REVIEW: X-Ray Crystallography of Bacteriorhodopsin and Its Photointermediates: Insights into the Mechanism of Proton Transport
J. K. Lanyi
Department of Physiology and Biophysics, University of California, Irvine, CA 92697, USA; E-mail: jlanyi@orion.oac.uci.edu
Received March 20, 2001; Revision received May 9, 2001
In the last few years, detailed structural information from high-resolution x-ray diffraction has been added to the already large body of spectroscopic and mutational data on the bacteriorhodopsin proton transport cycle. Although there are still many gaps, it is now possible to reconstruct the main events in the translocation of the proton and how they are coupled to the photoisomerization of the retinal chromophore. Future structural work will concentrate on describing the details of the individual proton transfer steps during the photocycle.
KEY WORDS: bacteriorhodopsin, x-ray diffraction, proton transport cycle, retinal
There is a huge amount of spectroscopic information, collected over many years, to describe the photoreaction cycle of wild type bacteriorhodopsin, as well as site specific mutants modified at critical resides [1-6]. It has resulted in a fairly complete assessment of the pathway of the transported proton across the membrane, and gave rise to many hypotheses for the conversion of the free energy gain in the excited state of the retinal into the movement of a proton and ultimately into a transmembrane electrochemical gradient. However, as with enzymes in general, many of the questions concerning the transformations of the protein in the reaction cycle are structural in nature, and for that reason the answers need to be framed in structural terms also. This short review will discuss insights gained into the transport mechanism from x-ray crystallography of the unilluminated bacteriorhodopsin and some of its photointermediates.
The acidic groups which play proton transfer roles in the transport are located in the interhelical cleft formed by the seven transmembrane helices, and are well known. The photoisomerization of the retinal from all-trans to 13-cis,15-anti drives these transfers during the cyclic reaction called the “photocycle”. Roughly speaking, the photocycle can be described as a linear sequence of the K, L, M, N, and O states [7-9]. Because they are defined by their spectra in the visible, infrared, etc. they are intermediates distinguished by the different states of the retinal and its immediate environment. Some are produced as several substates, the chromophore being the same but with additional changes elsewhere in the protein. The K to L transition reflects partial relaxation of the isomerized but twisted retinal, and many small changes in the protein and bound water that sets the stage for the L to M reaction, in which the retinal Schiff base becomes deprotonated and Asp-85 protonated. The M intermediate exhibits complex rise and decay kinetics, which must originate from several M substates that reflect the fact that numerous important events must occur in the protein before the Schiff base is reprotonated. During the lifetime of M, there will have to be a change in the accessibility of the Schiff base to a proton donor/acceptor from one side to the other. Related to this [10], there will be release of a proton to the extracellular surface, from a so-far undefined site, perhaps a hydrogen-bonded network of water molecules [11], upon protonation of Asp-85. The Schiff base had become deprotonated with a lowered pKa, but in the next step it will be reprotonated, and one would expect that its pKa will rise. The pKa of the proton donor from the cytoplasmic direction, Asp-96, on the other hand, will decrease from its initial value >11. Finally, a pathway for transferring the proton from Asp-96 to the Schiff base must be built.
After the Schiff base is reprotonated during the M to N reaction, Asp-96 becomes reprotonated from the cytoplasmic surface producing a late N substate. This requires a new protein conformation in which Asp-96 is accessible to the surface but no longer to the Schiff base [5, 12]. The retinal may now thermally reisomerize to all-trans, producing the O state. The decay of O, that regenerates the initial state, depends on deprotonation of Asp-85 and reprotonation of the extracellular proton release site [13-15].
STRUCTURE OF THE BR STATE
The latest and highest resolution structures, at 1.55 [16] and 1.9 Å [17], are from x-ray diffraction of crystals grown by the cubic phase method [18, 19]. The P63 space group arises by multiple stacking of the naturally occurring purple membrane sheets with P3 symmetry. This means that the environment of the protein in the crystals is very similar to that in the original membrane. Indeed, the entire complement of the archaeal lipids native to the purple membrane appear to be present in the crystals [16, 17], to the exclusion of detergent and the lipids used for the cubic lipid matrix. The photocycle of bacteriorhodopsin in the crystals is similar to that in membrane sheets [20].
