FTIR Emission Spectra of Bacteriorhodopsin in a Vibrational Excited State
E. L. Terpugov* and O. V. Degtyareva
Institute of Cell Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142290 Russia; fax: +7 (0967) 79-0509; E-mail: terpugov@ipc.psn.ru* To whom correspondence should be addressed.
Received April 10, 2001; Revision received July 6, 2001
Vibrational IR-emission spectra of bacteriorhodopsin (bR) were recorded under continuous illumination with visible light at room temperature. They contain selective information about the chromophore, Schiff base, and opsin. The spectral bands were identified by comparing the data with resonance Raman and IR absorption data. The IR-emission spectra were shown to contain a set of bands characteristic for both all-trans (bR568) and 13-cis conformations (K610-like intermediate) simultaneously. Variation of spectral composition and the intensity of visible light illumination influenced the spectral traces and intensity distribution between them. Greater intensity of deformational vibrations suggests distorted retinal structure in the vibrationally excited ground electronic state. The origin of the emitting species of bR is discussed.
KEY WORDS: FTIR-spectroscopy, IR-emission, vibrationally excited state, bacteriorhodopsin, K610-like intermediate
The membrane protein bacteriorhodopsin (bR) contains retinal as its chromophore. In light-adapted bR, retinal is in the all-trans form covalently linked to the apoprotein at Lys216 by a protonated Schiff base. Photon absorption isomerizes the retinal into the 13-cis-configuration. This primary event is responsible for both transmembrane proton transfer and subsequent re-isomerization of retinal (see for review [1-3]). These two processes are characterized by high quantum and thermodynamic efficiency [4]; thus, bR is an attractive model for structure-functional studies and for studies of solar energy transformation in biological systems.
The functioning of bR is a cyclic process that is characterized by sequential conversion of spectrally identified intermediates (J, K, L, M, N, and O); the exact number of intermediates and the pathways of their interconversion remains unclear. Primary light-induced stages are usually described by the following chain of events [5]: bR570~bR*~J625-->K610. They are characterized by accumulation of absorbed energy for subsequent transmembrane proton transfer. These stages include the changes in retinal and the protein and the changes in the interaction between the chromophore and the apoprotein. Spectral changes revealed that the K610 intermediate is the first stable photointermediate with 13-cis-like configuration of the retinal [6], but the final structure of this photointermediate has not yet been identified. In spite of recent intensive studies, our understanding of the photo-dynamics of the primary processes still remains incomplete. Perhaps this situation reflects the lack of adequate methods for such studies.
Vibrational spectroscopy (resonance Raman spectroscopy) and differential Fourier transformed infrared (FTIR) spectroscopy are the most popular methods employed for the molecular study of the structure and properties of bR. However, due to high contribution of water and protein backbone to bR absorption, it is very difficult to obtain the vibrational IR-spectrum of the chromophore complementary to the Raman spectrum. The use of IR-emission spectroscopy would overcome these difficulties; however, we do not know of results of such studies available in the literature.
Infrared emission spectroscopy has great potential. Due to the development of highly sensitive Fourier transform infrared techniques, it is widely used in physics and chemistry [7]. Until recently, it had limited applicability in biology because sample preparation required heating up to 100°C and above [8, 9]. Recently, an approach was developed that allows the use of FTIR emission spectroscopy under normal conditions. With this approach the IR-emission is excited by non-monochromatic visible light [10, 11]. It was shown that the FTIR technique is applicable for registration of the vibrational IR emission spectra of the bR chromophore in situ [11].
In the present work, we continue these studies; we were particularly interested in the study of the effect of excitation conditions (spectral composition and intensity of light) on vibrational IR-spectra of bR emission.
MATERIALS AND METHODS
Bacteriorhodopsin in purple membranes (PM) was isolated from the halophilic bacterium Halobacterium salinarum (strain ET-1001) using the standard method [12]. Purple membrane films were prepared using concentrated aqueous suspension, which was layered onto a KRS-5 plate (Carl Zeiss Yena, Germany) and dried under air at room temperature and ~60% humidity for a few hours. The films were characterized by absorbance of 0.6 at 568 nm.
Infrared vibrational spectra were recorded using an FS-02 double-beam double-channel Fourier infrared spectrometer equipped with a low temperature MCT detector with germanium window on the cryostat. This spectrometer was described earlier [13, 14]. It incorporates the original variant of the Michelson interferometer, which employs a scheme for optical compensation proposed by Genzel and Kuhl [15]. This scheme provides 5-10-fold increased sensitivity compared with a usual single-beam spectrometer. This is achieved by optical exclusion of noise caused by drift of the emission source, fluctuations of the interference contrast, unstable sensitivity of the detector, and medium scatter. The spectrometer can collect emission on both detectors or on either one of them. Emission spectra were recorded with one detector. Over the whole working range (400-4000 cm-1) the instrumental resolution of the spectrometer is 1 cm-1, but for better spectral reproducibility, we chose the resolution of 4 cm-1. For the recording of one interferogram the signal/noise ratio was 500. The duration of one scan was 12 sec. Each spectrum was recorded with accumulation of 600 scans. The resulting spectra were characterized by good reproducibility under the same experimental conditions.
