REVIEW: Internal Water Molecules as Mobile Polar Groups for Light-Induced Proton Translocation in Bacteriorhodopsin and Rhodopsin as Studied by Difference FTIR Spectroscopy
A. Maeda
Center for Biophysics and Computational Biology and Department of Biochemistry, University of Illinois at Urbana/Champaign, Urbana, IL 61801, USA; E-mail: amaeda@life.uiuc.edu, akimaeda@za2.so-net.ne.jp
Received April 8, 2001; Revision received May 9, 2001
FTIR spectroscopy is advantageous for detecting changes in polar chemical bonds that participate in bacteriorhodopsin function. Changes in H-bonding of Asp85, Asp96, the Schiff base, and internal water molecules around these residues upon the formation of the L, M, and N photo-intermediates of bacteriorhodopsin were investigated by difference FTIR spectroscopy. The locations and the interactions of these water molecules with the amino acid residues were further revealed by use of mutant pigments. The internal water molecules in the cytoplasmic domain probably work as mobile polar groups in an otherwise apolar environment and act to stabilize the L intermediate, and carrying a proton between the Schiff base and the proton acceptor or donor. Similar internal water molecules were shown to be present in bovine rhodopsin.
KEY WORDS: hydrogen bonding, FTIR, internal water molecules, bacteriorhodopsin, rhodopsin
The light-induced proton pump bacteriorhodopsin belongs to a family of seven-helix membrane proteins. Elucidation of its series of light-induced reactions will help understand not only proton transport in bacteriorhodopsin but also the analogous reactions in visual pigments, which belong to a different family, the G-protein coupled receptors involved in cellular information transfer.
Bacteriorhodopsin is one of the best known enzymes, particularly with regards to reaction mechanism. This knowledge is partly derived from the fact that the reaction can be started by light, which does not perturb samples by having to mix substrates with enzymes. Bacteriorhodopsin is one of a few proteins whose microsecond intermediates have been analyzed. A colored active center, which is formed by the retinal chromophore linked to Lys216 through a protonated Schiff base, is one of the advantages in using this protein. We can distinguish different intermediates by their distinct visible absorption spectra, and these intermediates are amenable to a variety of spectroscopic techniques. Bacteriorhodopsin and its mutants in purple membranes are easily purified and are quite stable to light and heat; its molecular size is small (246 residues). Moreover, crystallographic structures are available not only for the unphotolyzed state but also for some of the light-induced intermediates. The comparison with the chloride pump, halorhodopsin, which has a similar structure, also helps elucidate the mechanism of the proton pump of bacteriorhodopsin.
Bacteriorhodopsin transports protons from the inside of the cell to the extracellular medium by use of light energy absorbed by the retinal chromophore. The aliphatic chain of the retinal moiety lies roughly in the middle of the membrane, and conceptually divides the membrane into two domains. Those close to the extracellular surface and to the cytoplasm are called the extracellular domain and the cytoplasmic domain, respectively. The extracellular domain, which has the proton release machinery, is more abundant in polar amino acid residues and water molecules than the cytoplasmic domain. The cytoplasmic domain is mainly composed of hydrophobic residues and is more flexible than the extracellular domain [1].
In the dark the chromophore is present in an equilibrium mixture of the all-trans and 13-cis, 15-syn forms [2]. Light isomerizes the C13-C14 double bond in both forms. The species with all-trans retinal (denoted BR hereafter) undergoes a photochemical cycle, which consists of intermediates J, K, L, M, N, and O [3]. The initial state of BR is restored in the photocycle with the accompanied translocation of one proton from the cytoplasm to the extracellular medium. The retinal of these intermediates is 13-cis, except for the last intermediate, O, which again contains all-trans retinal. The other species of the dark-adapted initial mixture with 13-cis, 15-syn retinal is inactive and does not produce blue shifted intermediates like L and M. L is a crucial intermediate that leads to pumping in both bacteriorhodopsin and a related chloride pump, halorhodopsin [4].
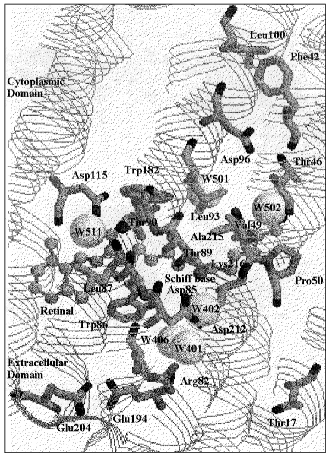
Intermediates in relation to proton pumping. The proton pumping process is composed of several steps: 1) proton transfer from the Schiff base to Asp85 in the extracellular domain in the L to M transition; 2) proton release from a complex that contains Glu194/Glu204 (proton release complex) during the lifetime of M [5-8]; 3) proton transfer from Asp96 in the cytoplasmic domain to the Schiff base in M to N; 4) proton uptake from the cytoplasm to Asp96 in N to O [9, 10]; 5) proton transfer from Asp85 to the proton release complex in O to BR [11, 12]. The locations of the residues discussed in this text are shown in Fig. 1 on the basis of the X-ray crystal structure by Luecke et al. [13], which is the highest resolution (1.55 Å) structure so far reported.
For unidirectional proton translocation, light energy stored in K is dissipated in a series of regulated steps [14], resulting in well-regulated changes in proton affinities of the Schiff base, Asp85, and Asp96 in L, M, and N. An early proposal [15] was a switching mechanism between two different M states. M1 or early M in equilibrium with L, has a connection to Asp85, while M2 or late M, in equilibrium with N, has a connection to Asp96. The unidirectional reaction between these two equilibrium states is due to a large free energy gap between them. This concept was interpreted as the local access mechanism, in which these connections flicker repeatedly during the lifetime of M [16, 17].Fig. 1. Protein residues and water molecules in the BR state built on the basis of the coordinate set in PDB 1C3W by Luecke et al. [13]. Water molecules are shown by space-filling model, and the retinal and Lys216 by ball and stick model.
Advantages of FTIR spectroscopy. Analysis of the structures of the photocycle intermediates should reveal the mechanism for proton pumping. FTIR spectroscopy is very useful in studying this photosensitive pigment as an alternative and complementary approach to the atomic level structures determined by X-ray crystallography. Infrared absorption is intensified by the polarization of a given chemical bond. Enzymatic reactions are largely mediated by changes in the polarity of chemical bonds, in the protonation states, and in the H-bonding. The FTIR technique is valuable for detecting such changes. In particular, FTIR technique is good in detecting changes in internal water molecules, in the peptide backbone (C=O and N-H), and in carboxylic acids (C=O) (reviewed in [18, 19]). All of these groups are polar and are often involved in protein function.
Using marker bands specific to particular intermediates, the difference spectra for the formation of the intermediates of K, L, M, and N from BR can be obtained from their photo-equilibrium states with BR at 80, 170, 230 and 273 K, respectively (reviewed in [18, 19]). The spectra of these intermediates at room temperature can also be extracted from a series of time-resolved spectra. Both methods gave almost the same spectra for L, M and N [20-22]. Changes of water molecules described below were detected by low temperature spectroscopy.
Water molecules around the Schiff base. Internal water molecules in general play a role in stabilizing charged groups and unsatisfied dipoles in proteins (reviewed in [23]). A structural role of the internal water molecules can be envisioned from the fact that they are highly conserved in homologous proteins. In bacteriorhodopsin, it is expected that water molecules are also involved in the proton translocation.
Proton transfer from the Schiff base to Asp85 initiates the light-driven proton pumping in bacteriorhodopsin. In BR, the Schiff base is protonated with an extraordinarily high pKa value of more than 13 [24, 25]. This value is enormously higher than the pKa value of ~7 for a protonated Schiff base in an aqueous methanol solution [26]. Asp85, the counterion, has a pKa of less than 3 [27-29]. Theoretical studies have predicted that water molecules present close to this charged pair help stabilize the separated charged groups of the Schiff base and the counterion in a hydrophobic environment [30-34].
The C=N stretching vibration band of the Schiff base at 1642 cm-1 in H2O in a resonance Raman spectrum [35] is wider than the corresponding band at 1624 cm-1 in D2O. This was attributed to energy transfer of the vibration of the Schiff base to the O-H in-plane bending vibration of water at 1635 cm-1. A neutron diffraction study subsequently detected several water molecules close to the Schiff base [36]. They were retained in the protein even at 0% humidity. Unusual chemical shift anisotropies of the Schiff base nitrogen in a 15N-solid state NMR experiment was attributed to a weak interaction with an intervening water molecule between the nitrogen and the counterion [37].
