REVIEW: Non-Isomerizable Artificial Pigments: Implications for the Primary Light-Induced Events in Bacteriorhodopsin
A. Aharoni1, B. Hou2, N. Friedman1, M. Ottolenghi2*, I. Rousso1, S. Ruhman2*, M. Sheves1*, T. Ye2, and Q. Zhong2
1Department of Organic Chemistry, The Weizmann Institute of Science, Rehovot 76100, Israel; fax: 972-8-9344142; E-mail: mudi.sheves@weizmann.ac.il2Department of Physical Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel; fax: 972-2-6524951, 972-2-5618033; E-mail: micott@chem.ch.huji.ac.il, sandy@fh.huji.ac.il
* To whom correspondence should be addressed.
Received June 10, 2001; Revision received June 29, 2001
The primary events in the photosynthetic retinal protein bacteriorhodopsin (bR) are reviewed in light of photophysical and photochemical experiments with artificial bR in which the native retinal polyene is replaced by a variety of chromophores. Focus is on retinals in which the “critical” C13=C14 bond is locked with respect to isomerization by a rigid ring structure. Other systems include retinal oxime and non-isomerizable dyes noncovalently residing in the binding site. The early photophysical events are analyzed in view of recent pump-probe experiments with sub-picosecond time resolution comparing the behavior of bR pigments with those of model protonated Schiff bases in solution. An additional approach is based on the light-induced cleavage of the protonated Schiff base bond that links retinal to the protein by reacting with hydroxylamine. Also described are EPR experiments monitoring reduction and oxidation reactions of a spin label covalently attached to various protein sites. It is concluded that in bR the initial relaxation out of the Franck-Condon (FC) state does not involve substantial C13=C14 torsional motion and is considerably catalyzed by the protein matrix. Prior to the decay of the relaxed fluorescent state (FS or I state), the protein is activated via a mechanism that does not require double bond isomerization. Most plausibly, it is a result of charge delocalization in the excited state of the polyene (or other) chromophores. More generally, it is concluded that proteins and other macromolecules may undergo structural changes (that may affect their chemical reactivity) following optical excitation of an appropriately (covalently or non-covalently) bound chromophore. Possible relations between the light-induced changes due to charge delocalization, and those associated with C13=C14 isomerization (that are at the basis of the bR photocycle), are discussed. It is suggested that the two effects may couple at a certain stage of the photocycle, and it is the combination of the two that drives the cross-membrane proton pump mechanism.
KEY WORDS: bacteriorhodopsin, retinal, isomerization, primary processes
Abbreviations: bR) bacteriorhodopsin; FC) Franck-Condon state; FS) fluorescent state; hR) halorhodopsin; CI) conical intersection (on energy diagram); PSB) protonated Schiff bases; EPR) electronic paramagnetic resonance.
A retinal isomer bound to the opsin via a protonated Schiff base linkage
with a lysine residue is the chromophore of all retinal proteins (see
[1-3] for a series of review
articles). Light absorption by rhodopsins triggers characteristic
photocycles extending into the ms time range. The photocycles reflect
structural transformations in the chromophore (primarily
cis-->trans or trans-->cis
isomerization) and in its surrounding protein matrix, which are
responsible for the biological activity of the pigments. In the
light-adapted form of the photosynthetic protein bacteriorhodopsin
(bR570), the chromophore is all-trans, and the
related photocycle, including several intermediates (see scheme), is
associated with transient isomerization to a 13-cis
configuration. A cross-membrane proton translocation is induced,
leading to ATP synthesis:
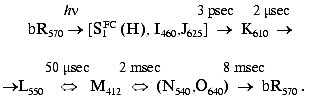
At the focus of retinal protein research in general, and of bR in particular, are the early light-induced ultrafast events occurring on femtosecond and picosecond timescales (for reviews see [4, 5]). A major problem has been the characterization of the early dynamics of both chromophore and protein following light absorption. More specifically, addressing the question as to when and how do the primary structural and electronic changes in the retinal trigger conformational changes in the protein, which subsequently lead to vectorial proton translocation. This review strives to answer these questions by addressing the following issues.
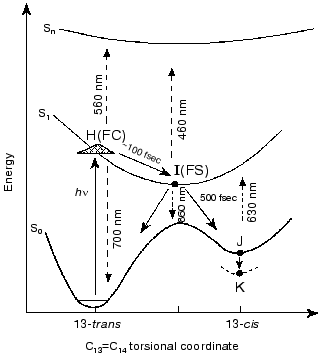
1. The prevailing models for the early events in retinal proteins have usually assumed that departure out of the Franck-Condon (FC) region towards the relaxed fluorescent state (the I460 intermediate or FS state), is controlled by substantial torsional motion about a “critical” double bond, C13=C14 in bR and in the closely related chloride ion pump halorhodopsin (hR) [1-17]. Accordingly, initiation of theall-trans-->13-cis isomerization is the very primary consequence of light absorption. This approach, first suggested for retinal proteins by Rosenfeld et al. [6] and Hurley et al. [7], is schematically presented in Fig. 1. The ~100 fsec relaxation from the FC to the FS state was invoked on the basis of pioneering time resolved pump-probe experiments with sub-picosecond time resolution [8-11]. In this schematic approach the fluorescent “common excited state”, I460, is partially (~90°) twisted about the C13=C14 coordinate, and isomerization is completed to the 13-cis (~180°) ground state J625 species. It should be noted that it was suggested that the model of Fig. 1 may be more complex due to the involvement of three (S0, S1, S2), rather than two (S0, S1) states [18, 19]. However, the three state approach was recently questioned on the basis of ab-initio quantum chemical computations [20].