For emission experiments, we modified the optical scheme of the single-beam spectrometer as described in [10]. Briefly, the standard source of infrared emission was removed and replaced by a wet bacteriorhodopsin film; the location of the Michelson interferometer was between the sample and detector (this is typical for emission measurements [9]). Such modification was possible due to the other characteristic feature of this spectrometer: in contrast to most commercial devices, the emission source is outside the interferometer. The film was continuously illuminated by a 100 W xenon lamp using a series of broadband glass filters. During sample illumination, visible light fell onto the sample normal to its surface and was focused within a spot of 2-mm diameter using a short focal length glass lens. The sample emission was collected by a spherical mirror and directed via the interferometer to the detector (Fig. 1). The geometry of illumination prevented direct incidence of visible light onto the detector.
All experiments were carried out at room temperature and ambient atmospheric pressure and the film was constantly bathed with humid air during the experiment.Fig. 1. Principal scheme of the device used for excitation of emission: 1) xenon lamp (100 W); 2) short focal length glass lens; 3) broadband glass filters; 4) sample (PM film); 5) spherical mirror; 6) Michelson interferometer.
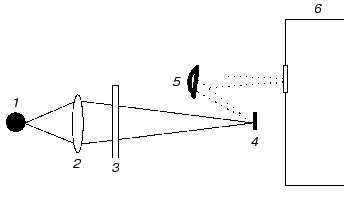
Under these conditions, background emission of the black body was negligible. However, in some experiments recorded spectra were corrected for background emission that was recorded under identical conditions but without the sample.
RESULTS
Dependence of emission spectra on spectral composition and intensity of illumination. Emission spectra were highly reproducible when they were recorded under the same experimental conditions. However, changes of illumination conditions significantly influences the spectral behavior (Fig. 2). There was marked intensity redistribution between separate bands. Changes in the set of bands or location of their maxima could also occur. The spectral composition changed over a wide range. In one series of experiments, we sequentially extended the spectral interval of actinic light to shorter wavelengths (from 550 to 360 nm). In another, we used a limited spectral interval in the region from 400 to 500 nm using various combinations of glass filters. In a third series of experiments, we changed the light intensity at constant spectral composition.
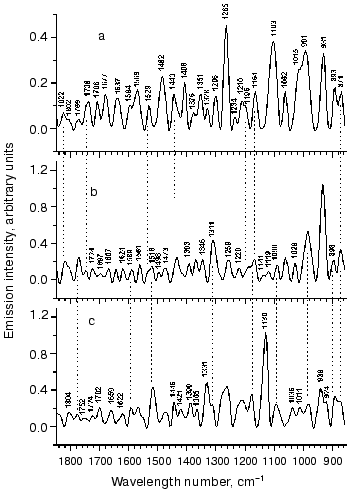
Figure 2 shows typical IR-emission spectra of bR in the middle infrared region recorded under various conditions. The upper spectrum was recorded during illumination with the widest part of the spectrum (>360 nm) without colored filters at 500 mW/cm2. The middle spectrum was recorded at 320 mW/cm2 but using another spectral composition, which was achieved using a broadband colored filter cutting the spectral part below 550 nm. The lower spectrum was recorded with illumination of the same spectral composition of actinic light as in the case of the upper spectrum, but with reduced (~2-fold) intensity. The latter was achieved using of a modulator operating with a frequency of light flow interruption of 100 Hz.
The spectra in Fig. 2 contain many resolved bands. For their identification we used resonance Raman and adsorption IR-spectroscopy data. We failed to find any corresponding spectral changes under these conditions. However, it was noted that the emission spectra contained bands with frequencies 1529 and/or 1516 cm-1 in the ethylenic C=C stretching region (Fig. 2, a-c).Fig. 2. Vibrational emission bR spectrum in the middle IR-range. The upper spectrum (a) was recorded at 500 mW/cm2 using a colorless glass filter (>360 nm). It contains a band of C=C vibrations with maximum at 1529 cm-1. The middle spectrum (b) was recorded at the illumination of 320 mW/cm2 and wavelength >550 nm. The extension of the line of C=C vibrations and the appearance of a “shoulder” at 1516 cm-1 suggests the presence of a K610-like intermediate in the mixture with bR568. The lower spectrum (c) was recorded at the illumination with the same spectral composition of acting light as in the case of upper spectrum (a), but with reduced (~2-fold) intensity (260 mW/cm2). (The latter was achieved using a light modulator with modulation frequency of 100 Hz.) A single band at 1516 cm-1 suggests the presence of a K610-like intermediate.