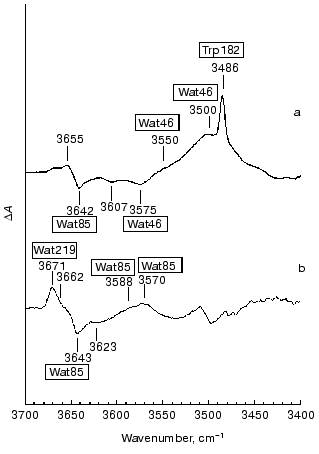
Internal water molecules detected by FTIR spectroscopy. FTIR studies can reveal structural changes in water. Maeda et al. [38] first detected unique bands in the L minus BR and M minus BR difference spectra in the 3700-3450 cm-1 region (Fig. 2). These bands, except for the 3486 cm-1 band due to the N-H stretching vibration of the indole N-H of Trp182 [39], were ascribed to water O-H stretching vibrations, because they are shifted by ~10 cm-1 toward lower frequency in H218O relative to those in normal H216O. The 3642 cm-1 band of BR disappears in the L minus BR spectrum in the blue form of D85N [40]. A similar band at 3637 cm-1 is restored in the purple form of D85N and D85T, if chloride is available as the counterion [41, 42]. The 3642 cm-1 band undergoes a shift by ~8 cm-1 toward lower frequencies in mutants of residues in the extracellular domain such as D212N [43], R82A [44], and D85N, and D85T in the presence of chloride [41, 42]. Hence, the 3642 cm-1 band is assigned to water molecules around Asp85, and they are called Wat85 in this text. Dehydration of bacteriorhodopsin changes the maximum absorbance from 570 to 530 nm [35]. In this process, a 3645 cm-1 band (probably due to Wat85) was lost or changed its state [45]. The results suggest the involvement of Wat85 in color regulation of bacteriorhodopsin.
Monomeric water molecules in a nitrogen matrix exhibit asymmetric and symmetric O-H stretching vibrations at 3725 and 3672 cm-1, respectively [46]. A model study on water molecules dissolved together with bases in an inert solvent like carbon tetrachloride showed that these frequencies decrease upon interaction with stronger bases [47]. The two O-H bonds of internal water molecules form different H-bonds with protein residues or other water molecules in the asymmetric environment in the protein. Hence the frequencies of the O-H stretching vibrations change in an unpredictable way depending on the coupling between the vibrations of the two O-H bonds. As a rule, each water molecule shows two O-H stretching vibrations and both move toward lower frequencies with increasing H-bond strength. We are unable to specifically label water molecules. However, we can plausibly infer their approximate positions by studying the FTIR spectra of mutants. In this case, we have to be cautious since mutants might affect the protein conformation at widely separated loci. However, the results predicted so far by FTIR spectroscopy analyzed by using mutants are consistent with those by subsequent X-ray studies. Some results will be described below.Fig. 2. The L minus BR (a) and M minus BR (b) difference spectra in the 3700-3450 cm-1 region. Wat85, Wat46, and Wat219 are assigned to water O-H stretching vibrations, which are affected by the D85N, T46V, and F219L, respectively. The others have not been specifically assigned.
X-Ray studies detected three water molecules around Asp85 [13, 48-53]. These water molecules, W401, W402, and W406 according to the notation by Luecke et al. [13], along with the oxygens of Asp85 and Asp212, form a pentagonal structure. Such a structure is similar to the pentagonal water cluster on the hydrophobic surfaces of proteins (reviewed in [23]). The Schiff base is H-bonded to W402.
Probably the 3642 cm-1 water O-H band of BR includes contributions from two or more water molecules around the Schiff base, as its intensity in the K minus BR spectrum is about half of that in the L minus BR spectrum [54, 55]. This 3642 cm-1 band along with other O-H stretching bands in the lower frequency region between 3600 and 2800 cm-1 were proposed to be due to this pentagonal structure [56]. Halorhodopsin exhibits a band at 3629 cm-1, which corresponds to the 3642 cm-1 band of BR [57]. The X-ray structure of halorhodopsin shows that chloride interacts directly with the Schiff base without an intervening water molecule like W402, but the two other water molecules corresponding to W401 and W406 are present [58].
Perturbations around the Schiff base in K. Absorption of light energy by the retinal chromophore causes isomerization about the C13-C14 double bond. This results in changes of the shape of the chromophore, presumably accompanied by a change in orientation of the N-H bond of the Schiff base. The extremely weak H-bonding of the Schiff base in K [59] suggests the disruption of its interaction with a water molecule. The intense C15-hydrogen out-of-plane vibration (HOOP) band at 956 cm-1 in K suggests a twisting around the terminal Schiff base region [60]. The red shift in the visible absorption spectrum indicates the disruption of the interaction of the Schiff base with the protein moiety, specifically the counterion. The initial pentagonal structure around the Schiff base is perturbed, as revealed by FTIR studies [54, 56]. A water molecule between the Schiff base and Asp85 disappeared in the X-ray structure of the K intermediate [61]. The amino acid residues linked to this cluster, Thr89, Thr90 (or Thr121) [62], and Asp115 [63] are perturbed in K.
Transition state-like structure in L. In enzyme reactions, the substrate of an enzyme first binds to the protein and forms an ES complex. In the next step, structural changes in surrounding protein residues are induced. For example in chymotrypsin, the target peptide bond in the substrate changes from a planar form to a tetrahedral form, which is induced by optimal interactions between strained substrates and strained protein residues. This tetrahedral intermediate has a higher free energy but is formed without large conformation alterations. Constraints are mainly provided by electric perturbations with local deformations. The tetrahedral intermediate is the starting structure for the catalytic reaction in chymotrypsin. The L intermediate in bacteriorhodopsin is the first intermediate in which the Schiff base forms moderately strong H-bonds with the protein just before the proton transfer from the Schiff base to Asp85, the first step in the proton pumping. FTIR studies on L detected constraints in many polar chemical bonds [64], but electron diffraction studies detected no gross conformation change [65, 66].
In BR the extremely high pKa value of the Schiff base is provided by a water-mediated electrostatic interaction with Asp85 and by geometrical interactions between the retinal and the surrounding protein moiety [67]. The H-bond of Thr89 to Asp85 also may contribute to the unprotonated state of Asp85 [68]. The rearrangement in the structure around the Schiff base upon photoisomerization disrupts a large part of these interactions. Asp85 enters into a more hydrophobic environment, and the hydration of the protonated Schiff base lowers its pKa [26]. The distorted structure of the chromophore around the Schiff base in L [59, 69-71] also is a factor in a lowing the pKa value of the Schiff base. Energy in the chromophore would be partitioned into the protein moiety in the K to L transition.
Protein and water changes in the cytoplasmic domain of L. The L minus BR FTIR spectrum in the 3700-3450 cm-1 region exhibits two features due to the O-H stretching vibrations on the positive (L) side. One is a small band at 3655 cm-1 with weak H-bonding and the other is a large, wide feature in the 3550-3450 cm-1 region (labeled at 3550 and 3500 cm-1 in Fig. 2a) with moderately strong H-bonding. The latter disappears almost completely in the T46V and V49M mutants of these cytoplasmic domain residues [72, 73]. These mutants do not affect the negative band at 3642 cm-1 of Wat85, but deplete a negative band at 3575 cm-1. Thus, this negative band in BR is one of the counterparts of the water molecules with moderately strong H-bonds in L. Water molecules responsible for these bands were named Wat46. The larger intensity of the positive band of Wat46 in L than the corresponding negative band in BR is due to a stronger H-bond, as shown by the model study [47].
The C=O stretching bands of the protonated carboxylic acids which appear in the L minus BR FTIR spectrum in the 1770-1700 cm-1 region are due to the perturbation of both Asp96 and Asp115 in L [63, 74-76]. The C=O stretching vibration frequencies of protonated carboxylic acids are increased by decreasing H-bond strength. A model study of propionic acid indicates that frequencies higher than 1745 cm-1 can be attained in non-H-bonding solvent with a low dielectric constant [77].
The C=O stretching band of Asp96 and Asp115 at 1741 and 1737 cm-1 in BR changes to 1748 and 1729 cm-1 in L [63, 75, 76], respectively. These perturbations upon the isomerization of the chromophore suggest the presence of connecting structures from the chromophore to these residues, both of which are more than 10 Å from the Schiff base. The C=O stretching band of Asp96 of L at 1748 cm-1 appeared in T46V at 1755 cm-1 [72], suggesting an H-bonding interaction between Thr46 and Asp96 in L. The same band of Asp96 also appeared in V49A at 1737 cm-1 [72], a frequency even lower than the 1742 cm-1 band for BR. These results suggest that Val49 is located at a position to interfere with the interaction between Thr46 and Asp96.