2. Early theoretical considerations [21-23] advocated that the earliest light-induced changes in a retinal protein are associated with charge redistribution in the excited retinal chromophore. Nevertheless, the prevailing working hypothesis has assumed that protein changes are induced by isomerization of the chromophore around a “critical” double bond. In other words, that a conformational change in the protein host can only result from a conformational change in the embedded retinal chromophore.Fig. 1. A generally accepted model accounting for the primary light-induced events in bR. Wavelengths denote absorption and stimulated emission maxima.
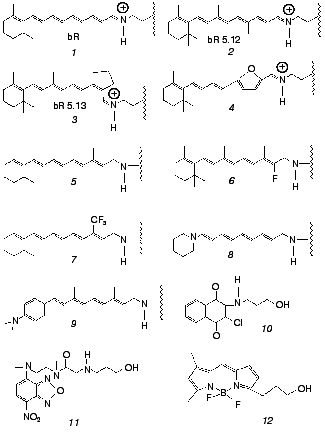
Both issues have recently been addressed by us in work that makes use of artificial bR pigments in which the native retinal chromophore is replaced by a suitable synthetic analog (for reviews on synthetic retinal proteins see [24, 25]). The principal tool is one of employing pigments in which the “critical” C13=C14 bond is locked into trans or cis configurations by rigid ring structures. The applied experimental methodologies are based on one hand on ultrafast pump-probe excitation and, on the other hand, on chemical methods aiming at detecting light-induced changes in protein structure. The ultrafast behavior of bR and its locked analogs were compared to those of the corresponding free protonated Schiff bases (PSB) in solution. In the chemical experiments, use was also made of retinal Schiff bases that are not covalently bound to the protein, as well as of non-retinal, non-isomerizable, chromophores, both residing in the binding site. Chromophores discussed in this Review are shown in Fig. 2. The emerging picture is that both basic postulates (1) and (2) should be thoroughly revised. In the following sections we review the major findingsand discuss their implications to the early light-induced events in bacteriorhodopsin.
Fig. 2. Native (1) and artificial (2-12) chromophores employed for probing the primary events in bR. Chromophores 1-9 are covalently bound to the protein, while 10-12 noncovalently reside in the retinal binding site. The “free” PSB chromophores--trans PSB, trans PSB 5.12 and 13-cis PSB 5.13--consist of molecules 1,2, and3 with n-butylamine replacing the lysine-protein protonated Schiff base.
I. ULTRAFAST PHENOMENA IN NATIVE AND ARTIFICIAL bR AND IN MODEL PSB IN SOLUTION
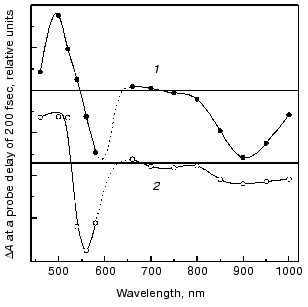
The fluorescent state (FS). Spectral and decay features. The fluorescent state of bacteriorhodopsin, also denoted as the I460 intermediate, is characterized by an intense absorption around 460 nm and a stimulated emission band around 900 nm [8-10]. The discrepancy between the stimulated emission spectrum and that of the spontaneous fluorescence (lambdamax = 730 nm) was accounted for in terms of an additional intense absorption band peaking around 720 nm that partially cancels the stimulated emission [19, 26, 27]. The nature of the FS state was approached in experiments with femtosecond time resolution, in which the early events in native bR (1) were compared to those of the two artificial pigments bR5.12 (2) and bR5.13 (3) (see Fig. 2) in which a synthetic retinal with a rigid five membered ring is locked into C13=C14 trans or cis configurations, respectively [28, 29]. Both artificial pigments were shown to lack the characteristic photocycle of native bR, stressing the crucial importance of isomerization in the photoreactivity of bR. Figure 3 depicts the transient changes of both free and locked trans systems at a probe delay of 200 fsec, when the absorption and emission bands of the FS attain their maximum value. The two spectra are essentially the same as is that observed for the 13-cis molecule bR5.13 (not shown). This suggests that highly similar fluorescent states (I460, I5.12 and I5.13, respectively) are generated in the case of the three molecules, excluding a twisted (~90°) C13=C14 configuration for the I460 state, as postulated by the model of Fig. 1.
Despite the above spectral similarities, the native and locked fluorescent states markedly differ in their emission lifetimes. While in the native system I460 decays in about 500 fsec (best represented by a bi-exponential function with 250 fsec and 1 psec components), giving rise to the 625 nm absorption of the J625 intermediate, I5.12 and I5.13 exhibit long lived decays of 18 and 11 psec, respectively, regenerating their original ground states.Fig. 3. Combined transient changes in absorbance, and in stimulated emission, recorded 200 fsec after pulsed laser excitation of light-adapted native bR (bR570) (2) and the C13=C14 (trans) locked pigment bR5.12 (1).