The seven transmembrane helices span the membrane at various angles not far from the membrane normal, as known already from cryo-electron microscopy [21-23]. The cytoplasmic half of the protein projects out of the lipid bilayer by about 5 Å farther than the extracellular half. This, and the greater mobility of the cytoplasmic region, as reflected in the higher temperature factors [16], is consistent with the fact that it is near the cytoplasmic surface where large-scale conformational changes occur in the photocycle [24-27].
The active site comprises the protonated retinal Schiff base, Asp-85, Asp-212, and their hydrogen bonds to water 402, which separates the retinylidene cation and the two aspartate anions [28]. The site is connected through chains of interaction to both extracellular and cytoplasmic regions. It is through these chains that long-range effects occur in the protein during the photocycle. In the extracellular region, an extended 3-dimensional hydrogen-bonded chain, that involves Arg-82, Glu-194, Glu-204, and at least seven water molecules, leads to the protein surface [16, 17]. Mutation of any of these three residues abolishes or strongly inhibits the release of a proton upon protonation of Asp-85. Hydrogen bonds with Thr-89, Tyr-185, Tyr-57, and Ser-193 lend additional stability to the network.
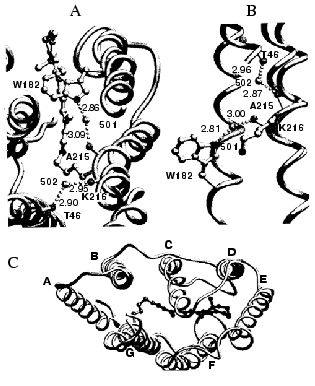
The cytoplasmic region contains no such polar network, but it does contain a movable structural element. The alpha-helical repeat of helix G is interrupted by a pi-bulge at Ala-215 and Lys-216 [16]. It is caused by hydrogen bonds of the two main-chain C=O groups to water molecules 501 and 502, as shown in Fig. 1. This feature offers the possibility of conformational change near the location on helix G where the retinal is bound, through shuttling between a more pi-helical and a more alpha-helical arrangement. Water 501 is further hydrogen bonded to the indole nitrogen of Trp-182, a residue that contacts the retinal polyene chain. Water 502, in turn, is hydrogen bonded to the C=O of Thr-46, whose side-chain is hydrogen bonded to Asp-96. This forms a chain of covalent and hydrogen bonds, with Trp-182, water 501, Ala-215, Lys-216, water 502, Thr-46, and Asp-96 as participants. The chain connects helices C, F, and G together. Breaking and remaking it would offer the possibility of communication between the isomeric state of the retinal and the protonation state of the cytoplasmic proton donor.
Fig. 1. Irregularities in the alpha-helices of bacteriorhodopsin. A and B represent views, along the c-axis and the a-axis, respectively, of the pi-bulge in helix G, and the two water molecules, 501 and 502, that stabilize it. The chain that extends from the vicinity of the retinal to Thr-46 near the cytoplasmic surface is evident. C is a view down the c-axis, to show the three proline-associated kinks, on helix B, C, and F, as well as the pi-bulge on helix G. In B and C, helix G is shown with an additional helix that represents a perfect alpha-helix aligned with the extracellular segment. Coordinates used, 1CW3. Reprinted with permission from [16].