X-Ray results on BR ([13, 53], see Fig. 1) show that a water molecule (W502 or W720), which forms H-bonds to both the peptide carbonyls of Thr46 and Lys216, is present in distances of about 3.5 Å to both the methyl groups of Thr46 and Val49. The side chain of Val49 is present close to the side chain of Lys216. This is evidenced by that the Schiff base N-H in-plane bending vibration at 1312 cm-1 is affected by its mutants, V49A and V49M [73]. These results open the possibility that a change of orientation of the Schiff base induces a geometrical change in Val49 through its interaction with Lys216, and the effect is transmitted to Asp96 through W502 and Thr46. A long-range interaction from Asp85 to Val49 to Thr46 in the BR state was also suggested by 13C-solid state NMR studies [78].
Perturbations of L are not confined to these residues. An intense L band at 3486 cm-1 was assigned to the indole N-H stretching vibration of Trp182 [39, 79]. This band arises from intensification of this stretching band by the interaction of the indole moiety with the methyl moiety of the retinal. This band is not observed in the N intermediate, probably reflecting the relaxation of the distorted Schiff base in L. A bilobe with 1625(+)/1618(-) cm-1 bands was ascribed to the peptide carbonyl (amide I) of Val49 by isotope labeling of the peptide carbonyl of valine [73] and by the absence of a labeled band at these frequencies in V49A [80]. This peptide carbonyl is present close to the Schiff base-Asp85 region. The perturbations of these residues close to the Schiff base may be brought about by the distortion of the Schiff base in L.
Two structural units in L with different connections to the chromophore. K is produced by illumination of BR with yellow light at 80 K, and completely returns to the initial BR state by illumination with red light at the same temperature. On the other hand, the illumination of L at 80 K does not restore the original BR but rather forms a blue shifted pigment. L´ is a form that has an all-trans chromophore; it returns to BR only upon warming above 200 K [81, 82]. The L minus L´ FTIR spectrum was compared with the L minus BR spectrum, in order to reveal which part of the pigment returns to BR state in L´ [83]. Asp115, Wat85, and Trp182 return to the states that they had in BR. The X-ray results for the BR state ([13], see Fig. 1) show that Asp115 has two interactions, one to Thr90 and the other to W511. Thr90 is a residue next to Thr89, which connects to the Schiff base through Asp85 and W402 by H-bonds. The other partner for the interaction of Asp115 is W511, which interacts by H-bonding to the peptide C=O of Leu87, a neighboring residue of Trp86, which is next to Asp85. The restoration of the Schiff base orientation toward Asp85 and W402 in L´ may restore Asp115 through these linkages.
The L minus L´ difference spectrum does not exhibit the C=O stretching vibration of the carboxylic acid of Asp96 and the peptide carbonyl vibration of Val49, both of which are perturbed in L. This result indicates that these residues do not change in the L to L´ photoreaction [83]. The 1348 cm-1 band, due to the coupled mode for the Schiff base N-H and lysine Calpha-H bending vibrations [84], is also absent in this spectrum, indicating the persistence of the backbone conformations of Lys216 in the L to L´ transition [83]. Meanwhile, Wat46 appears as a sharp intense band at 3549 cm-1 in L´ instead of negative bands at 3607 and 3575 cm-1 in BR (see Fig. 2a). Merging of these separate bands into a single intense band at 3549 cm-1 is probably the result of the formation of a cluster by H-bonding water molecules. Thus, in L´ Wat46 stays in a different state from those in BR and L.
These results indicate that W502 is relocated in L from its initial position (between the peptide carbonyls of Thr46 and Lys216) to another place, presumably closer to the Schiff base, and forms a cluster with other water molecules. As a result, Asp96 is disconnected from the chromophore and stays in the same state even upon the isomerization of the chromophore.
Stabilization of L by internal water molecules. The equilibrium constant between L and M, which was determined from the forward and backward rate constants, is not far from unity [15]. This equilibrium is governed not only by the pKa shifts of the Schiff base and Asp85 but also by other protein changes. Conformational change in the extracellular domain makes an important contribution. The fact that the L to M process is accelerated in the mutants of E204Q [5] and R82A [44, 85, 86] suggests there are interactions between Asp85 and the proton-releasing complex in M. The pKa value of Asp85 decreases to below 10 in the M of E194Q [87].
The equilibrium is also controlled from the Val49-Thr46-Asp96 structural unit in the cytoplasmic domain (see Fig. 1 and above). The L to M transition is accelerated in T46V, but the additional mutation of D96N in T46V/D96N restores the original rate. These mutations diminish and restore the perturbation of the peptide carbonyl of Val49, and in parallel deplete and restore Wat46 for some unknown reasons [73]. The results suggest that Wat46 water molecules are involved in the stabilization of L through the perturbation of the peptide backbone of Val49. The V49A mutation, which strengthens H-bonding of Wat46 in L, decelerates M formation. Another mutant of Val49, V49M, depletes Wat46 almost completely as T46V does, while shifting the equilibrium toward L [73, 88]. In this case, strong H-bond of Asp96 in V49A may be responsible for the L to M transition. Probably either Wat46, Asp96, or both together (part of the structural unit of Lys216-Val49-W502-Thr46-Asp96) work to stabilize the protonated Schiff base in L.
Protein changes in M are different from those in L. M is an intermediate with an unprotonated Schiff base and protonated Asp85. Protonation of a carboxylic acid was first discovered as a band at 1762 cm-1 appearing in the FTIR difference spectrum upon the formation of M [89, 90]. Later the carboxylic acid was identified by using mutants at Asp85 by FTIR [63, 74, 91] and solid state NMR studies [92]. The high C=O stretching frequency of the carboxylic acid suggests that the C=O part of Asp85 is in a low dielectric environment without H-bonding [77]. The pKa value of Asp85 in M was estimated to be more than 11 [93]. In the photocycle, the protonation of Asp85 precedes proton release from the proton release complex to the extracellular medium. The resulting negative charge in this complex in turn causes stabilization of the protonated Asp85, preventing the reverse flow of the proton to the Schiff base [17]. M formation is greatly accelerated at alkaline pH because the proton release complex (pKa ~ 9) is already deprotonated even in BR [6, 7].
Only a few changes were revealed in the protein part of the cytoplasmic domain in M by FTIR spectroscopy. The C=O of Asp96 that was perturbed in L (with its stretching frequency at 1748 cm-1) largely returns to the initial state as in BR (1742 cm-1) [94], but a small fraction changes to another state (1736 cm-1) [95]. The perturbation of the peptide carbonyl of Val49 that occurred in L is extinguished in M [80]. The 3486 cm-1 band of Trp182, which resulted from its interaction with the retinal in L, reduces in intensity in M [96]. These changes are probably related to the relaxation of the distorted structure in L due to the deprotonation of the Schiff base. The carboxylic C=O of Asp115, which forms a stronger H-bond in L (1729 cm-1), loses the H-bond in M (1742 cm-1) [95], presumably as a result of the disruption of the structural unit of the Schiff base-W402-Asp85-Thr89 upon deprotonation of the Schiff base (see Fig. 1 and above).
The perturbation of the peptide carbonyl of Lys216 at 1620 cm-1, as detected in an FTIR study, was interpreted to be due to the hydration of the peptide carbonyl group with a water molecule [97]. The X-ray results show H-bonding of the peptide carbonyl of Lys216 with a water molecule (W502) in BR [13, 50, 53], which in turn forms a H-bond with the peptide carbonyl of Thr46. These two peptide carbonyl groups change relative locations in the BR to M transition [48, 50, 53].
Interaction of a water molecule with Asp85 in M. Strongly H-bonding water molecules in the cytoplasmic domain as observed in L are no longer detected in M, and water O-H bands that are influenced by the residues in the cytoplasmic domain appear only with very weak H-bonding. The positive side of the M minus BR spectrum exhibits a band at 3671 cm-1 with its shoulder at 3662 cm-1 and a wide feature which is composed of two bands at 3588 and 3570 cm-1 (Fig. 2b) [80, 94]. All of these bands are shifted in H218O and thus are due to water O-H stretching vibrations of M.
M is produced by illumination at 230 K by yellow light, and can be reversed to BR by blue light at the same temperature. However, photo-isomerization of the chromophore back to the all-trans form with blue light at lower temperatures does not lead to complete reversal back to the initial BR state [81, 82, 98]. These low temperature photoproducts of M are collectively named M´. Their structures differ depending on the temperature at which M was illuminated. When M is illuminated at 80 and 100 K, even the reprotonation of the Schiff base does not occur, and Wat85 preserves its stronger H-bonds (water bands at 3588 and 3570 cm-1) as in M [94]. The reprotonation occurs by half at 133 K, but without the restoration of Wat85. The complete reprotonation of the Schiff base at 173 K is still accompanied by incomplete restoration of Wat85 (to 3640 vs. 3643 cm-1 in BR). These results suggest that the reprotonation of the Schiff base from Asp85 may occur via a different mechanism than the initial deprotonation process, which causes a change in an intervening Wat85 molecule.