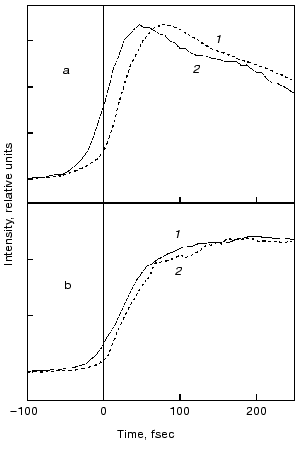
Evolution of the FS (I) state. A crucial mechanistic aspect concerns the rate of appearance of the FS state from its primary FC precursor. Figure 4 presents high time resolution data for both locked and native molecules leading to the following observations. In native bR evolution of both 460 nm absorption and 800 nm emission is nearly instantaneous, i.e., below the 15-20 fsec resolution of the experimental system. The rise of the stimulated emission at 950 nm is delayed by 15-20 fsec with respect to the 460 and 800 nm signals (Fig. 4a). In bR5.12 the rise in emission at both 800 and 950 nm is delayed with respect to the native system (Fig. 4b). Thus, a sub-20-fsec evolution of both locked and unlocked I states is followed by a structural reorganization, on the order of tens of femtoseconds, that Stokes shifts the FS emission. The identity in the time scales of the evolution of I460, I5.12, and I5.13, provides further support for the conclusion that the primary FC-->FS relaxation is not associated with a substantial C13=C14 torsional motion.
The notion that, independently of any specific mechanism (see below), coordinates other than torsion about the isomerizing C=C bond should play a role in the early events in bR, hR, and visual rhodopsins, finds support in recent independent studies. Thus, a Fourier transform analysis of the absorption spectrum of native bR and bR5.12 indicates analogous relaxation times out of the FC state in both systems [30]. The nature of such coordinates has been discussed in a multi-mode analysis identifying vibrational degrees of freedom of the retinal chromophore, as well as of the surrounding protein (dielectric relaxation) coordinates, which might be associated with departure of the wave packet out of the FC region [31]. A specific model in this respect was provided by the general ab-initio approach for protonated Schiff bases [32]. The model leads to the conclusion that in retinal proteins the initial motion out of the FC state is dominated by in-plane C=C stretching rather than by out of plane twisting modes. A time resolved resonance Raman study in bR is indeed indicative of changes in the C=C stretching frequency in the I460 FS [33].Fig. 4. Rise of the stimulated emission at 800 nm (dashed lines) and 950 nm (solid lines) following 600 nm excitation of bR (a) and bR5.12 (b).
Dynamic spectral features in the intermediate 500-700 nm range. The ultrafast features between 500 and 700 nm are of crucial mechanistic importance since they represent the absorption range of the primary photoproducts of native bR, J625, and K610. Characteristic traces at 660 nm where J625 absorbs (see Fig. 5), are indicative of three distinct dynamic phases: 1) an instantaneous (<30 fsec) generation of a stimulated emission; 2) a slower (40-80 fsec) reversion to net absorption; 3) a gradual rise in gain, on the order of 400-600 fsec. Analogous but inverted features are observed at 520-560 nm: a prompt initial absorbance excess (“phase 1”) is followed by a net bleach which corresponds to “phase 2”. This phase has been previously interpreted [8, 9] in terms of the FC-->FS relaxation, including C13=C14 torsion and vibrational dephasing. However, this is incompatible with the <30 fsec rise of the FS as discussed above, as well as with the fact that essentially identical patterns characterize the native and the C13=C14 locked molecules (Fig. 5). More interestingly, the observation of the same “phase 3” process in the locked systems that lack the J625 and later photointermediates, implies that in variance with previous assertions, the 400-600 fsec process does not reflect evolution of the J625 species. Moreover, it cannot reflect substantial trans-->13-cis isomerization.
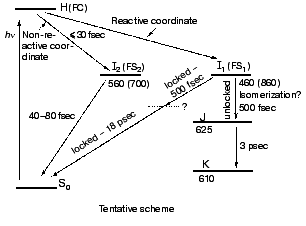
The high similarity between the primary light-induced patterns of the native and C13=C14 locked bR exclude (see “Discussion” in [29]) the model of Fig. 1, as well as later modifications [11-14] that include branching at the FC state into a reactive (isomerization) channel and a non-reactive (back-reaction) channel. The major inconsistency lies with the basic hypothesis shared by all of such models, that the primary dynamic consequence of light absorption involves substantial torsional motion about the “critical” C13=C14 bond. This is in keeping with previous photochemically and spectroscopic data that point to the deficiency of the simplified one-dimensional curve of Fig. 1 [34, 35]. A plausible model, accounting for the <30 fsec evolution of I460 (FS), along with the 40-80 fsec changes in the 500-700 nm range, is shown in Fig. 6. It adopts the concept of two molecular pathways branching at the FC state. One leading back to the relaxed all-trans ground state, the other going forward along the photocycle. In this model the 40-80 fsec process is attributed to the decay of the second non-reactive excited state (I2 or FS2) rather then to the FC-->FS relaxation of the single path models. Although the scheme of Fig. 6 is in keeping with the presently available data, it still awaits direct verification, e.g., by three pulse experiments currently in progress in our laboratory.Fig. 5. Characteristic traces showing the early evolution of the absorbance changes at 660 (a, c) and 520-560 nm (b, d) for native bR (bR570) (a, b) and bR5.12 (c, d).