Chromophore vibrations in emission spectra. The recorded spectra include the range from 850 to 1850 cm-1 that is characterized by the presence of the most intense bands. Spectral lines were also found in the regions below 850 and above 1850 cm-1 including the working range up to 4000 cm-1; they were weak (or poorly identified) and therefore we leave them out of consideration here.
The region between 850 and 1680 cm-1 contains information about the retinal chromophore. Besides bands attributed to C=C stretching vibrations and located between 1500 and 1580 cm-1, this region also contains chromophore bands (above 1600 cm-1) attributed to vibrations of the aldimine (C=NH) group of the Schiff base. C-C and C-C-H stretching vibrations are localized in the fingerprint region (between 1100 and 1400 cm-1). Frequencies of all types of deformational vibrations are located in the region below 1000 cm-1.
As stated above, IR-emission spectra (Fig. 2, a-c) contain bands at 1529 and/or 1516 cm-1 in the region of C=C stretching vibrations. Frequencies of these vibrations may characterize a certain intermediate since they are in linear relationship with the maxima of electronic absorbance bands [16]. These bands coincide with vibrations of the main form of bR568 (nuC=C = 1530 cm-1) and K610-intermediate (nuC=C = 1518 cm-1) in Raman spectra [6, 16-19]. However, they may also be attributed to N (lambdamax = 560 nm) and O (lambdamax = 640 nm) intermediates possessing similar frequencies nuC=C 1532 and 1521 cm-1, respectively [19]. Let us consider which retinal states and bR intermediates may account for vibrational frequencies in the emission spectra.
It should be noted that the characteristic band of C=C stretching vibrations in the emission spectra has relatively low intensity compared with Raman spectra, where these bands are the most pronounced bands in the spectra. We cannot explain this discrepancy now. Perhaps it will be possible in the future on the basis of an understanding of the physical mechanism of the origin of the emission line intensity.
The region between 1100 and 1400 cm-1 is the characteristic fingerprint region. It is especially sensitive to changes in the configuration of the C=C double bonds of the polyenic chain. The light adapted form (bR568) and photocycle intermediates are characterized by sets of lines in their Raman spectra. Many lines are also found in all emission spectra (Fig. 2). In spectra (a) and (b) (see Fig. 2) there are groups of 10 bands (1103, 1164, 1196, 1210, 1234, 1265, 1296, 1328, 1351 and 1376 cm-1) and (1119, 1141, 1164, 1196, 1220, 1259, 1311, 1346, 1376 and 1393 cm-1), respectively. In spectrum (c) there are fewer bands (1130, 1164, 1207 with a shoulder at 1220, 1259, 1311, 1331, 1365 and 1390 cm-1). All these bands can be identified by comparing them with resonance Raman and IR-absorption data, which were recently supplemented with data obtained by kinetic FTIR-difference spectroscopy [20, 21].
Analysis of bands observed in the fingerprint region revealed that many bands are consistent with vibrations of bR568 and a K610-like intermediate. For example, the position of six maxima (of 10) of the bands at 1164, 1196, 1210, 1328, 1351 and 1376 cm-1 is consistent with vibrational frequencies 1169, 1201, 1214, 1330, 1348 and 1378 cm-1 of the bR568 form in the Raman spectrum [6, 18]. A group of bands with maxima at 1164, 1194, 1210, 1234, 1265, 1296, and 1351 cm-1 almost completely coincides with corresponding bands at 1164, 1194, 1207, 1239, 1267, 1298, and 1348 cm-1, characterizing primary intermediates J625 and/or K610 [6, 18-20, 23-25]. However, some of these bands, such as 1164, 1234, 1328, and 1376 cm-1 do overlap with corresponding bands (1169, 1234, 1328, and 1378 cm-1) of the dark-adapted form, bR548 (with 13-cis-chromophore) [22]. Frequencies 1164, 1194, and 1328 cm-1 were also found in Raman spectra of the O640-intermediate [26], which contains the chromophore in all-trans-configuration. Thus, it is impossible to identify a certain intermediate by using a set of bands because bR and its intermediates share many of them. Identification of bR forms also requires information on the intensity of bands and their relative distribution. Unfortunately, we do not have the possibility to analyze the intensity of bands of emission spectra. We can only conclude that the emission may be attributed to the chromophore simultaneously resembling all-trans and 13-cis conformation.
The spectrum shown at Fig. 2b contains a different set of bands; there were only three bands (1164, 1346, and 1376 cm-1), which were also observed at the same position of maxima as in Fig. 2a. The distribution of the intensity of these lines differs from the previous one. There are several bands characteristic for bR568 and the K610-intermediate. For example, bands 1119, 1141, 1164, 1220, 1259, 1348, and 1376 cm-1 well correspond to the vibration frequencies at 1122, 1135, 1169, 1216, 1255, 1348, and 1378 cm-1 of bR568. Vibration frequencies at 1164, 1196, 1348, 1378, and 1393 cm-1 may be also detected in Raman spectra and differential IR-absorbance spectra of the K-intermediate [23-26].
