and
Tyulina, O. (1995) Comp. Biochem. Physiol., 112B,
546-548.
21.Boldyrev, A., Song, R., Lawrence, D., and
Carpenter, D. (1999) Neuroscience, 94, 571-577.
22.Trombley, P. Q., Horning, M. S., and Blakemore,
L. J. (2000) Biochemistry (Moscow), 65, 807-816.
23.Stvolinsky, S., Kukley, M., Dobrota, D.,
Mezesova, V., and Boldyrev, A. (2000) Brain Res. Bull.,
53, 445-448.
24.Yuneva, M., Bulygina, E., Gallant, S.,
Kramarenko, G., Stvolinsky, S., Semyonova, M., and Boldyrev, A. (1999)
J. Anti-Aging Med., 2, 337-342.
25.Halliwell, B., and Gutteridge, J. M. C. (1999)
Free Radicals in Biology and Medicine, Oxford University Press,
p. 135.
26.Alper, G., Girgin, F. K., Ozgonul, M., Mentes,
G., and Erzos, B. (1999) Eur. Neuropsychopharmacol., 9,
247-252.
27.Choi, S., Kwon, H., Kwon, O., and Kang, J. (1999)
Biochim. Biophys. Acta, 1472, 651-657.
28.Sureda, F. X., Camins, A., Trullas, R., Camarasa,
J., and Escubedo, E. (1996) Brain Res., 723, 110-114.
29.Boldyrev, A. A. (2000) Byul. Eksp. Biol.
Med., 130, 244-251.
30.Raha, S., and Robinson, B. H. (2000) Trends
Biochem. Sci., 25, 502-508.
31.Park, J. W., Choi, C. H., Kim, M. S., and Chung,
M. H. (1996) J. Gerontol., 51A, B337-B345.
32.Tsay, H. J., Wang, P., Wang, S. L., and Ku, H. H.
(2000) J. Biomed. Sci., 7, 466-474.