In the photoreaction of M at 80 K, one of the Wat85 molecules comprising part of the 3570 cm-1 band (Fig. 2b) changes its frequency to 3550 cm-1, and Asp85 also undergoes the perturbation from 1762 cm-1 in the M state to 1754 cm-1. Both perturbations of Wat85 and Asp85 relax in parallel upon increasing temperature to 100 and 133 K. The results suggest that the O-H stretch band of water at 3570 cm-1 is due to a water molecule that is interacting with Asp85 in M.
The X-ray results show that, among three resolved water molecules around Asp85 in BR, two are invisible in M, either diffusing away from this site or being disordered but in a similar position [50, 53]. These changes may create an apolar environment around Asp85 and the Schiff base, stabilizing the protonated form of Asp85 and the unprotonated form of the Schiff base. The other water molecule remains interacting with the C-OH part of Asp85 and stabilizes Asp85 in a protonated state. The 3570 cm-1 water molecule may correspond to this water molecule (W401). On the other hand, the C=O part of protonated Asp85, which is responsible for the 1762 cm-1 band of Asp85, is free from water and present in an environment with a low dielectric constant [77]. The stabilization of protonated Asp85 with an accompanied rearrangement of water molecules around it drives the proton movement from the Schiff base to Asp85 in the L to M process.
Different interactions of water molecules in the cytoplasmic domain in L, M, and N. In a variety of mutants of the cytoplasmic domain the M minus BR spectra show that the 3671 cm-1 band is depleted in F219L [80]. Water molecules responsible for this band are called Wat219 in this text. The N minus BR spectrum (not shown in figures) shows a 3654 cm-1 band instead of the 3671 cm-1 band due to Wat219 on M. These bands of M and N similarly decrease their intensities in V49M and F219L. Hence, the same water molecule is responsible for the 3671 cm-1 band of M and the 3654 cm-1 band of N [80].
The 3671 cm-1 band of M shifted to 3666 cm-1 in T46V, while the 3654 cm-1 band of N did not. In contrast, the V49A mutation shifted the 3654 cm-1 band of N, but not the 3671 cm-1 band of M. Thus, the Wat219 molecule moves closer to Val49 in N relative to M. In the M to N transition, the 1618 cm-1 band of the peptide carbonyl of Val49 increases intensity, and the imide II band due to the Val49-Pro50 peptide amide at 1418 cm-1 changes to 1432 cm-1. These changes around the peptide bond of Val49 could be induced by the interaction with the protonated Schiff base of N, probably through a Wat219 molecule. These interactions may be one of the factors helping to stabilize the protonated Schiff base of N. In MN (see below) the 3671 cm-1 band due to Wat219 shifted toward a lower frequency at 3660 cm-1, though not as large as in N. The lower frequency shift of Wat219 in N is not the result of the protonation of the Schiff base, but due to the protein changes characteristic of N [80].
In the M to M´ reactions at 133 and 173 K, Wat219 molecules return to the initial states they had in BR [94]. However, at 80 K a Wat219 molecule responsible for the 3671 cm-1 band (Fig. 2b) is not restored to its initial state and changes its O-H stretching frequency only slightly to 3662 cm-1. Also the return of the other Wat219 molecule responsible for the 3662 cm-1 band is inhibited in V49A, a mutant of the cytoplasmic residue close to the Schiff base. These inhibitions were weakened at 100 K and almost removed at 133 K. Such temperature dependent perturbation was also observed for Asp96. The C=O stretching vibration of the protonated Asp96 is located at 1742 cm-1 in M and change to 1748 cm-1 upon the photo-isomerization of the retinal at 80 K. This perturbation is diminished by elevating the temperature to 100 K. Thus, the perturbations of Asp96 and Wat219 that occur in the M to M´ reaction at 80 K reverse in parallel upon elevating the temperatures to 100-133 K, similarly to the perturbations of Asp85 and Wat85 described above. These results suggest that the photoreaction of M at 80 K produces an unstable structure around Schiff base that accommodates a water molecule relocating from the cytoplasmic domain.
The X-ray results [13, 53] show that, in M one or two water molecules are present in the region between Trp182, peptide carbonyls (C=O) of Ala215 and Phe219, and three mutually interacting water molecules in the region surrounded by Thr46, Asp96, Ala215 and the peptide carbonyl (C=O) of Lys216. These two groups are separated by the peptide carbonyl of Ala215. Each site that accommodates only one water molecule in BR (Fig. 1) expands in M to allow more water molecules. The L intermediate shows many changes in electrostatic interactions as detected by FTIR spectra, but no gross structural changes as assayed from electron diffraction experiments [65, 66]. This electrostatic perturbation in L is replaced in M by a conformation change with accompanied enlargements of cavities, which allow the accommodation of more water molecules as polar residues in an otherwise apolar environment. An electron diffraction study [99] showed that the conformational change in M is restricted to the cytoplasmic domain. A H-bonding network in the cytoplasmic domain involving water molecules was suggested from the effects of mutations on the photocycle [88]. Such an extended water-containing structure may play a role in stabilizing protonated Asp96 in M.
No water molecules directly interacting with the Schiff base were detected in M by X-ray studies. The closest water molecule (W704) is located at 5.5 Å apart from the Schiff base [53]. This water molecule is located relatively far (3.5 Å) from the closest potential H-bond partner, the peptide carbonyl of Ala215, and orients its O-H roughly parallel to the membrane plane [53]. This may correspond to the weak H-bonding of Wat219, whose O-H bond is responsible for the 3671 cm-1 band and is oriented roughly parallel to the membrane plane [100]. The X-ray work on the wild type revealed another water molecule (W740) in the region surrounded only by the hydrophobic residues between Phe42 and Leu100, outside of the pair of Thr46, Asp96, and Phe219. This water molecule does not form any H-bonds [53].
Structure changes of N in the cytoplasmic domain.The M to N transition is caused by a proton transfer from Asp96 to the Schiff base. This could result from a decrease in the pKa value of Asp96 to ~7 [22] along with an increase in the pKa value of the Schiff base. The N minus BR FTIR spectrum exhibits a negative band at 1742 cm-1 and a corresponding positive band at 1402 cm-1 due to the deprotonation of Asp96 [69, 76, 101]. In D212N, the deprotonation of Asp96 can occur without going through the M state. In other words, Asp96 can deprotonate even in the absence of the deprotonated Schiff base as a proton acceptor [102, 103]. Thus, the cause of the decrease in the pKa value of Asp96 could be explained as an issue between L and N, rather than one between M and N. The pKa value of the Schiff base is very high in N, as envisaged from the fact that N persists at pH 10, in contrast to L, which decays more rapidly at alkaline pH than at neutral pH. The Schiff base region of the chromophore in N is not distorted as it is in L, as revealed by FTIR [69] and 15N-solid sate NMR [70] studies. The relaxed form of the Schiff base could be one of the reasons for the high pKa value of the Schiff base in N. A state like N with a neutral Asn85 residue, anionic Asp96, and protonated Schiff base can be created in the F42C/D85N mutant even in the dark. Its retinal takes a 13-cis, 15-trans configuration like N. These facts suggest that in N the light energy is completely transferred to the protein [104].
Other changes in the M to N process, or differences of N from L, are detected in the peptide backbone as revealed by changes of 1692 and 1672 cm-1 amide I bands of BR (in place of the 1658 cm-1 band of BR in the conversion to L and M) to 1647 cm-1 in N and the appearance of amide II band at 1548 cm-1 [69, 105-107]. Part of the changes in the 1672 cm-1 band was attributed to the peptide carbonyl of Tyr185 [108].
One noticeable change in N relative to M is the perturbation of Asp85, whose C=O stretching vibration is altered from 1762 cm-1 in M to 1754 cm-1 in N [69, 101]. A similar downshift of Asp85 from 1761 to 1754 cm-1 occurred in the M to M´ transition at 80 K [94], which is supposed to be due to rearrangement of water molecules around Asp85 with the accompanied relocation of Wat219 toward the Schiff base. The changes of the C=O stretching frequency of Asp85 in the M to N transition might result from the relocation of water molecules carrying a proton (hydronium ion) from the cytoplasmic domain toward a site close to Asp85 [94]. A similar idea was presented by Sass et al. [53]. This further suggests that even the proton transfer from the Schiff base to Asp85 in the L to M process is carried out by relocation of a water molecule with an associated proton (hydronium ion). Molecular dynamics calculations on an artificial ion channel [109] suggest that internal water molecules can be relocated between different loci in a hydrophobic pore with a very small kinetic barrier.
MN as N with an unprotonated Schiff base. It is known that the D96N mutant forms an MN state at alkaline pH, in which changes in protein part of the pigment (Asp85, amide I and amide II) are as in N, but the Schiff base remains unprotonated as in M [105]. Asn96 exhibits a large depletion peak due to BR at 1704 cm-1, instead of the 1742 cm-1 band seen upon deprotonation of Asp96 in N of the wild type. MN is produced as a photosteady state only at alkaline pH values for the purple membrane [105], and is present together with M, as N is also found with M in the wild type [69]. MN is probably an alkaline form of N with the deprotonation of the Schiff base. The pKa value to the Schiff base of D96N was determined to be 8.3 [110]. This low pKa value of the Schiff base in D96N might be a result of the absence of the negative charge at position 96.