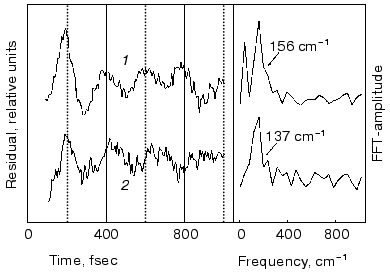
Spectral modulations. Wave packet formation through impulsive excitation should give rise to periodical motions along displaced nuclear coordinates, detectable through spectral modulations of the excited state emission. In bR, impulsive excitation was shown to give rise to spectral modulations in the ground state absorption band [36], while in visual rhodopsin periodical motions were detected in the bathorhodopsin photoproduct [37]. However, in variance with other biopigments, no oscillations corresponding to the S1 state were detected in a retinal protein, in spite of the resonance Raman activity of a large number of vibrational coordinates [36-38]. Such absence was attributed to critical damping or over-damping. Recently [39], using 20 fsec pulses and a high signal-to-noise ratio in pump-probe experiments, we have been able to observe modulations in the stimulated fluorescence of both bR and bR5.12, which are associated with vibronic wavepacket motion on the S1 surface (see Fig. 7). An FFT analysis clearly shows one major peak at 157 and 137 cm-1 for the native and locked molecules, respectively. The observed frequency is below the C-C stretching frequency and is also inconsistent with C=C torsion [40]. Most likely it represents a skeletal motion of the retinal along a bound coordinate in the excited state. A future challenge will be that of identifying the active mode and establishing if it is associated with accessing the conical intersection leading to the ground state.Fig. 6. Tentative scheme accounting for the ultrafast primary events in bR in terms of two fluorescent states: I1 (FS1) and I2 (FS2). Numbers refer to approximate wavelengths of maximum absorption and (in parenthesis) maximum emission.
Role of the protein: comparison of the ultrafast bR dynamics with those of the free protonated Schiff bases in solution. The basic process in retinal proteins is activation of the protein by light-induced changes in the polyene. On the other hand, the protein controls the selectivity, yield, and time scales of the primary photoreactions of the chromophore. Thus, understanding the early events in a pigment such as bR, and the way they are controlled by chromophore-protein interactions, requires a comparison with the photophysical behavior of the corresponding free protonated Schiff base (PSB) in solution.Fig. 7. Modulations and respective FFT residuals observed in the stimulated emission of bR (1) and bR5.12 (2).
The ultrafast spectroscopy of model PSB isomers in solution has been investigated by fluorescence up-conversion [41, 42] and by pump-probe [43-45] techniques. In the case of the all-trans isomer, the primary feature is the prompt formation of a broad absorption between 490 and 650 nm attributed to the FC state [44, 45]. After 200 fsec the transient spectrum consists of a stimulated emission at about 670 nm and by an absorption peaking at 500 nm. Both features, decaying over several ps, are associated with the FS state [43-45]. Analogous patterns are exhibited by 13-cis PSB [43]. These features have been interpreted in terms of two approaches.
1. That of the model of Fig. 1 (see also [44]) in which the initial ~100 fsec decay of the FC state is attributed to a ~90° torsion about the C13=C14, or C11=C12, bond. Subsequent motion along the same reaction coordinate occurs on the S0 ground state surface.
2. A second approach, based on ab-initio quantum chemical calculations on model compounds [32, 40] and on temperature effects on the FS lifetime [38], attributes the picosecond decay component to a thermally activated C13=C14 torsion on the S1 surface, to a 90° conical intersection (CI) point. In this model the primary FC-->FS transition is due to in-plane C=C stretching modes, while the CI(S1)-->S0 step is fast and non rate determining.
Experiments have been recently carried out [45] comparing the ultrafast spectroscopy of the two locked analogs trans PSB5.12 and 13-cis PSB5.13 (molecules 2 and 3 in Fig. 2, but with n-butylamine replacing the lysine protein residue) with those of the “native” all-trans and 13-cis PSB. Interestingly, the basic spectroscopic and dynamic features were found to be almost unaffected by locking of the C13=C14 (trans or cis) bond, explicitly: a) C13=C14 locking influences the FS decay time negligibly. This is in variance with bR where the FS (I460) 500 fsec decay is much faster than that of bR5.12 (18 psec); b) the 50-100 fsec evolution of the FS state is somewhat slower than in bR (unresolved, <30 fsec), but is the same for the two locked and all-trans, unlocked PSBs; c) the free PSBs exhibit spectral modulations (e.g., at 117 cm-1) that are similar, but not identical, to those observed in bR.
These results in the detached prosthetic group demonstrate that the protein catalyzes relaxation to the ground state, and that the protein matrix is instrumental in directing the pathway of curve crossing to include the crucial C13=C14 torsion. Our results do not identify the alternative reaction pathways which lead to CI in the various PSBs, and render the C13=C14 torsion superfluous in achieving this goal. Point (c) suggests that the spectral modulations in the stimulated emission, which are interpreted to reflect wave packet motions in the reactive excited state, are primarily taking place in the retinal moiety in the case of bR as well. Point (b) shows that the primary response in the PSB alone is consistent with that in the bR. Accordingly, insensitivity of FC relaxation to the locking implies that as in bR, the primary relaxation out of the FC region in PSB is not controlled by substantial out of plane C13=C14 motions, excluding model (1) (considered in the introductory section and presented in Fig. 1). The fact that in all PSBs this process is slower than its counterpart in bR or bR5.12 may indicate that this stage of evolution is also somewhat catalyzed by the opsin pocket.
II. PROTEIN CONFORMATIONAL CHANGES INDUCED VIA NON-ISOMERIZABLE
CHROMOPHORES
The biological functions of retinal proteins, such as vision, photosynthesis, and phototaxis, are based on changes in protein structure induced by light absorption by the retinal chromophore [1]. Such changes in the protein are interpreted in terms of a primary isomerization of the retinal about the “critical” C=C bond. Accordingly, light provides the necessary energy for overcoming the C=C, trans-->cis or cis-->trans, barrier. The corresponding change in retinal conformation leads, in a key-lock like process, to a sustainable change in protein structure that drives the biological activity. In spite of the general consensus as to the fundamental role of C=C isomerization, theoretical considerations have pointed out the possibility that along with such steric effects, light-induced electronic polarization of the retinal chromophore as well, may induce a structural reorganization of the protein environment [22, 23]. Such electronic effects are intimately related to the very nature of the FC, FS, and CI excited states, as discussed in the previous section.