STRUCTURAL CHANGES AT THE RETINAL
In solution, photoisomerization of the retinal from all-trans to 13-cis,15-anti drastically changes the shape of the polyene chain and rotates the Schiff base N-H bond. To a degree that is not yet quite clear, the retinal binding pocket will oppose these changes in geometry. The resolution of the structures for the K [29] and L [30] states determined so far does not reveal the kind of distortion this conflict will produce at the retinal. The low occupancy of these states is also a serious problem. On the other hand, comparison of the structures of the two M states, the “early” M from the E204Q mutant [31] where proton release is blocked, and the “late” M from the D96N mutant [32] where reprotonation of the Schiff base is blocked, indicates progressive relaxation of an initially distorted retinal. In the “early” M the distortion is mainly of an increase of the bond angle at C13 because the polyene chain is still straight like all-trans, and less bent at C13 than in the “late” M. The relaxation therefore consists of C13, and therefore the 13-methyl group, buckling upward. In both M states the angle of the Schiff base nitrogen has inverted so that the N: points toward the cytoplasmic direction. If the small difference observed [31] in this angle in the two M states is significant (and this is not yet clear), the direction of the rotation is clockwise (sweeping past Asp-85).
Because the geometry of the retinal in the L intermediate is not yet known, the mechanism for the deprotonation of the Schiff base is unclear. If the N-H bond in L continues to point toward Asp-85, there will be direct proton transfer to the aspartate. If it points toward the cytoplasmic direction, direct transfer is less likely, and the participation of bound water near this location might need to be invoked. One possibility [33] would be the dissociation of a water molecule, with the H+ moving to Asp-85 and the OH- moving to the Schiff base and receiving its proton. This would be tantamount to hydroxyl ion transport in the opposite direction, analogous to chloride transport by halorhodopsin [2, 34, 35] and the D85T and D85S mutants of bacteriorhodopsin [36, 37].
RELEASE OF A PROTON TO THE EXTRACELLULAR SURFACE
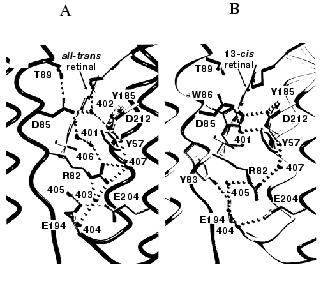
Protonation of Asp-85 induces proton release from a poorly defined site near the extracellular surface, which has been suggested to be Glu-204 [38], the Glu-194/Glu-204 pair [39], Tyr-185 [40], or the water molecules in the hydrogen-bonded network [11]. Lack of a negative C=O stretch band [11], as well as UV-Raman [41] and NMR [42] evidence argue against the first three alternatives. The pKa of Asp-85 and a second proton binding site, that must correspond to the release site, are coupled in the BR state already [10, 43]. Figure 2 compares the structure of the BR state (Asp-85 anionic, proton release site protonated) with the structure of the M state (Asp-85 protonated, proton release site unprotonated). The results for M produced by illumination of the D96N mutant, as shown [32], as well as for M of the wild type and the E204Q mutant [31, 44], strongly suggest that the means for this coupling is the shuttling of the positively charged side-chain of Arg-82 between the “up” position where it is connected to Asp-85 through water 406, and the “down” position where it is connected to the Glu-194/Glu-204 pair through water 405.
The link between the protonation of Asp-85 and the release of a proton to the surface will influence the reversibility of the deprotonation of the Schiff base. Free energy is dissipated at the release of the proton, proportional to the difference between the pH and the pKa for proton release. This will raise the pKa of Asp-85 and thereby block the return of the Schiff base proton. Indeed, double-perturbation experiments indicated [45] that with increasing pH, and with an apparent pKa of 6 (i.e., the pKa for proton release), the protonation equilibrium of the Schiff base in the M state is drawn away from Asp-85. From this observation it appears that shifts of proton affinity during the photocycle play a greater part in the protonation switch than changes in the geometry of the groups involved in the proton transfer. This is in accord with the “local-access” mechanism for the transport switch [46].Fig. 2. Structural changes in the extracellular domain upon formation of the M intermediate. A and B are views of selected features of the BR and M states of the D96N mutant, respectively. The protonation of Asp-85 results in downward movement of the side-chain of Arg-82, and the breaking hydrogen bonds that connect the Schiff base region to the extracellular surface. Coordinates used 1C8R and 1C8S. Reprinted with permission from [32].