MN is produced even at neutral pH from glucose-embedded wild type purple membrane [111] and from wild type membrane in 2 M guanidine-hydrochloride [112]. These are, however, clearly perturbed conditions. X-Ray diffraction studies [113] have shown that MN is also similar to N, which has its global conformation changes mainly in the F and G helices and definitely is different from those of M, which has its changes in the B and G helices [114]. The atomic structure of M was determined for the D96N mutant, and this form of M was proposed to be MN [48]. The atomic structure of MN for D96N [48] is different from that of M determined for the wild type [53] and for E204Q [50]. The decrease in the distance between Phe219 and Trp182 is compensated for by an increase in the distance between Ala215 and Trp182 in the M of D96N relative to the M of the wild type or E204Q. This perturbation may cause the relocalization of water molecules from the positions in M closer to the Schiff base. This MN does not exhibit the large movement of helix F expected from electron diffraction studies on the purple membranes [115]. It has been argued that such a large movement is inhibited in the crystals [116].
Similarity of rhodopsin to bacteriorhodopsin. The visual pigment rhodopsin is a transmembrane seven-helix protein like bacteriorhodopsin, and the chromophore, 11-cis retinal, is linked through a protonated Schiff base to a lysine residue (Lys296) in the seventh helix. Light causes the isomerization of the cis C11-C12 bond to the trans form. The cytoplasmic domain near the active site is highly hydrophobic, like bacteriorhodopsin. The difference FTIR spectra for the formation of a series of intermediates, bathorhodopsin (Batho), lumirhodopsin (Lumi), metarhodopsin I (Meta I) and metarhodopsin II (Meta II) were obtained in the photoequilibrium states by illumination of rhodopsin (Rho) at 80, 200, 240 and 290 K [117-122], respectively, for hydrated films of bovine rhodopsin. The Schiff base is deprotonated in Meta II, but the unprotonated Schiff base is not a prerequisite for Meta II binding transducin. Meta II-like states of the protein structure similar to Meta II but with a protonated Schiff base were observed in the E113Q/A117E mutant (123) and in a transient state for the binding of transducin [124].
Rhodopsin is not a proton pump, but at some pH values up to two protons are taken up from the cytoplasm into the protein after the formation of Meta II [125, 126]. As a reflection of this, at acidic pH values the equilibrium between Meta I and Meta II favors Meta II with its unprotonated Schiff base [122, 127]. Meta II is known to interact with transducin [128] and catalyzes the release of GDP from transducin and allows the binding of GTP [129]; the now activated transducin can subsequently activate a phosphodiesterase cascade ([130], reviewed in [131]). It is known that Meta II is not formed in dehydrated rhodopsin [132, 133], similar to M not converting to N in dehydrated bacteriorhodopsin [35]. One interesting question is to address how water is involved in the formation of Meta II.
Water molecules around the Schiff base of rhodopsin.H-Bonding changes in internal water molecules of rhodopsin were observed in the difference FTIR spectra of several bleaching intermediates at low temperature. Extensive studies were done at 80 K, where one can use the photoreactions between Rho, Batho, and isorhodopsin (Iso) to interconvert them [120]. The Batho minus Rho spectrum exhibits three bilobes, 3573(-)/3591(+) cm-1 (Wat-3), 3564(-)/3542(+) cm-1 (Wat-2), and 3538(-)/3525(+) cm-1 (Wat-1) [134]. The change of Wat-1 was extinguished in E113Q, in which the counterion of Glu113 was replaced by a chloride [134]. The E113Q mutation also abolishes several changes associated with the peptide backbone, suggesting that the water molecule is also interacting with several peptide carbonyls. It is known that the Schiff base forms a strong H-bond in Rho and this H-bond does not change in strength upon formation of Batho [135]. These results suggest that a water molecule is present between Glu113 and the protonated Schiff base in the wild type and they move together in the isomerization event. This is in contrast to the BR to K transition of bacteriorhodopsin, in which the Schiff base seems to be separated from the water molecule after isomerization. The Wat-1 band was not detected in the spectra between Rho and Lumi or Meta I, indicating that Wat-1 returns to its initial state interacting with Glu113 in these intermediates [136]. The E113Q mutant in the presence of chloride shows a bilobe with a positive composite band between 3040 and 2880 cm-1 and with a similar negative band between 2880 and 2740 cm-1. This pair was assigned tentatively to the N-H+ stretching vibration of the protonated Schiff base, and changes H-bonding only in the absence of Wat-1 [134].
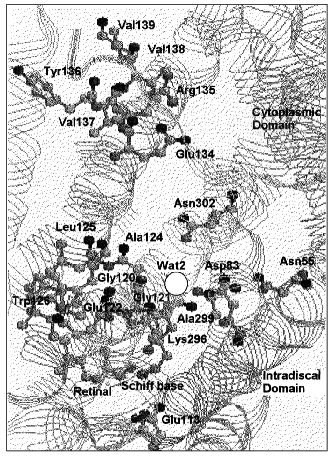
The presence of a water molecule close to the Schiff base was also suggested by the rate of deuteron exchange of the Schiff base [137]. Solid state NMR studies show a larger distance between the Schiff base and the counterion than that expected for their direct interaction [138, 139]. However, a recent X-ray structure (Fig. 3) does not show enough of a gap to allow a water molecule between the Schiff base and Glu113 [140]. Wat-1 may be present in another position close to this pair.
Water molecules in the cytoplasmic domain of rhodopsin. Wat-2 is affected by the mutation of Asp83 and Gly120. These two residues are close to each other since the carboxylic acid C=O stretching vibration of Asp83 is affected by the G120A mutation and replacement of Asp83 or Gly120 affects the same peptide carbonyl groups. Wat-2 is also affected by E113Q. Thus, Wat-2 is present between Asp83 and Gly120 and is connected to Glu113 via the peptide backbone [141]. Wat-2 undergoes perturbations upon formation of Lumi and Meta I [136]. Upon formation of Meta II another water molecule with much weaker H-bonds (3661 and 3615 cm-1) undergoes H-bonding changes with the accompanying appearance of large positive bands at 3575 and 3540 cm-1. These bands obscure possible negative bands of Wat-1 and Wat-2 in the Meta II minus Rho spectrum.Fig. 3. Protein residues and a water molecule in the unphotolyzed state of bovine rhodopsin based on the coordinate set in PDB 1F88 by Palczewski et al. [140].
FTIR studies on the HOOP bands of the retinal chromophore have shown that the perturbation around C12-H of retinal in Batho, which causes a large decrease in the frequency of the C12-HOOP [142], is removed in Lumi and Meta I, giving rise to a coupled mode for the C11-HOOP and C12-HOOP bands at 947 and 950 cm-1, respectively [143]. The fact that a C14-HOOP band in C12-deuterated rhodopsin was observed at 883 cm-1 in Meta I but not in Lumi was interpreted as being due to a geometrical strain between C14-H and N-H of the Schiff base in Lumi. This strain relaxes in Meta I. Lumi resembles the L intermediate of bacteriorhodopsin in that both have a distorted Schiff base. Lumi is then transformed to an equilibrium mixture of Meta I (protonated Schiff base) and Meta II (unprotonated Schiff base), as L is transformed to a mixture of the M (unprotonated Schiff base) and N (protonated Schiff base) intermediates of bacteriorhodopsin. Dehydrated rhodopsin, which retains Wat-1 but not Wat-2 [136], stays in a Meta I-like state and does not form Meta II. In the Meta I-like state, the strain of the Schiff base in Lumi was preserved. Thus, Wat-2 helps relax the strain around the Schiff base.
Wat-2 is present in a nonpolar region in the cytoplasmic domain, similar to the Wat46 or Wat219 water molecule of bacteriorhodopsin. The X-ray structure of rhodopsin revealed a water molecule between Asp83 and Gly120 ([140], see Fig. 3). Asp83 is protonated in Rho and throughout all the photo-intermediates as well. The C=O stretching vibration of Asp83 at 1767 cm-1 shows its presence in a quite hydrophobic environment with a dielectric constant of ~2 [77]. The environment of the C=O part of Asp83 becomes more influenced by the electric field in Meta II, as revealed by the shift of the C=O stretching band to 1746 cm-1 [144]. This could be the result of water molecules approaching this region upon Meta II formation [136].