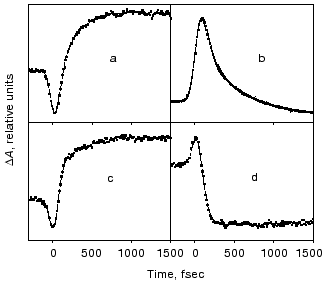
The first experimental indication for light-inducing conformational change in a protein, with no isomerization of an embedded chromophore, came from a study based on atomic force sensing (AFS) [46]. The methodology monitors the vertical deflection of the tip of an atomic force microscope, in contact with the bR purple membrane, as a result of a laser pulse that is absorbed by the pigment. Unexpectedly, following optical excitation, C13=C14 locked bR pigments that do not undergo a photocycle, exhibited transient AFS signals, analogous to those of native bR. The effect was attributed to changes in protein structure that are associated with the excited state rather than with the characteristic photointermediates of bR. Recently, independent evidence for a light-induced structural transformation on the locked bR5.12 pigment (2, Fig. 2) following optical excitation was obtained from photoacoustics experiments [47]. More direct evidence for the occurrence of structural changes in the bR protein that do not necessitate a photocycle, was subsequently derived from light-induced changes in the chemical activity of a variety of bR and artificial bR pigments. These are reviewed in some detail in the subsequent sections.
Light-induced cleavage of the C=NH+ bond: hydroxylamine reactions. The protonated Schiff base bond between retinal and the lysine residue in the binding site of retinal proteins (K216 in bR) is stable to hydrolysis in the dark, but undergoes cleavage (to retinal oxime and the lysine residue) in the presence of hydroxylamine. The reaction is catalyzed by light [48], yielding the standard procedure for the replacement of retinal with synthetic chromophores. It has been assumed that, at the stage of the M [49] or L [50] photocycle intermediates, the protein undergoes a conformational change that catalyzes the reaction by exposing the C=NH+ bond to the reagent or to a suitable catalytic protein moiety.
This assumption was tested by studying the light effect on the hydroxylamine reaction in the C13=C14 locked pigments, bR5.12 (2, Fig. 2), bR5.13 (3, Fig. 2) and bR11-epoxy (4, Fig. 2), that do not exhibit a photocycle [51]. Surprisingly, in all three cases light altered the reaction kinetic. Since the bR photocycle is associated with C13=C14 isomerization of the retinal, it was concluded that the chemical activity of the protein (with respect to the hydroxylamine reaction) is affected by light even when no C13=C14 isomerization takes place.
Two alternative mechanisms were suggested, accounting for this conclusion: 1) in the locked molecules the light-catalyzed hydroxylamine reaction takes place in the excited state, e.g., in the FS,or possibly even earlier; 2) the reaction occurs after complete relaxation of the chromophore to the ground state, due to a conformational change in the protein that is not associated with an (optical) photocycle.
The first alternative was excluded on the basis of considerations based on the quantum yield of the light-induced reaction and on the FS lifetime, that in the case of, e.g., bR5.13 were determined as 2*10-3 and 11 psec, respectively. It was thus concluded that in the locked molecules, the reaction is due to a protein conformational change persisting on a timescale that is longer than hundreds of nanoseconds and that is not accompanied by a conformational change of the retinal chromophore.
Chemical reactivity outside of the binding site: reduction-oxidation of spin labels covalently bound to the protein. While the hydroxylamine reactions described above detect protein structural alteration in the region of the retinal binding site, methods were developed to monitor light-induced conformational alterations in other protein domains [52]. This was achieved by applying a site-directed spin labeling technique in bR that is widely used to obtain information on the label environment and its alterations during the photocycle [53-61]. Our experiments revealed that the spin label can be reduced by hydroxylamine and subsequently (after hydroxylamine removal) reoxidized by molecular oxygen to form the original radical. Both reduction and oxidation reactions, followed by EPR spectroscopy, were used as tools to monitor light-induced protein conformational alterations in a variety of artificial and chemically modified chromophores:
a. The reduction reaction with retinal and synthetic retinals as chromophores. The spin label reduction reaction was found to be catalyzed by light in the A103C-labeled pigment but not when labeling was at the E74 or E163 positions. These experiments clearly indicated that the protein experiences light-induced conformational alterations that selectively affect the label reactivity in the cytoplasmic side of the protein, in the vicinity of the A103 residue. Moreover, analogous catalytic effects were observed in artificial pigments 2-4 (Fig. 2) in which the C13=C14 double bond isomerization cannot take place. This indicates that the effect does not require isomerization about the “critical” double bond and with the bR photocycle.
b. The oxidation reaction with retinal oxime as chromophore. The oxidation reaction of the spin label was studied in hydroxylamine-bleached membranes that carry a retinal oxime non-covalently trapped in the binding site. Light absorption by the oxime chromophore was found to considerably accelerate the reaction when the label was at the A103 position. Again, the same phenomenon is observed in the case of the oximes derived from molecules 2-4 (Fig. 2). These results show that the catalytic effects, and thus the associated changes in protein conformation, are not specific to covalently attached (protonated) Schiff bases of retinal.