REPROTONATION OF THE RETINAL SCHIFF BASE
In the M intermediate produced at 230 K, that should correspond to a state immediately after loss of the Schiff base proton, the O-H stretch band of Thr-89 indicates that its hydrogen bond to Asp-85 is retained [47]. In the M state produced at room temperature, however, this bond is broken [31]. The gradual changes at the retinal cause upward displacement of the 13-methyl group of the retinal after deprotonation of the Schiff base pushes the indole ring of Trp-182 in the cytoplasmic direction [31]. Water 501 loses its hydrogen bond to the C=O of Ala-215 on helix G and forms a new one to Thr-178 on helix F. This breaks the connection of Trp-182 on helix F to helix G through water 501. Partial recovery of an alpha-helical structure at the pi-bulge, displacement of the side-chain of Lys-216, and repacking of the side-chains between helices F and G cause Asp-96 and Thr-46 to move apart [31]. In the M state already, this allows the entry of water into the cytoplasmic region, either from the bulk or from other locations in the protein. Thus, a hydrogen-bonded network of water molecules is assembled that leads from a water that now bridges Asp-96 and Thr-46 in the direction of the Schiff base. Presumably, completion of this network is the rate-limiting step in the reprotonation of the Schiff base in the M to N reaction.
In the D96N mutant, where it could be easily determined [48], the pKa of the Schiff base at this stage of the photocycle is 8, and proton transfer will not occur unless the pKa of Asp-96 is lowered correspondingly. The water molecule intercalated between Asp-96 and Thr-46, and the generally less hydrophobic character of the now well-hydrated cytoplasmic region should suitably decrease the initially very high pKa (>11) [49] of Asp-96.
THE LAST STEPS OF THE PHOTOCYCLE
Return to the initial state requires several steps after the N state: reprotonation of Asp-96 from the cytoplasmic surface, reisomerization of the retinal to all-trans, deprotonation of Asp-85, and conformational recovery [5, 6]. There is no direct structural evidence for how these occur, but there are clues. The properties of a mutant bacteriorhodopsin indicate that in the dark the thermal equilibrium between the 13-cis,15-anti and all-trans isomeric states is shifted toward 13-cis,15-anti when Asp-96 is deprotonated [50]. This implies a second functional coupling in the protein during the photocycle, between the retinal and the cytoplasmic region, perhaps through reversal of the events that had mobilized the proton of Asp-96 for the reprotonation of the Schiff base.
It was recently suggested that a single protein conformation can account for proton transfer between Asp-96 and the Schiff base and the cytoplasmic surface and Asp-96 [24]. Nevertheless, it is clear that Asp-96 cannot be in protonation equilibrium with the Schiff base and the cytoplasmic surface at the same time [5], because reprotonation of the Schiff base is largely independent of pH. Because the two protonation reactions proceed with not greatly different time-constants, an additional conformational change in the cytoplasmic region is needed, which is distinct from the one that allows deprotonation of Asp-96. The nature of this conformation change is still uncertain.
Deprotonation of Asp-85 will be the consequence of the reestablishment of the initial geometry at the Schiff base. Loss of the Asp-85 proton limits the rate of the final photocycle step [14], and its irreversibility must reflect recovery of the very low pKa (2.5) of Asp-85.
PERSPECTIVES
High-resolution x-ray crystallography will continue to contribute unique and essential information to uncover the transport mechanism in bacteriorhodopsin. The main unsolved questions are about the exact path of the proton transfer reactions. However, the limitations of the crystallographic method are also becoming evident. States or substates of the photocycle, which arise in rate limiting steps, might be so transient that they can never be stabilized. Visualizing the most interesting changes at the retinal, which occur in the K and L states, will need crystals that diffract much better than the current resolution. Describing the large-scale conformational changes in the second half of the photocycle will require new approaches which avoid disorder in the tightly packed crystals. Successful solution of the still outstanding problems will no doubt utilize many experimental strategies and much ingenuity.
REFERENCES
1.Lanyi, J. K. (1993) Biochim. Biophys. Acta,
1183, 241-261.
2.Haupts, U., Tittor, J., and Oesterhelt, D. (1999)
Annu. Rev. Biophys. Biomol. Struct., 28, 367-399.