The spectrum shown at Fig. 2c was recorded under illumination with the same spectral composition of actinic light as in the case of the upper spectrum, but with reduced intensity. The latter did not cause proportional reduction of the intensity of emission lines. This spectrum was characterized by qualitative changes: redistribution of the intensity between spectral lines and the change of the spectral line set. For example, in the low frequency region there was only one line at 1130 cm-1, whereas the spectrum in Fig. 2a contained several bands: at 931, 991, 1103, and 1265 cm-1. Interestingly, the 1516 cm-1 band appeared instead of the 1529 cm-1 band (at Fig. 2a). This band (1516 cm-1) is characteristic for C=C vibrations of the chromophore of the K610-intermediate; together with the other bands (1164, 1207, 1259, 1331 and 1390 cm-1) it is typical for K-intermediate vibrations. However, this region also contains vibrations (e.g., bands 1164, 1207, 1220, 1259, 1271, 1311, and 1331 cm-1) that may be attributed to the chromophore in bR568. It is possible that these changes were induced by the modulator. The latter employed instead of attenuating filters (for reduction of the intensity of actinic light) could influence the photodynamic equilibrium reached under these conditions.
Let us consider the region between 1000 and 1100 cm-1; in Raman spectra and IR-absorbance spectra this region is characterized by one strong band at ~1010 cm-1. This vibration is attributed to torsion vibration of methyl groups. It is active in resonance Raman combinational light scattering and IR and is present in all retinal isoforms and in the chromophore of all bR forms. Other vibrations in this region are very weak and can be determined only theoretically. According to calculations, (non-planar) vibrations of methyl groups have zero intensity in the region between 1020 and 1040 cm-1 [26]. The other type of nonplanar vibrations of methyl groups located between 1040 and 1060 cm-1 also has very low intensity in resonance Raman combinational light scattering and IR. However, emission spectra are characterized by strong lines in this region. For example, in Fig. 2a these are bands at 1015 cm-1 (overlapping with a band at 991 cm-1), 1062, and 1103 cm-1. The latter is the strongest band in this emission spectrum.
In Fig. 2b there are three bands within this region: 1029, 1062, and 1090 cm-1; they are characterized by rather low but equal intensity. This part of the spectrum differs from that shown in Fig. 2a. Nevertheless, it contains some bands, which are less intensive than in Fig. 2a. These are complex bands (doublet 1011 and 1036 cm-1 with the shoulder at ~1062 cm-1) and a single (asymmetric at the low frequency side) band with maximum at 1090 cm-1. These bands exhibit equal intensity.
We suggest that the chromophore emission band at 1103 cm-1 may correspond to vibration (1111 cm-1) of the all-trans-isomer of retinal. The other emission band (1062 cm-1) (Fig. 2b) may correspond to vibration (1060 cm-1) of the 13-cis isomer of retinal. Both bands were found in IR-absorbance spectra of retinal isomers [26].
It is possible that two other bands (1029 and 1130 cm-1) can be attributed to vibrations of the beta-ionone ring. The band at 1130 cm-1 is one of the strongest in the spectrum (Fig. 2c). It can be decomposed into two bands of 1123 and 1134 cm-1, which coincide with vibration frequencies of the beta-ionone ring. These vibrations were shown to be IR-active [27].
The region below 1000 cm-1 (where deformational vibrations are localized) contains several bands, including the strongest ones. The comparison with literature data revealed that all vibrations observed in the region between 1000 and 850 cm-1 can be attributed to certain components. They almost completely coincide with vibrations that were observed in direct experiments (e.g., in Raman spectrum of bR568) or obtained on the basis of theoretical considerations [27]. (As a rule, Raman spectra of bR568 or other intermediates possessing planar configuration in this region is characterized by weak lines. According to theoretical calculations the actual number of lines should be greater [27].) These include the following bands: 871, 893, 931 and 991 cm-1 in spectrum (a); 871, 898, 931 cm-1, and a wide band with maximum at ~985 cm-1 possessing a shoulder at 1003 cm-1 in (b); 871 cm-1 and two complex bands, 989 cm-1 (with shoulder at 924 cm-1) and 939 cm-1 (with shoulder at 985 cm-1) in spectrum (c).
The doublet (871 and 893 cm-1) was found in all spectra; the 871 cm-1 band always occupies the same position, whereas the second may be shifted (e.g., to higher frequencies by 5 cm-1 as shown in Fig. 2, b and c). The frequency at 871 cm-1 is consistent with the frequency at 869 cm-1, and the frequency at 893 cm-1 may correspond to frequencies at 890 or 898 cm-1. All these vibrations were calculated for the bR568 chromophore [18].