c. The oxidation reaction with a reduced retinal Schiff base as chromophore. To test the hypothesis that the light-induced change in protein conformation occurs as a respond to charge redistribution in the excited state [22, 23, 46], the label oxidation reaction was studied in several reduced bR systems [52]. Reduction of the retinal-protein Schiff base linkage with sodium borohydride leads to a symmetric polyene, covalently bound to the protein via a -CH2-NH-bond (molecule 5, Fig. 2). In keeping with the above hypothesis, this pigment did not show any light-catalyzed oxidation reaction. Further support for the light-induced charge redistribution mechanism was gained by experiments with substituted (reduced) chromophores. Substitution with electron withdrawing groups (F, CF3) close to the Schiff base linkage (molecules 6 and 7, Fig. 2), that reinstate charge delocalization, was found to restore light-induced acceleration. Similarly, electron donating groups replacing the beta-ionone ring (molecules 8 and 9, Fig. 2) initiated the protein response as well. All such experiments are in keeping with the charge redistribution mechanism, excluding alternative explanations, e.g., attributing the effect to excess light energy dissipated as heat.
d. The oxidation reaction in non-retinal, non-isomerizable, chromophores. An inherent difficulty with all of the above systems arises from the fact that the retinal analogs employed carried isomerizable C=C or C=N bonds, other than the “critical” C13=C14 bond. Therefore, although unlikely, isomerization around other double bonds could replace that of the locked C13=C14 as a cause of the light catalytic effect. To circumvent this difficulty, several dye molecules (10-12, Fig. 2), characterized by a completely rigid structure in which no double bond isomerization can occur, were incorporated into the binding site of the spin labeled opsin [62]. Incorporation occurs via noncovalent trapping in the binding site that displaces the retinal oxime. It was revealed (see table) that in the case of A103C labeling (but not in the case of A74C) the oxidation reaction was significantly accelerated by light. It is thus evident that non-isomerizable chromophores that carry no resemblance to retinals are as effective in inducing the light-induced conformational changes in the A103C region of the protein.
Relative photocatalytic efficiency
(relative to non-illuminated solution) of native and artificial
bacteriorhodopsin pigments (values are relative to native
bacteriorhodopsin)

aDark and light reactions were carried out in the presence of
0.2 M hydroxylamine.
bReduction of the radical attached to 103C residue, followed
by EPR spectroscopy.
cSpontaneous reoxidation of the reduced radical by molecular
oxygen.
dThe dyes were illuminated with a 450 nm cut-off filter,
whereas native bR was illuminated with white light.
III. CONCLUDING DISCUSSION
It is appropriate to compare the effects of locking of the C13=C14 bond on the early time-resolved photophysical phenomena in bR, as revealed by ultrafast spectroscopy, with those associated with the light-induced conformational alterations in the protein. We have seen that such alterations, reflected by changes in the chemical reactivity of the Schiff base or of an attached spin label, are essentially insensitive to locking of the “critical” C=C bond. This implies that the alterations are triggered prior to the stage at which the photoreactions of the native pigment and those of the locked pigments diverge. As discussed above this occurs at the stage of the FS (I intermediate) that in the locked molecules exhibits a lifetime of 10-20 psec. In the native pigment I460 decays in less than 1 psec giving rise to the subsequent photocycle that lasts for several msec. Since the light-catalytic efficiency does not appear to depend on the excited state lifetime it seems reasonable to conclude that protein activation is triggered on a sub-picosecond time scale, as early as during, or after, the FC-->FS relaxation. Moreover, the data discussed above clearly show that during this time range the retinal chromophore does not undergo a substantial torsion about a C=C bond.
At present, the accumulated evidence points to charge delocalization in the excited state as the cause of the light-induced conformational changes in the protein. In some cases [63-65] the light catalytic effect can be directly correlated with the presence of an induced dipole (as determined for the vertically excited FC state) by second harmonic generation, or by two photon double resonance spectroscopy [66]. In other cases (see above) the presence of an excited state dipole is postulated on the basis of molecular symmetry considerations. For the noncovalently bound dyes 10-12 (Fig. 2) the presence and size of charge delocalization still awaits direct experimental verification. A major challenge will be that of defining on a molecular level the structural reorganization of the protein. It will primarily be understanding the mechanisms that affect the reactivity of the Schiff base and transmits structural alterations to other protein domains. However, independently of such quantitative knowledge, it is evident that optical excitation of an appropriately embedded chromophore, may induce transient structural changes in the opsin that affect its chemical reactivity. This is a general conclusion that may also apply to visual pigments, to other proteins, and possibly to other macromolecules.
It should be pointed out that additional questions related to the mechanism of protein activation and its consequences are still open. For example, the time scale of the chemical reaction. Does the reaction occur during the excited state lifetime or on longer time scales, in which the protein structural change persists after complete relaxation of the chromophore to its original ground state? In case of the hydroxylamine bond cleavage described above, the reaction was shown to occur on time scales that are much longer than that of the FS. However, such discrimination between the two alternatives is not yet available in the case of the spin label reduction and reoxidation processes.
A second point concerns the question as to whether and how are the structural changes that do not require isomerization related to the bR photocycle and to its biological activity. A simple approach may invoke that such changes are unrelated to the photocycle and (as it is usually assumed) all photocycle phenomena are exclusively triggered by isomerization, as early as at the stage of the J or K intermediates. It is tempting to suggest, however, that the above changes do couple to those triggered by isomerization. In such a case, there appear to be two alternative coupling mechanisms. a) The early changes, induced most probably by charge delocalization in the excited state, are a pre-requisite for isomerization of the chromophore at the J or K stages. Note that although C13=C14 isomerization at the K stage is well established, a recent CARS study of bR [67] suggests that J still maintains the original all-trans configuration of light-adapted bR. This study points that J is a twisted non-isomerized either excited or ground state. b) Alternatively, the changes induced by charge redistribution and those induced by isomerization are triggered independently, coupling only at later stages of the photocycle. According to either mechanism the molecular changes occurring during the photocycle that are responsible for the function of bR require both kinds of protein structural transformations. Discrimination between the various alternatives, clarifying the exact mechanism of retinal-protein activation by light, remains a challenging objective of future studies.