Asp83 of rhodopsin is similar to Asp96 of bacteriorhodopsin with respect to its position in the cytoplasmic domain. Asp83 of rhodopsin is a residue conserved in most G-protein coupled receptors [145]. The carboxyl oxygen of Asp83 in the B helix also forms a H-bond with the peptide carbonyl of Asn55 in A helix and of Ala299 in G-helix, connecting these different helices [140]. The D83N mutant, which affects Wat-2, is known to form Meta II even in the absence of transducin at alkaline pH, where the wild type does not form Meta II [146]. This uncoupling is similar to the G-protein-uncoupled agonist binding [147]. Thus, Asp83 comprises an agonist-binding site in rhodopsin. It is also known that Ser124 in place of Ala124 in rhodopsin comprises a sodium binding site in many G-protein coupled receptors [145], suggesting the feasibility of accommodating a positive electric charge in this region for rhodopsin. This could be one of the sites that takes up a proton as a hydronium cation when Meta II is formed. The Glu134/Arg135 pair, which is separated from the cytoplasm by hydrophobic residues, is thought to be the site where a proton is taken up from the cytoplasm upon Meta II formation [148]. Protonated water molecules could be translocated from the cytoplasm to a binding site close to Asp83 through a hydrophobic channel in the cytoplasmic domain. This idea should be clarified in future experiments.
Water as a mobile mediator of the function. A central part of the proton pumping of bacteriorhodopsin consists of the proton transfer from the protonated Schiff base to unprotonated Asp85 in the L to M transition, and then proton transfer from protonated Asp96 to the unprotonated Schiff base in the M to N transition. H-bonding interactions with water molecules stabilize the protonated Schiff base in L, protonated Asp85 and Asp96 in M, and again the protonated Schiff base in N. The water molecule could be involved as mobile polar groups by their H-bonding interactions. Proton transfer may be carried out with relocation of water molecules even in the absence of the H-bonding networks, by use of affinity difference of the proton between two residues. A similar role for internal water molecules is suggested in activating rhodopsin after light absorption.
The author is thankful to Tom Ebrey, Sergei Balashov, and Farol Tomson for their critical suggestions.
REFERENCES
1.Réat, V., Patzelt, H., Ferrand, M., and
Pfister, C. (1998) Proc. Natl. Acad. Sci. USA, 95,
4970-4975.
2.Harbison, G. S., Herzfeld, J., and Griffin, R. G.
(1983) Biochemistry, 22, 1-4.
3.Lozier, R. H., Bogomolni, R. A., and Stoeckenius,
W. (1975) Biophys. J., 15, 955-962.
4.Lanyi, J. K., Duschl, A., Váró, G., and
Zimányi, L. (1990) FEBS Lett., 265, 1-6.
5.Brown, L. S., Sasaki, J., Kandori, H., Maeda, A.,
Needleman, R., and Lanyi, J. K. (1995) J. Biol. Chem.,
270, 27122-27126.
6.Richter, H.-T., Brown, L. S., Needleman, R., and
Lanyi, J. K. (1996) Biochemistry, 35, 4054-4062.
7.Balashov, S. P., Imasheva, E. S., Ebrey, T. G.,
Chen, N., Menick, D. R., and Crouch, R. K. (1997) Biochemistry,
36, 8671-8676.
8.Dioumaev, A. K., Richter, H. T., Brown, L. S.,
Tanio, M., Tuzi, S., Saitô, H., Kimura, Y., Needleman, R., and
Lanyi, J. K. (1998) Biochemistry, 37, 2496-2506.
9.Zimányi, L., Cao, Y., Needleman, R.,
Ottolenghi, M., and Lanyi, J. K. (1993) Biochemistry, 32,
7669-7678.
10.Cao, Y., Brown, L. S., Needleman, R., and Lanyi,
J. K. (1993) Biochemistry, 32, 10239-10248.
11.Kandori, H., Yamazaki, Y., Hatanaka, M.,
Needleman, R., Brown, L. S., Richter, H.-T., Lanyi, J. K., and Maeda,
A. (1997) Biochemistry, 36, 5134-5141.
12.Balashov, S. P., Lu, M., Imasheva, E. S.,
Govindjee, R., Ebrey, T. G., Othersen, B., III, Chen, Y., Crouch, R.
K., and Menick, D. R. (1999) Biochemistry, 38,
2026-2039.
13.Luecke, H., Schobert, B., Richter, H.-T.,
Cartailler, J.-P., and Lanyi, J. K. (1999) J. Mol. Biol.,
291, 899-911.
14.Váró, G., and Lanyi, J. K. (1991)
Biochemistry, 30, 5016-5022.
15.Váró, G., and Lanyi, J. K. (1991)
Biochemistry, 30, 5008-5015.
16.Brown, L. S., Dioumaev, A. K., Needleman, R., and
Lanyi, J. K. (1998) Biochemistry, 37, 3982-3993.
17.Brown, L. S., Dioumaev, A. K., Needleman, R., and
Lanyi, J. K. (1998) Biophys. J., 75, 1455-1465.
18.Maeda, A. (1995) Isr. J. Chem.,
353, 387-400.
19.Kandori, H. (2000) Biochim. Biophys. Acta,
1460, 177-191.
20.Heâling, B., Souvignir, G., and Gerwert, K.
(1993) Biophys. J., 65, 1929-1941.
21.Rödig, C., Chizhov, I., Weidlich, O., and
Siebert, F. (1999) Biophys. J., 76, 2687-2701.
22.Zscherp, C., and Heberle, J. (1997) J. Phys.
Chem., 101B, 10542-10547.
23.Teeter, M. M. (1991) Annu. Rev. Biophys.
Biophys. Chem., 20, 577-600.
24.Druckmann, S., Ottolenghi, M., Pande, A., Pande,
J., and Callender, R. H. (1982) Biochemistry, 21,
4953-4959.
25.Sheves, M., Albeck, A., Friedman, N., and
Ottolenghi, M. (1986) Proc. Natl. Acad. Sci. USA, 83,
3262-3266.
26.Baasov, T., and Sheves, M. (1986)
Biochemistry, 25, 5249-5258.
27.Jonas, R., and Ebrey, T. G. (1991) Proc. Natl.
Acad. Sci. USA, 88, 149-153.
28.Balashov, S. P., Imasheva, E. S., Govindjee, R.,
and Ebrey, T. G. (1996) Biophys. J., 70, 473-481.
29.Balashov, S. P., Imasheva, E. S., Govindjee, R.,
Sheves, M., and Ebrey, T. G. (1996) Biophys. J., 71,
1973-1984.
30.Scheiner, S., and Duan, X. (1991) Biophys.
J., 60, 874-883.
31.Beppu, Y., Kakitani, T., and Tokunaga, F. (1992)
Photochem. Photobiol., 56, 1113-1117.
32.Humphrey, W., Lugonov, I., Schulten, K., and
Sheves, M. (1994) Biochemistry, 33, 3668-3678.
33.Harosi, F. I., and Sandorfy, C. (1995)
Photochem. Photobiol., 61, 510-517.
34.Murata, K., Fujii, Y., Enomoto, N., Hata, M.,
Hoshino, T., and Tsuda, M. (2000) Biophys. J., 79,
982-991.
35.Hildebrandt, P., and Stockburger, M. (1984)
Biochemistry, 23, 5539-5548.
36.Papadopoulos, G., Dencher, N. A., Zaccai, G., and
Bült, G. (1990) J. Mol. Biol., 214, 15-19.
37.De Groot, H. J. M., Harbison, G. S., Herzfeld,
J., and Griffin, R. G. (1989) Biochemistry, 28,
3346-3353.
38.Maeda, A., Sasaki, J., Shichida, Y., and
Yoshizawa, T. (1992) Biochemistry, 31, 462-467.
39.Yamazaki, Y., Sasaki, J., Hatanaka, M., Kandori,
H., Maeda, A., Needleman, R., Shinada, T., Yoshihara, K., Brown, L. S.,
and Lanyi, J. K. (1995) Biochemistry, 34, 577-582.
40.Maeda, A., Sasaki, J., Yamazaki, Y., Needleman,
R., and Lanyi, J. K. (1994) Biochemistry, 33,
1713-1717.
41.Sasaki, J., Brown, L. S., Chon, Y.-S., Kandori,
H., Maeda, A., Needleman, R., and Lanyi, J. K. (1995) Science,
269, 73-75.
42.Chon, Y.-S., Sasaki, J., Kandori, H., Brown, L.
S., Lanyi, J. K., Needleman, R., and Maeda, A. (1996)
Biochemistry, 35, 14244-14250.
43.Kandori, H., Yamazaki, Y., Sasaki, J., Needleman,
R., Lanyi, J. K., and Maeda, A. (1995) J. Am. Chem. Soc.,
117, 2118-2119.
44.Hatanaka, M., Sasaki, J., Kandori, H., Ebrey, T.
G., Needleman, R., Lanyi, J. K., and Maeda, A. (1996)
Biochemistry, 35, 6308-6312.
45.Renthal, R., Gracia, N., and Regalado, R. (2000)
Photochem. Photobiol., 72, 714-718.