We have already mentioned that bands of C=C stretching vibrations attributed to vibrations of conjugated double bonds are located between 1500 and 1580 cm-1. In all retinal isomers (with rare exception) as in all bR forms this region contains one characteristic band which is attributed to one double bond, C13=C14. However, the polyene chain of retinal contains five double bonds. Other bands may contribute to the intensity of this main band; they may also be responsible for the appearance of weak bands in Raman spectra of geometric retinal isomers [27] or native bR [18-22]. For example, the Raman spectrum of bR568 is characterized by three resolved bands at 1527, 1581, and 1600 cm-1. Decomposition of the 1527 cm-1 band revealed two other bands, at 1533 and 1550 cm-1 [27].
BR548 has two resolved bands, at 1536 and 1599 cm-1. Decomposition of the band at 1536 cm-1 resulted in the appearance of less intensive bands at 1515, 1550, and 1570 cm-1. Vibrations of this region in the emission spectra are given in Fig. 3 (a-c) with an expanded scale.
Figure 3a shows fragments of Fig. 2a. Besides the characteristic band at 1529 cm-1, this region contains two bands at 1569 and 1594 cm-1. They can be attributed to bR568 and bR548, respectively. In the spectrum of all-trans retinal similar vibrations at 1569 and 1594 cm-1 were found. These suggest that the bands (1569 and 1594 cm-1) may be explained by the chromophore vibration in bR568 and bR548.Fig. 3. Illustration of some parts of spectral regions of Fig. 2 (with expanded scale) attributed to the opsin part of the molecule. a-c) Vibration region of Amide I and Amide II; d-f) the region between 1400 and 1500 cm-1; a, d) fragments of Fig. 2a; b, e) fragments of Fig. 2b; c, f) fragments of Fig. 2c.
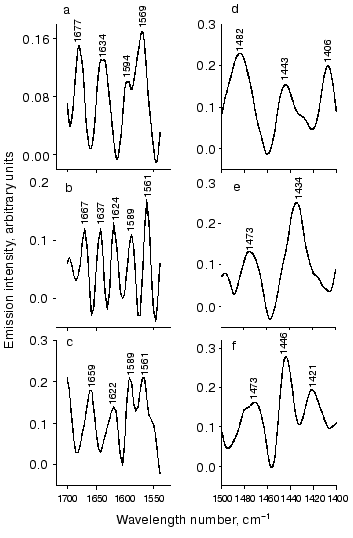
The spectrum in Fig. 3b (which is an enlarged fragment of Fig. 2b) also contains two bands (1561 and 1589 cm-1) that are shifted to lower frequencies compared to Fig. 3a by 8 and 6 cm-1, respectively. The position of these maxima is also close to vibration frequencies of the chromophore. However, they may also overlap with the Amide II band located in the same region [28].
The spectrum in Fig. 3c (which is an enlarged fragment of Fig. 2c) also contains two bands that coincide with the position of maxima shown in Fig. 3b. However, in contrast to Fig. 3b these bands merge together and form a doublet. The 1561 cm-1 band can be recognized together with a shoulder at ~1549 cm-1, which may correspond to the vibration (at 1550 cm-1) of the bR548 chromophore [18].
It is generally accepted that high frequency chromophore vibrations are manifested around 1640 cm-1. Vibrations of the protonated Schiff base C=NH+ were observed at 1642 cm-1. During deprotonation this band shifts to 1624 cm-1 [16-18]. According to literature data, vibrations in this region and with the same frequency may be attributed to vibration of the opsin part of the molecule [29].
The number of bands in the emission spectra depends on excitation conditions (Fig. 3, a-c). For example, the spectrum in Fig. 3a contains two equally intensive bands at 1637 and 1677 cm-1. The latter coincides with band 1677 cm-1 in the Raman spectrum of all-trans retinal [27]. The band at 1637 cm-1 is consistent with the vibration frequency 1639 cm-1 of the bR568 Raman spectrum [18, 22]. However, it is also possible that this band represent the sum of two overlapping bands, 1642 and 1624 cm-1 (Fig. 3b). The first of these can be attributed to Schiff base vibration of bR568 [16-18], whereas the latter can be attributed to vibrations of the C=NH group of K610 [24]. It is also possible that these bands have protein origin (see above). The last spectrum shown at Fig. 3c contains one complex band with a maximum at 1622 cm-1, which may include higher-frequency vibrations.
Vibrations of the opsin part of the bR molecule in the emission spectrum. Vibrations arising from the opsin part of the molecule can be recognized in the region of chromophore vibration at frequencies close to those of the C=C and C=NH vibrations. However, protein bands may also be present in other parts of the spectral range (see below).
Vibrations related to Amide I might be present in the region between 1620 and 1690 cm-1. Various bands were observed in this region in the emission spectra (Fig. 3, a and b). For example, besides the 1637 cm-1 band the spectrum of Fig. 3a contains another band at 1677 cm-1. The latter is consistent with the amide band at 1680 cm-1 [29]. In addition to the vibrations considered above, the spectrum of Fig. 3b also contains bands at 1667 and 1697 cm-1 that can be attributed to Amide I [29]. There is a distinct band in Fig. 3c (1659 cm-1) that is consistent with Amide I vibrations at 1658 cm-1 [29].