The research was supported by grants from the A. M. N. Fund for the Promotion of Science, Culture and Arts in Israel, the Israel National Science Foundation, the US-Israel Binational Science Foundation, and the Human Frontier Science Program.
REFERENCES
1.Ottolenghi, M., and Sheves, M. (eds.) (1995) The
Photophysics and Photochemistry of Retinal Proteins. Isr. J. Chem.,
35, 193-515.
2.Lanyi, J. (ed.) (2000) Biochim. Biophys. Acta
Bioenerg., 1460, 1-239.
3.Haupt, U., Tittor, J., and Oesterhelt, D. (1999)
Ann. Rev. Biophys. Biomol. Struct., 21, 367-399.
4.Kochendoerfer, G. G., and Mathies, R. A. (1995)
Isr. J. Chem., 35, 211-226.
5.Stuart, J. A., and Birge, R. R. (1996)
Biomembranes, 2A, 33-139.
6.Rosenfeld, T., Honig, B., Ottolenghi, M., Hurley,
J., and Ebrey, T. G. (1977) Pure Appl. Chem., 49,
341-351.
7.Hurley, J., Ebrey, T. G., Honig, B., and
Ottolenghi, M. (1977) Nature (London), 270,
540-542.
8.Sharkov, S., Pakulev, A., Chekalin, S., and
Matveetz, Y. (1985) Biochim. Biophys. Acta, 808,
94-102.
9.Mathies, R. A., Brito-Cruz, C. H., Pollard, T. W.,
and Shank, C. V. (1988) Science, 240, 777-779.
10.Dobler, J., Zinth, W., Kaiser, K., and
Oesterhelt, D. (1988) Chem. Phys., 144, 215-220.
11.Kobayashi, T., Terauchi, M., Kouyama, T.,
Yoshizawa, M., and Taiji, M. (1990) SPIE, 1403,
407-416.
12.Kobayashi, T., Kim, M., Taiji, M., Iwasa, T.,
Nakagawa, M., and Tsuda, M. (1998) J. Phys. Chem. B.,
102, 272-280.
13.Gai, F., Hasson, H. C., McDonald Kooper, J., and
Anfinrud, P. A. (1998) Science, 279, 1886-1891.
14.Arlt, T., Schmidt, S., Zinth, W., Haupts, U., and
Oesterhelt, D. (1995) Chem. Phys. Lett., 241,
559-565.
15.Kandori, H., Yoshihara, K., Tomioka, H., and
Sasabe, H. (1992) J. Phys. Chem., 96, 6066-6071.
16.Loppnow, G. R., and Mathies, R. A. (1988)
Biophys. J., 54, 35-43.
17.Palings, I., Pardoen, J. A., van den Berg, E.,
Winkel, C., Lugetenburg, J., and Mathies, R. A. (1987)
Biochemistry, 26, 2544-2556.
18.Hasson, K. C., Gai, F., and Anfinrud, P. A.
(1996) Proc. Natl. Acad. Sci. USA, 93, 16124-16129.
19.Humphrey, W., Lu, H., Logunov, I., Werner, H.,
and Schulten, K. (1998) Biophys. J., 75, 1689-1699.
20.Gonzalez-Luque, R., Garavelli, M., Bernardi, F.,
Merchan, M., Robb, M., and Olivucci, M. (2000) Proc. Natl. Acad.
Sci. USA, 17, 9379-9384.
21.Salem, L., and Bruckmann, P. (1975) Nature
(London), 258, 526-529.
22.Lewis, A. (1978) Proc. Natl. Acad. Sci.
USA, 75, 543-547.
23.Xu, D., Martin, D., and Schulten, K. (1996)
Biophys. J., 70, 453-460.
24.Ottolenghi, M., and Sheves, M. (1989) J.
Membr. Biol., 112, 193-212.
25.Nakanishi, K., and Crouch, R. (1995) J. Isr.
Chem., 35, 253-272.
26.Haran, G., Wynne, K., Xie, A., He, Q., Chance,
M., and Hochstasser, R. M. (1996) Chem. Phys. Lett.,
26,389-395.
27.Gai, G., McDonald, P. A., and Anfinrud, P. A.
(1997) J. Am. Chem. Soc., 119, 6201-6202.
28.Zhong, Q., Ruhman, S., Ottolenghi, M., Sheves,
M., Friedman, N., Atkinson, G. H., and Delaney, J. K. (1996) J. Am.
Chem. Soc., 118, 12828-12829.
29.Ye, T., Friedman, N., Gat, Y., Atkinson, G.,
Sheves, M., Ottolenghi, M., and Ruhman, S. (1999) J. Phys. Chem.
B, 103, 5122-5130.
30.Akiyama, R., Yoshimori, A., Kakitani, T.,
Imamoto, Y., Shichida, Y., and Hatamo, Y. (1997) J. Phys. Chem.
A, 101,412-417.
31.Myers, A., Harris, R., and Mathies, R. (1983)
J. Phys. Chem., 79, 603-613.
32.Garavelli, M., Celani, P., Bernardi, F., Robb, M.