3.Lanyi, J. K. (1999) Int. Rev. Cytol.,
187, 161-202.
4.Lanyi, J. K. (2000) Biochim. Biophys. Acta,
1459, 339-345.
5.Balashov, S. P. (2000) Biochim. Biophys.
Acta, 1460, 75-94.
6.Kaulen, A. D. (2000) Biochim. Biophys. Acta,
1460, 204-219.
7.Lozier, R. H., Xie, A., Hofrichter, J., and Clore,
G. M. (1992) Proc. Natl. Acad. Sci. USA, 89,
3610-3614.
8.Mathies, R. A., Lin, S. W., Ames, J. B., and
Pollard, W. T. (1991) Annu. Rev. Biophys. Biophys. Chem.,
20, 491-518.
9.Lanyi, J. K., and Váró, G. (1995)
Israel J. Chem., 35, 365-386.
10.Balashov, S. P., Imasheva, E. S., Govindjee, R.,
and Ebrey, T. G. (1996) Biophys. J., 70, 473-481.
11.Rammelsberg, R., Huhn, G., Lubben, M., and
Gerwert, K. (1998) Biochemistry, 37, 5001-5009.
12.Radionov, A. N., and Kaulen, A. D. (1999) FEBS
Lett., 451, 147-151.
13.Kandori, H., Yamazaki, Y., Hatanaka, M.,
Needleman, R., Brown, L. S., Richter, H. T., Lanyi, J. K., and Maeda,
A. (1997) Biochemistry, 36, 5134-5141.
14.Richter, H. T., Needleman, R., Kandori, H.,
Maeda, A., and Lanyi, J. K. (1996) Biochemistry, 35,
15461-15466.
15.Balashov, S. P., Lu, M., Imasheva, E. S.,
Govindjee, R., Ebrey, T. G., Othersen, B., Chen, Y., Crouch, R. K., and
Menick, D. R. (1999) Biochemistry, 38, 2026-2039.
16.Luecke, H., Schobert, B., Richter, H. T.,
Cartailler, J. P., and Lanyi, J. K. (1999) J. Mol. Biol.,
291, 899-911.
17.Belrhali, H., Nollert, P., Royant, A., Menzel,
C., Rosenbusch, J. P., Landau, E. M., and Pebay-Peyroula, E. (1999)
Structure, 7, 909-917.
18.Landau, E. M., and Rosenbusch, J. P. (1996)
Proc. Natl. Acad. Sci. USA, 93, 14532-14535.
19.Rummel, G., Hardmeyer, A., Widmer, C., Chiu, M.
L., Nollert, P., Locher, K. P., Pedruzzi, I., Landau, E. M., and
Rosenbusch, J. P. (1998) J. Struct. Biol., 121,
82-91.
20.Heberle, J., Buldt, G., Koglin, E., Rosenbusch,
J. P., and Landau, E. M. (1998) J. Mol. Biol., 281,
587-592.
21.Grigorieff, N., Ceska, T. A., Downing, K. H.,
Baldwin, J. M., and Henderson, R. (1996) J. Mol. Biol.,
259, 393-421.
22.Kimura, Y., Vassylyev, D. G., Miyazawa, A.,
Kidera, A., Matsushima, M., Mitsuoka, K., Murata, K., Hirai, T., and
Fujiyoshi, Y. (1997) Nature, 389, 206-211.
23.Mitsuoka, K., Hirai, T., Murata, K., Miyazawa,
A., Kidera, A., Kimura, Y., and Fujiyoshi, Y. (1999) J. Mol.
Biol., 286, 861-882.
24.Kamikubo, H., Oka, T., Imamoto, Y., Tokunaga, F.,
Lanyi, J. K., and Kataoka, M. (1997) Biochemistry, 36,
12282-12287.
25.Subramaniam, S., Lindahl, M., Bullough, P.,
Faruqi, A. R., Tittor, J., Oesterhelt, D., Brown, L., Lanyi, J., and
Henderson, R. (1999) J. Mol. Biol., 287, 145-161.