46.Van Thiel, M., Becker, E. D., and Pimentel, G. C.
(1957) J. Chem. Phys., 27, 486-490.
47.Mohr, S. C., Wilk, W. D., and Barrow, G. M.
(1965) J. Am. Chem. Soc., 87, 3048-3052.
48.Luecke, H., Richter, H.-T., and Lanyi, J. K.
(1998) Science, 280, 1934-1937.
49.Luecke, H., Schobert, B., Richter, H.-T.,
Cartailler, J.-P., and Lanyi, J. K. (1999) Science, 286,
255-260.
50.Luecke, H., Schobert, B., Cartailler, J.-P.,
Richter, H.-T., Rosengarth, A., Needleman, R., and Lanyi, J. K. (2000)
J. Mol. Biol., 300, 1237-1255.
51.Belrhali, H., Nollert, P., Royant, A., Menzel,
C., Rosenbusch, J. P., Landau, E. M., and Pebay-Peyroula, E. (1999)
Structure, 7, 909-917.
52.Sato, H., Takeda, K., Tani, K., Hino, T., Okada,
T., Nakasako, M., Kamiya, N., and Kouyama, T. (1999) Acta
Crystallogr., D55, 1251-1256.
53.Sass, H. J., Büldt, G., Gessenich, R., Hehn,
D., Neff, D., Schlesinger, R., Berendzen, J., and Ormos, P. (2000)
Nature, 406, 649-653.
54.Fischer, W. B., Sonar, S., Marti, T., Khorana, H.
G., and Rothschild, R. J. (1994) Biochemistry, 33,
12757-12762.
55.Hatanaka, M., Kashima, R., Kandori, H., Friedman,
N., Sheves, M., Needleman, R., Lanyi, J. K., and Maeda, A. (1997)
Biochemistry, 36, 5493-5498.
56.Kandori, H., and Shichida, Y. (2000) J. Am.
Chem. Soc., 122, 11745-11746.
57.Chon, Y.-S., Kandori, H., Sasaki, J., Lanyi, J.
K., Needleman, R., and Maeda, A. (1999) Biochemistry, 29,
9449-9455.
58.Kolbe, M., Besir, H., Essen, L.-O., and
Oesterhelt, D. (2000) Science, 288, 1390-1396.
59.Maeda, A., Sasaki, J., Pfefferlé, J.-M.,
Shichida, Y., and Yoshizawa, T. (1991) Photochem. Photobiol.,
54, 911-921.
60.Weidlich, O., Uji, L., Jäger, F., and
Atkinson, G. H. (1997) Biophys. J., 72, 2329-2341.
61.Edman, K., Nollert, P., Royant, A., Belrhali, H.,
Pebay-Peyroula, E., Hajdu, J., Neutze, R., and Landau, E. M. (1999)
Nature, 401, 822-826.
62.Kandori, H., Kinoshita, M., Yamazaki, Y., Maeda,
A., Shichida, Y., Needleman, R., Lanyi, J. K., Bizounok, M., Herzfeld,
J., Raap, J., and Lugtenburg, J. (2000) Proc. Natl. Acad. Sci.
USA, 97, 4643-4648.
63.Braiman, M. S., Mogi, T., Marti, T., Stern, L.
S., Khorana, H. G., and Rothschild, K. J. (1988) Biochemistry,
27, 8516-8520.
64.Maeda, A., Kandori, H., Yamazaki, Y., Nishimura,
S., Hatanaka, M., Chon, Y.-S., Sasaki, J., Needleman, R., and Lanyi, J.
K. (1997) J. Biochem., 121, 399-406.
65.Hendrickson, F. M., Buckard, F., and Glaeser, R.
M. (1998) Biophys. J., 75, 1446-1454.
66.Subramaniam, S., Lindahl, M., Bullough, P.,
Faruqi, A. R., Tittor, J., Oesterhelt, D., Brown, L., Lanyi, J., and
Henderson, R. (1999) J. Mol. Biol., 287, 145-161.
67.Rousso, I., Friedman, N., Sheves, M., and
Ottolenghi, M. (1995) Biochemistry, 34, 12059-12065.
68.Russell, T. S., Coleman, M., Rath, P., Nilsson,
A., and Rothschild, K. J. (1997) Biochemistry, 36,
7490-7497.
69.Pfefferlé, J.-M., Maeda, A., Sasaki, J.,
and Yoshizawa, T. (1991) Biochemistry, 30, 6548-6556.
70.Hu, T. G., Griffin, R. G., and Herzfeld, J.
(1997) Biochemistry, 36,9316-9322.
71.Griffiths, J. M., Bennett, A. E., Engelhard, M.,
Siebert, F., Raap, J., Lugtenburg, J., Herzfeld, J., and Griffin, R. G.
(2000) Biochemistry, 39, 362-371.
72.Yamazaki, Y., Hatanaka, M., Kandori, H., Sasaki,
J., Karstens, W., Raap, J., Lugtenburg, J., Bizounok, M., Herzfeld, J.,
Needleman, R., Lanyi, J. K., and Maeda, A. (1995) Biochemistry,
34, 7088-7093.
73.Yamazaki, Y., Tuzi, S., Saitô, H., Kandori,
H., Needleman, R., Lanyi, J. K., and Maeda, A. (1996)
Biochemistry, 35, 4063-4068.
74.Engelhard, M., Gerwert, K., Hess, B., Kreutz, W.,
and Siebert, F. (1985) Biochemistry, 24, 400-407.
75.Gerwert, K., Hess, B., Soppa, J., and Oesterhelt,
D. (1989) Proc. Natl. Acad. Sci. USA, 86, 4943-4947.
76.Maeda, A., Sasaki, J., Shichida, Y., Yoshizawa,
T., Chang, M., Ni, B., Needleman, R., and Lanyi, J. K. (1992)
Biochemistry, 31, 4684-4690.
77.Dioumaev, A. K., and Braiman, M. S. (1995) J.
Am. Chem. Soc., 117, 10572-10574.
78.Tanio, M., Inoue, S., Yokota, K., Seki, T., Tuzi,
S., Needleman, R., Lanyi, J. K., Naito, A., and Saitô, H. (1999)
Biophys. J., 77, 431-442.
79.Maeda, A., Sasaki, J., Ohkita, Y. J., Simpson,
M., and Herzfeld, J. (1992) Biochemistry, 31,
12543-12545.
80.Yamazaki, Y., Kandori, H., Needleman, R., Lanyi,
J. K., and Maeda, A. (1998) Biochemistry, 37,
1559-1564.
81.Litvin, F. F., and Balashov, S. P. (1977)
Biofizika, 22, 1111-1114.
82.Hurley, J. B., Becher, B., and Ebrey, T. G.
(1978) Nature, 272, 87-88.
83.Maeda, A., Tomson, F. L., Gennis, R. B., Ebrey,
T. G., and Balashov, S. P. (1999) Biochemistry, 38,
8800-8807.
84.Gat, Y., Grossjean, M., Pinevsky, I., Takei, H.,
Rothman, Z., Sigrist, H., Lewis, A., and Sheves, M. (1992) Proc.
Natl. Acad. Sci. USA, 89, 2434-2438.
85.Balashov, S. P., Govindjee, R., Kono, M.,
Imasheva, E. S., Lukashev, E., Ebrey, T. G., Crouch, R. K., Menick, D.
R., and Feng, Y. (1993) Biochemistry, 32,
10331-10343.
86.Balashov, S. P., Govindjee, R., Imasheva, E. S.,
Misra, S., Ebrey, T. G., Feng, Y., Crouch, R. K., and Menick, D. R.
(1995) Biochemistry, 34, 8820-8834.
87.Lazarova, T., Sanz, C., Querol, E., and Padros,
E. (2000) Biophys. J., 78, 2022-2030.
88.Brown, L. S., Yamazaki, Y., Maeda, A., Sun, L.,
Needleman, R., and Lanyi, J. K. (1994) J. Mol. Biol.,
239, 401-414.
89.Rothschild, K. J., Zagaeski, M., and Canter, W.
A. (1981) Biochem. Biophys. Res. Commun., 103,
483-489.
90.Siebert, F., M äntele, W., and Kreutz,
W. (1982) FEBS Lett., 141, 82-87.
91.Fahmy, K., Weidlich, O., Engelhard, M., Tittor,
J., Oesterhelt, D., and Siebert, F. (1992) Photochem.
Photobiol., 56, 1073-1083.
92.Metz, G., Siebert, F., and Engelhard, M. (1992)
FEBS Lett., 303, 237-241.
93.Braiman, M. S., Dioumaev, A. K., and Lewis, J. R.
(1996) Biophys. J., 70, 939-947.
94.Maeda, A., Tomson, F. L., Gennis, R. B., Kandori,
H., Ebrey, T. G., and Balashov, S. P. (2000) Biochemistry,
39, 10154-10162.