Let us consider the region between 1400 and 1500 cm-1, which is shown on an expanded scale in Fig. 3 (d-f). This region is less studied because chromophore vibrations are inactive in resonance Raman combinational light scattering.
The emission spectrum of this region is characterized by quite intense vibrations. Instead of one (at 1446 cm-1) or two (at 1446 and 1401 cm-1) bands seen in IR-absorbance spectra of all-trans and 13-cis-retinal, respectively [27], the emission spectra contain several bands. However, only a few of them can be identified. For example, two bands at 1406 and 1443 cm-1 are close to those observed in retinal (see above). They also overlap with vibrations (1400 and 1454 cm-1) from the opsin part of bR [30]. Other emission bands, e.g., 1421 and 1435 cm-1 can also be identified as protein bands. They coincide with vibrations that were observed in FTIR-spectra at 1418 and 1437 cm-1 and were attributed to vibrations of proline residues (Pro50 and/or Pro91 or Pro186) [30]. It is possible that the emission bands at 1482 cm-1 (Fig. 3, d and f), 1473, and 1496 cm-1 (Fig. 3e) are also due to the protein.
Now let us consider the region above 1680 cm-1. It was shown that side chains of aspartate residues may be responsible for vibrations in this region; four Asp residues were identified: Asp85, Asp96, Asp115, and Asp212 [31-33]. Emission spectra of this region contain more bands than FTIR-difference spectra. For example, there are bands at 1706, 1738, 1769, 1802, and 1822 cm-1 in the emission spectrum (Fig. 2a). According to differential IR-Fourier spectroscopy data, two of them (1738 and 1769 cm-1) can be attributed to Asp115 or Asp212 and Asp85, respectively (two others were not attributed to particular components). The intensity of these bands may be comparable with the intensity observed in the C=C and C=NH vibrations (e.g., the intensity of bands at 1706 and 1738 cm-1).
A similar set of bands (with the exception of the band at 1724 cm-1), 1738, 1769, 1822 with shoulder 1802 cm-1 was observed in the spectrum shown in Fig. 2b. However, in contrast to the previous spectrum there was a different distribution of the intensities of these bands. Under these conditions higher-frequency bands (1769 and asymmetric 1822 cm-1) demonstrated higher intensity. The spectrum recorded under different conditions of illumination (Fig. 2c) also differed from previous ones in this part. Thus, emission spectra contain information about vibrations of the opsin part of the bR molecule. Variations of illumination conditions significantly influence the protein and chromophore bands.
DISCUSSION
The major goal of this study was the development of FTIR-emission spectroscopy for monitoring changes in a pigment chromophore structure in an excited vibrational state. The applicability of the approach was tested using the photocycling molecule, bR. No previous studies have employed this method, which differs from another highly sensitive FTIR-technique based on thermally stimulated emission (see for review [7] and references given there).
With our method, infrared emission of the bR molecule is excited by non-monochromatic light of the visible range. In contrast to thermally stimulated emission employing heating (above 100°C and more), our method requires only normal conditions, room temperature and atmospheric pressure. The sample can be in the form of a film (dry or wet) or in solution, including aqueous solutions. In contrast to background absorbance in adsorption measurements, background emission of water or steam has a minor influence, and this is especially important for experimentation with biological samples.
IR-emission spectra of bR contain selective information about the retinal chromophore in situ. Very often it has bands possessing weak intensity in Raman spectra (or predicted by theoretical considerations).
Besides bands attributed to the retinal chromophore, IR-emission spectra also have bands which can be attributed to the protein part of the bR molecule, its particular groups or vibrations of Amide I and Amide II.
The structure of the retinal chromophore in the excited (vibrational) state resembles all-trans and 13-cis configurations simultaneously. This situation can occur if the excitation of retinal is accompanied by distortion of its structure resulting in an intermediate configuration (between all-trans and 13-cis). The existence of such intermediate configuration is supported by the spectral data of Fig. 2a, where spectral bands (in the region of the hydrogen out-of plane wag vibrations) were especially intense. The presence of numerous protein bands in the emission spectra suggests strong interaction between the retinal and the apoprotein in the excited state. This is consistent with results of theoretical studies indicating that electrostatic and steric interactions control both early and late stages of the photocycle [34].
Emission states of bR. The problem of the origin of vibrational states emitting in the IR-region is rather complex; therefore, we shall consider it within a framework that allows the interpretation of the data presented here.
When the initial bR568 absorbs a quantum of visible light, 65% of bR molecules are converted to the K-intermediate (within 2-3 psec) and 35% of bR molecules return to the initial state (bR568) [4]. The energy of electron excitement is partially (in the case of K-intermediate formation) or completely (when the pigment returns to the initial state, bR568) dissipated in vibrational sublevels of the ground electron state.