A., and Olivucci, M. (1997) J. Am. Chem. Soc., 119,
6891-6901.
33.Song, L., and El-Sayed, M. (1998) J. Am. Chem.
Soc., 120, 8889-8890.
34.Govindjee, R., Balashov, V. S., and Ebrey, T.
(1990) Biophys. J., 58, 597-608.
35.Balashov, S. P., Karneeva, N. V., Litvin, F. F.,
and Sineshchekov, V. A. (1987) in Retinal Proteins (Ovchinnikov,
Yu. A., ed.) VNU Science Press, Utrecht, The Netherlands, pp.
505-517.
36.Dexheimer, S. L., Wang, Q., Peteanu, L. A.,
Pollard, W. T., Mathies, R. A., and Shank, C. V. (1992) Chem. Phys.
Lett., 188, 61-67.
37.Wang, Q., Schoenlein, R., Peteanu, L., Mathies
R., and Shank, C. (1994) Science, 266, 422-424.
38.Althaus, T., Weisfeld, R., Lohrmann, M., and
Stockburger, M. (1995) Isr. J. Chem., 35,227-251.
39.Ye, T., Gershgoren, E., Friedman, N., Ottolenghi,
M., Sheves, M., and Ruhman, S. (1999) Chem Phys., 314,
429-434.
40.Garavelli, M., Negri, F., and Olivucci, M. (1999)
J. Am. Chem. Soc., 121, 1023-1029.
41.Kandori, H., and Sasabe, H. (1993) Chem. Phys.
Lett., 216, 126-132.
42.Kandori, H., Katsuta, Y., Ito, M., and Sasabe, H.
(1995) J. Am. Chem. Soc., 117, 2669-2670.
43.Logunov, S. L., Song, L., and El-Sayed, M. A.
(1996) J. Phys. Chem., 100, 18586-18591.
44.Hamm, P., Zurek, M., Röschinger, T.,
Patzelt, H., Oesterhelt, D., and Zinth, W. (1996) Chem. Phys.
Lett., 263, 613-621.
45.Hou, B., Friedman, N., Ruhman, S.,
Sheves,M., andOttolenghi, M. (2001) J. Phys. Chem., in
press.
46.Rousso, I., Khachatryan, E., Gat, Y., Brodsky,
I., Ottolenghi, M., Sheves, M., and Lewis, A. (1997) Proc. Natl.
Acad. Sci. USA, 94, 7937-7941.
47.Losi, A., Michler, J., Gartner, W., and
Braslavsky, S. (2000) Photochem. Photobiol., 72,
590-597.
48.Oesterhelt, D., Meetzen, M., and Schumann, L.
(1973) Eur. J. Biochem., 40, 453-463.
49.Oesterhelt, D., Shumann, L., and Gruber, H.
(1974) FEBS Lett., 44, 257-261.
50.Subramaniam, S., Marti, T., Rösselet, S. J.,
Rothschild, K. J., and Khorana, H. G. (1991) Proc. Natl. Acad. Sci.
USA, 88, 2583-2587.
51.Rousso, I., Gat, Y., Lewis, A., Sheves, M., and
Ottolenghi, M. (1998) Biophys. J., 75, 413-417.
52.Aharoni, A., Weiner, L., Ottolenghi, M., and
Sheves, M. (2000) J. Biol. Chem., 275, 21010-21016.
53.Feix, Y. B., and King, C. (1998)
Spin-Labeling, Plenum Press, New York, pp. 251-281.
54.Hubbell, W., Gross, A., Langen, R., and Leitzow,
M. (1998) Curr. Opin. Struct. Biol., 8, 649-656.
55.Altenbach, C., Fitsch, L., Khorana, H. G., and
Hubbell, W. (1989) Biochemistry, 28, 7806-7812.
56.Altenbach, C., Marti, T., Khorana, H. G., and
Hubbell, W. (1990) Science, 248, 1088-1092.
57.Steinhoff, H., Mollaaghababa, R., Altenbach, C.,
Hideg, K., Krebs, M., Khorana, H. G., and Hubbell, W. (1994)
Science, 266, 105-107.
58.Hubbell, W., and Altenbach, C. (1994) Curr.
Opin. Struct. Biol., 4, 566-573.
59.Thorgeirsson, T., Xiao, W., Brown, L., Needleman,
R., Lanyi, J., and Shin, Y. (1997) J. Mol. Biol., 273,
951-957.
60.Rink, T., Riesk, J., Oesterhelt, D., Gerwert, K.,
and Steinhoff, H. (1997) Biophys. J., 73, 983-993.
61.Pfeiffer, M., Rink, T., Gerwert, K., Oesterhelt,
D., and Steinhoff, H. (1999) J. Mol. Biol., 275,
163-171.
62.Aharoni, A., Weiner, L., Ottolenghi M., and
Sheves, M. (2001) J. Am. Chem. Soc., 123, 6612-6616.
63.Huang, J., Chen, Z., and Lewis, A. (1989) J.
Phys. Chem., 93, 3314-3320.
64.Birge, R., and Zheng, X. (1990) J. Chem.
Phys., 94, 7178-7179.
65.Clays, K., Hendrick, E., Triest, M., Verhiest,
T., Persoons, A., Dehu, C., and Bredas, J. (1993) Science,
262, 1419-1422.
66.Birge, R., and Zhang, C. (1990) J. Chem.
Phys., 92, 7178-7195.
67.Atkinson, G. H., Ujj, L., and Zhou, Y. (2000)
J. Phys. Chem., 104, 4130-4139.