26.Subramaniam, S., and Henderson, R. (2000)
Nature, 406, 653-657.
27.Oka, T., Yagi, N., Fujisawa, T., Kamikubo, H.,
Tokunaga, F., and Kataoka, M. (2000) Proc. Natl. Acad. Sci. USA,
97, 14278-14282.
28.Luecke, H., Richter, H. T., and Lanyi, J. K.
(1998) Science, 280, 1934-1937.
29.Edman, K., Nollert, P., Royant, A., Belrhali, H.,
Pebay-Peyroula, E., Hajdu, J., Neutze, R., and Landau, E. M. (1999)
Nature, 401, 822-826.
30.Royant, A., Edman, K., Ursby, T., Pebay-Peyroula,
E., Landau, E. M., and Neutze, R. (2000) Nature, 406,
645-648.
31.Luecke, H., Schobert, B., Richter, H. T.,
Cartailler, J.-P., Rosengarth, A., Needleman, R., and Lanyi, J. K.
(2000) J. Mol. Biol., 300, 1237-1255.
32.Luecke, H., Schobert, B., Richter, H. T.,
Cartailler, J. P., and Lanyi, J. K. (1999) Science, 286,
255-261.
33.Luecke, H. (2000) Biochim. Biophys. Acta,
1460, 133-156.
34.Lanyi, J. K. (1990) Physiol. Rev.,
70, 319-330.
35.Oesterhelt, D. (1995) Isr. J. Chem.,
35, 475-494.
36.Sasaki, J., Brown, L. S., Chon, Y., Kandori, H.,
Maeda, A., Needleman, R., and Lanyi, J. K. (1995) Science,
269, 73-75.
37.Brown, L. S., Needleman, R., and Lanyi, J. K.
(1996) Biochemistry, 35, 16048-16054.
38.Brown, L. S., Sasaki, J., Kandori, H., Maeda, A.,
Needleman, R., and Lanyi, J. K. (1995) J. Biol. Chem.,
270, 27122-27126.
39.Essen, L., Siegert, R., Lehmann, W. D., and
Oesterhelt, D. (1998) Proc. Natl. Acad. Sci. USA, 95,
11673-11678.
40.Wang, J., and El Sayed, M. A. (2001) Biophys.
J., 80, 961-971.
41.Ames, J. B., Bolton, S. R., Netto, M. M., and
Mathies, R. A. (1990) J. Am. Chem. Soc., 112,
9007-9009.
42.McDermott, A. E., Thompson, L. K., Winkel, C.,
Farrar, M. R., Pelletier, S., Lugtenburg, J., Herzfeld, J., and
Griffin, R. G. (1991) Biochemistry, 30, 8366-8371.
43.Richter, H. T., Brown, L. S., Needleman, R., and
Lanyi, J. K. (1996) Biochemistry, 35, 4054-4062.
44.Sass, H. J., Buldt, G., Gessenich, R., Hehn, D.,
Neff, D., Schlesinger, R., Berendzen, J., and Ormos, P. (2000)
Nature, 406, 649-653.
45.Brown, L. S., Dioumaev, A. K., Needleman, R., and
Lanyi, J. K. (1998) Biophys. J., 75, 1455-1465.
46.Brown, L. S., Dioumaev, A. K., Needleman, R., and
Lanyi, J. K. (1998) Biochemistry, 37, 3982-3993.
47.Kandori, H., Yamazaki, Y., Shichida, Y., Raap,
J., Lugtenburg, J., Belenky, M., and Herzfeld, J. (2001) Proc. Natl.
Acad. Sci. USA, 98, 1571-1576.
48.Brown, L. S., and Lanyi, J. K. (1996) Proc.
Natl. Acad. Sci. USA, 93, 1731-1734.
49.Száraz, S., Oesterhelt, D., and Ormos, P.
(1994) Biophys. J., 67, 1706-1712.
50.Dioumaev, A. K., Brown, L. S., Needleman, R., and
Lanyi, J. K. (1998) Biochemistry, 37, 9889-9893.