A negligible portion of absorbed energy is re-emitted via direct transformation from the first singlet (S1) state into the ground state (S0) with emission of a visible quantum. This is the case of fluorescent molecules. BR is characterized by low quantum efficiency (from 10-4 to 2*10-5 [35, 36]).
Some molecules may enter the photocycle (the cycle of dark-adapted bR548 [37]) unrelated with transmembrane proton transfer. Red light may induce transition of bR568 into the dark-adapted state, bR548. Reduction of humidity potentiates this transition [38].
Each of these bR states can emit IR-quanta, but their relative contribution to the emission will be determined by the ratio between these forms under given experimental conditions.
Under these conditions, there was a multicomponent mixture of bR photoproducts detected by various experimental techniques. Results of adsorption and fluorescent measurements in the visible region indicate that this mixture strongly depended on illumination conditions [39, 40]. However, under these conditions FTIR-emission spectroscopy revealed only a limited number (one or two) bR states. Using frequencies of C=C (1529 and/or 1516 cm-1) and other characteristic bands they may be identified as bR568 or a K610-like intermediate. The reasons underlying domination in vibrational IR-emission spectra of bands of bR and the K-like intermediate and possible contribution of other state(s) remain unclear. This is the task for further studies. It requires elucidation of the mechanism responsible for the formation of the IR-emitting states.
Assuming that the emission occurs not from an equilibrium completely relaxed state but is caused by transitions between vibrational sublevels of some “hot” states that are located above the equilibrium ground electron state S0, it can be concluded that the emission spectra can be attributed to the vibrational excited state S0 of the chromophore. There may be several such states. Such unstable states precede the formation of stable products, which are usually studied by traditionally employed methods. This represents the principal difference between our approach and other methods, in which spectral information is collected from the system existing in stable, equilibrium (and as a rule completely relaxed) state.
Infrared emission is registered from quasi-equilibrium states. The set of emitting states depends on excitation conditions. It is likely that the emission by itself is due to the competition between radiationless relaxation and deactivation of the excited vibrational state by induced energy output in the form of IR-quanta. Both this inducible output and the generation of a populated state can be initiated by the resonant combined action of optical photons with frequencies omegai and omegaj, whose differences omegai - omegaj = ij coincide with the frequencies of the vibrational modes of BR. Using simply organized molecules we demonstrated that such a mechanism might operate under similar conditions of illumination [41].
The number of emitting states and their nature depend on excitation conditions. Changes of vibrational emission spectra during variation of the spectral composition as well as intensities of the actinic light possibly reflect the formation of a series of quasi-equilibrium states similar to the states already detected at low temperatures [39, 42].
The data of the present report clearly demonstrate the applicability of the FTIR-technique for emission studies of such complex biological systems as bR. The spectra presented here provide information additional to that provided by Raman and adsorption IR-spectroscopy. We believe that together with data from other laboratories our results can be used to create a comprehensive picture of the structure of vibrationally excited pigment and pathways of its vibrational relaxation.
The first discussion of the bacteriorhodopsin data was at a seminar with the late Professor A. D. Kaulen. We never forget him, his attention to our work, and his valuable advise. We are also very grateful to S. P. Balashov for his valuable comments and useful discussion of the results presented here and generous help in the revision of this paper.
REFERENCES
1.Stoeckenius, W., Lozier, R. H., and Bogomolni, R.
A. (1979) Biochim. Biopys. Acta, 505, 215-278.
2.Mathies, R. A., Lin, S. W., Ames, J. B., and
Pollard, W. T. (1991) Annu. Rev. Biophys. Biophys. Chem.,
20, 491-518.
3.Lanyi, J. K. (1992) J. Bioenerg. Biomembr.,
24, 169-179.
4.Govinjee, R., Balashov, S. P., and Ebrey, T. G.
(1990) Biophys. J., 58, 597-608.
5.Lozier, R. H., Bogomolni, R. A., and Stoeckenius,
W. (1975) Biophys. J., 15,614-622.
6.Braiman, M. A., and Mathies, R. A. (1982) Proc.
Natl. Acad. Sci. USA, 79, 403-407.
7.DeBlase, F. J., and Compton, S. (1991) Appl.
Spectrosc., 45, 611-622.
8.Mink, J., and Keresztury, G. (1993) Appl.
Spectrosc., 47, 1446-1459.
9.Willis, H. A., van der Maas, J. H., and Miller, R.
G. J. (eds.) (1987) Laboratory Methods in Vibrational
Spectroscopy, J. Willey and Sons, New York.
10.Terpugov, E. L., and Degtyareva, O. V. (2000)
Proc. SPIE, 4129, 97-81.
11.Terpugov, E. L., and Degtyareva, O. V. (2001)
J. Mol. Struct., 565/566, 287-292.
12.Oesterhelt, D., and Stoechenius, W. (1974)
Meth. Enzymol., 31, 667.