95.Sasaki, J., Lanyi, J. K., Needleman, R.,
Yoshizawa, T., and Maeda, A. (1994) Biochemistry, 33,
3178-3184.
96.Liu, X., Lee, M. J., Coleman, M., Rath, P.,
Nilsson, A., Fischer, W. B., Bizounok, M., Herzfeld, J., Karstens, W.
F., Raap, J., Lugtenburg, J., and Rothschild, K. J. (1998) Biochim.
Biophys. Acta, 1365, 363-372.
97.Takei, H., Gat, Y., Rothman, Z., Lewis, A., and
Sheves, M. (1994) J. Biol. Chem., 269, 7387-7389.
98.Takei, H., Gat, Y., Sheves, M., and Lewis, A.
(1992) Biophys. J., 63, 1643-1653.
99.Subramaniam, S., Gerstein, M., Oesterhelt, D.,
and Henderson, R. (1993) EMBO J., 12, 1-8.
100.Hatanaka, M., Kandori, H., and Maeda, A. (1997)
Biophys. J., 73, 1001-1006.
101.Bousché, O., Braiman, M. S., He, Y.-W.,
Marti, T., Khorana, H. G., and Rothschild, K. J. (1991) J. Biol.
Chem., 266, 11063-11067.
102.Needleman, R., Chang, M., Ni, B.,
Váró, G., Fornes, J., White, S. H., and Lanyi, J. K. (1991)
J. Biol. Chem., 266, 11478-11484.
103.Cao, Y., Váró, G., Klinger, D. M.,
Czajkowsky, M. S., Braiman, M. S., Needleman, R., and Lanyi, J. K.
(1993) Biochemistry, 32, 1981-1990.
104.Dioumaev, A. K., Brown, L. S., Needleman, R.,
and Lanyi, J. K. (1999) Biochemistry, 38,
10070-10078.
105.Sasaki, J., Shichida, Y., Lanyi, J. K., and
Maeda, A. (1992) J. Biol. Chem., 267, 20782-20786.
106.Ormos, P., Chu, K., and Mourant, J. (1992)
Biochemistry, 31, 6933-6937.
107.Lazarova, T., and Padros, E. (1996)
Biochemistry, 35, 8354-8358.
108.Ludlam, C. F. C., Sonar, S., Lee, C.-P.,
Coleman, M., Herzfeld, J., RajBhandary, U. L., and Rothschild, K. J.
(1995) Biochemistry, 34, 2-6.
109.Qi, Z., and Sokabe, M. (1998) Biophys.
Chem., 71, 35-50.
110.Brown, L. S., and Lanyi, J. K. (1996) Proc.
Natl. Acad. Sci. USA, 93, 1731-1734.
111.Vonck, J., Han, B.-G., Burkhard, F., Perkins,
G. A., and Glaeser, R. M. (1994) Biophys. J., 67,
1173-1178.
112.Sass, H. J., Schachowa, I. W., Rapp, G., Koch,
M. H. J., Oesterhelt, D., Dencher, N. A., and Büldt, G. (1997)
EMBO J., 16, 1484-1491.
113.Kamikubo, H., Kataoka, M., Váró,
G., Oka, T., Tokunaga, F., Needleman, R., and Lanyi, J. K. (1996)
Proc. Natl. Acad. Sci. USA, 93, 1386-1390.
114.Oka, T., Yagi, N., Fujisawa, T., Kamikubo, H.,
Tokunaga, F., and Kataoka, M. (2000) Proc. Natl. Acad. Sci. USA,
97, 14278-14282.
115.Vonck, J. (1996) Biochemistry,
35, 5870-5878.
116.Subramaniam, S., and Henderson, R. (2000)
Nature, 406, 653-657.
117.Siebert, F., M äntele, W., and
Gerwert, K. (1983) Eur. J. Biochem., 136, 119-127.
118.Ganter, U. M., Gärtner, W., and Siebert,
F. (1988) Biochemistry, 27, 7480-7488.
119.Maeda, A., Ohkita, Y. J., Sasaki, J., Shichida,
Y., and Yoshizawa, T. (1993) Biochemistry, 32,
12033-12038.
120.Kandori, H., and Maeda, A. (1995)
Biochemistry, 34, 14220-14229.
121.Rath, P., DeLange, F., DeGrip, W. J., and
Rothschild, K. J. (1998) Biochem. J., 329, 713-717.
122.Bartl, F., Ritter, E., and Hofmann, K. P.
(2000) FEBS Lett., 473, 259-264.
123.Fahmy, K., Siebert, F., and Sakmar, T. P.
(1994) Biochemistry, 33, 13700-13705.
124.Nishimura, S., Kandori, H., and Maeda, A.
(1998) Biochemistry, 37, 15816-15824.
125.Arnis, S., and Hofmann, K. P. (1993) Proc.
Natl. Acad. Sci. USA, 90, 7849-7853.
126.Szundi, I., Mah, T. L., Lewis, J. W.,
Jäger, S., Ernst, O. P., Hofmann, K. P., and Kliger, D. S. (1997)
Biochemistry, 37, 14237-14244.
127.Mathews, R. G., Hubbard, R., Brown, P. K., and
Wald, G. (1963) J. Gen. Physiol., 47, 215-240.
128.Emeis, D., Kühn, H., Reichert, J., and
Hofmann, K. P. (1982) FEBS Lett., 143, 29-34.
129.Bornancin, F., Pfister, C., and Chabre, M.
(1989) Eur. J. Biochem., 184, 687-698.
130.Fukada, Y., and Yoshizawa, T. (1981)
Biochim. Biophys. Acta, 675, 195-200.
131.Helmreich, E. J. M., and Hofmann, K. P. (1996)
Biochim. Biophys. Acta, 1286, 285-322.
132.Wald, G., Durrel, J., and St. Goerge, R. C. C.
(1950) Science, 111, 179-181.
133.Nishimura, S., Sasaki, J., Kandori, H.,
Lugtenburg, J., and Maeda, A. (1995) Biochemistry, 34,
16758-16763.
134.Nagata, T., Terakita, A., Kandori, H., Kojima,
D., Shichida, Y., and Maeda, A. (1997) Biochemistry, 36,
6164-6170.
135.Mathies, R. A., Smith, S. O., and Palings, I.
(1987) in Biological Applications of Raman Spectroscopy (Spiro,
T. G., ed.) Vol. 2, John Wiley & Sons, New York, pp. 59-109.
136.Nishimura, S., Kandori, H., and Maeda, A.
(1997) Photochem. Photobiol., 66, 796-801.
137.Deng, H., Huang, L., Callender, R. H., and
Ebrey, T. G. (1994) Biophys. J., 66, 1129-1136.
138.Eilers, M., Reeves, P. J., Ying, W., Khorana,
H. G., and Smith, S. O. (1999) Proc. Natl. Acad. Sci. USA,
96, 487-492.
139.Creemer, A. F. L., Klaasen, C. H. W.,
Bovee-Geurtz, P. H. M., Kelle, R., Kragl, U., Raap, J., DeGrip, W. J.,
Lugtenburg, J., and DeGroot, H. J. M. (1999) Biochemistry,
38, 7195-7199.
140.Palczewski, K., Kumasaka, T., Hori, T., Behnke,
C. A., Motoshima, H., Fox, B. A., le Trong, I., Teller, D. C., Okada,
T., Stenkamp, R. E., Yamamoto, M., and Miyano, M. (2000)
Science, 289, 739-745.
141.Nagata, T., Terakita, A., Kandori, H.,
Shichida, Y., and Maeda, A. (1998) Biochemistry, 37,
15815-15824.
142.Palings, I., van den Berg, E. M. M.,
Lugtenburg, J., and Mathies, R. A. (1989) Biochemistry,
28, 1498-1507.
143.Ohkita, Y. J., Sasaki, J., Maeda, A.,
Yoshizawa, T., Groesbeek, M., Verdegem, P., and Lugtenburg, J. (1995)
Biophys. Chem., 56, 71-78.
144.Fahmy, K., Jäger, F., Beck, M., Zyvaga, T.
A., Sakmar, T. P., and Siebert, F. (1993) Proc. Natl. Acad. Sci.
USA, 90, 10206-10210.
145.Pogozheva, I. D., Lomize, A. L., and Mosberg,
H. I. (1997) Biophys. J., 70, 1963-1985.
146.Weitz, C. J., and Nathans, J. (1993)
Biochemistry, 32, 14176-14182.
147.Fraser, C. M., Chung, F.-Z., Wang, C.-D., and
Venter, J. C. (1988) Proc. Natl. Acad. Sci. USA, 85,
5478-5482.
148.Arnis, S., Fahmy, K., Hofmann, K. P., and
Sakmar, T. P. (1994) J. Biol. Chem., 269,
23879-23881.