13.Balashov, A. A., Vagin, V. A., Viskovatich, A.
V., Lazarev, Yu. A., Terpugov, E. L., and Grishkovshi, G. A. (1991)
Proc. SPIE, 1575, 182-183.
14.Terpugov, E. L., Viskovatykh, A. V., Degtyareva,
O. V., and Fesenko, E. E. (1998) Biofizika, 43,
1002-1011.
15.Genzel, L. L., and Kuhl, J. (1978) Infrared
Phys., 18, 113-120.
16.Aton, B., Doukas, A. G., Callender, R. H.,
Becher, B., and Ebrey, T. G. (1977) Biochemistry, 16,
2995-2999.
17.Lewis, A., Spoonhower, J. P., Bogomolni, R. A.,
Lozier, R. H., and Stoeckenius, W. (1974) Proc. Natl. Acad. Sci.
USA, 76, 4462-4466.
18.Smith, S. O., Braiman, M. S., Myers, A. B.,
Pardoen, J. A., Courtin, J. M. L., Winkel, C., Lugtenburg, J., and
Mathies, R. A. (1987) J. Am. Chem. Soc., 109,
3108-3125.
19.Terner, J., Hsieh, C.-L., Burns, A. R., and
El-Sayed, M. A. (1979) Proc. Natl. Acad. Sci. USA, 76,
3046-3050.
20.Dioumaev, A. K., and Braiman, M. S. (1997) J.
Phys. Chem. B, 101, 1655-1662.
21.Hage, W., Kim, M., Frei, H., and Mathies, R. A.
(1996) J. Chem. Phys., 100, 16026-16033.
22.Smith, S. J., Pardoen, J. A., Lugtenburg, J., and
Mathies, R. A. (1987) J. Am. Chem. Soc., 91, 804-819.
23.Van den Berg, R., Jang, Du-J., Bitting, H. C.,
and El-Sayed, M. (1990) Biophys. J., 58, 135-141.
24.Siebert, F., and Mantele, W. (1983) Eur. J.
Biochem., 130, 565-573.
25.Bagley, K., Dollinger, G., Eisenstein, L., Singh,
A. K., and Zimanyi, L. (1982) Proc. Natl. Acad. Sci. USA,
79, 4972-4976.
26.Smith, S. O., Pardoen, J. A., Mulder, P. P. J.,
Curry, B., Lugtenburg, J., and Mathies, R. A. (1983)
Biochemistry, 22, 6141-6148.
27.Curry, B., Palings, H., Brock, A. D., Padroen, J.
A., Lughenburg, J., and Mathies, R. A. (1985) in Advances in
Infrared and Raman Spectroscopy (Clark, R. J. H., and Hester, R.
E., eds.) Vol. 12, J. Willey and Sons, N. Y., pp. 115-177.
28.Ormos, P. (1991) Proc. Natl. Acad. Sci.
USA, 88, 473-477.
29.Clareda, J., Sabes, M., and Padros, E. (1993)
Biochemistry, 31, 12363-12368.
30.Gerwert, K., Hess, B., and Engelhard, M. (1990)
FEBS Lett., 261, 449-454.
31.Rothshild, K., Gray, D., Mogi, T., Marti, T.,
Braiman, M. A., Stern, L. G., and Khorana, H. G. (1989)
Biochemistry, 28, 7052-7059.
32.Rothshild, K. J., He, Y. W., Gray, D., Roepe, P.,
Pelltien, S. L., Brown, R. S., and Herzfeld, J. (1987) Proc.
Natl. Acad. Sci. USA, 84, 5221-5225.
33.Gerwert, K., Hess, B., Soppa, J., and Oesterhelt,
D. (1989) Proc. Natl. Acad. Sci. USA, 86, 4943-4949.
34.Zhou, F., Windemuth, A., and Schulten, K.
(1993) Biochemistry, 32, 2291-2306.
35.Alfano, R. R., Govindjee, R., Becker, B., and
Ebrey, T. G. (1976) Biophys. J., 16, 541-545.
36.Sineshchekov, V. A., and Litvin, F. F. (1976)
Biofizika, 21, 313-320.
37.Dencher, N., and Wilms, M. (1975) Biophys.
Struct. Mechanism, 1, 259-271.
38.Kouyma, T., Bogomolni, R. A., and Stoeckenius, W.
(1985) Biophys. J., 48, 201-208.
39.Balashov, S. P., and Litvin, F. F. (1981)
Biofizika, 26, 557-568.
40.Sineshchekov, V. A., Balashov, S. P., and Litvin,
F. F. (1981) Biofizika, 26, 964-972.
41.Terpugov, E. L., and Degtyareva, O. V. (2001)
Pisma v ZhETF (Moscow), 6, 320-323.
42.Balashov, S. P. (1995) Isr. J. Chem.,
35, 415-428.