Synthesis and Properties of Bacteriorhodopsin Analogs Containing Electron-Density Labels in the Chromophore Moiety
E. V. Mironova1, A. Yu. Lukin1, S. V. Shevyakov1, S. G. Alexeeva1, V. I. Shvets1, O. V. Demina2, A. A. Khodonov1*, and L. V. Khitrina3
1Lomonosov State Academy of Fine Chemical Technology, pr. Vernadskogo 86, Moscow, 117571 Russia; fax: (095) 434-8233; E-mail: biotechnology@mtu-net.ru2Emanuel Institute for Biochemical Physics, Russian Academy of Sciences, ul. Kosygina 4, Moscow, 117977 Russia; E-mail: chembio@sky.chph.ras.ru
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia, fax: (095) 939-3181; E-mail: khitr@phtbio.genebee.msu.su
* To whom correspondence should be addressed.
Received April 4, 2001; Revision received June 13, 2001
A method for synthesis of retinal analogs labeled with electron-density groups is suggested. The interaction of these polyene compounds with bacterioopsin in apomembrane of Halobacterium salinarum was tested. A retinal analog containing a crown-ether receptor group is able to interact readily with bacterioopsin giving rise to rapid formation of a pigment with absorption maximum at 460 nm. This pigment is capable of undergoing cyclic photoconversion. The crown-bacteriorhodopsin photocycle is extremely slow and its quantum efficiency is very low (~3% of that in native bacteriorhodopsin). This photocycle includes an M-like intermediate with a differential absorption maximum at 380 nm. A retinal analog in which the beta-ionone ring is replaced by ferrocene moiety forms a stable chromoprotein with the main absorption band at 483 nm and a shoulder near 590-610 nm.
KEY WORDS: bacteriorhodopsin, bacterioopsin, chromophore, apomembranes, photocycle, M-intermediate, electron-density labels, retinal, bacteriorhodopsin analogs, synthesis and photochemical properties, crown ether, ferrocene
Abbreviations: BR) bacteriorhodopsin; BO) bacterioopsin; PM) purple membrane; THF) tetrahydrofuran; Mes) (2-[N-morpholino]ethanesulfonic acid; DIBAH) diisobutylaluminumhydride; Et2O) diethyl ether; OS) opsin shift; SB) Schiff base; SBH+) protonated Schiff base.
Investigation of molecular mechanisms of light energy conversion into
chemical energy in living cells is one of the most important problems
of modern biochemistry. It seems that the structure of
bacteriorhodopsin (BR) is substantially simpler than the structure of
presently known chromoprotein complexes mediating light energy
conversion in photosynthetic systems. Bacteriorhodopsin is a
light-driven proton pump. It was discovered in 1971 [1]. Bacteriorhodopsin is a membrane protein with
molecular weight of 26.5 kD [2]. The protein moiety
of BR, bacterioopsin (BO), is a single polypeptide chain. The
chromophore group of BR is a protonated retinal aldimine linked to the
epsilon-amino group of Lys216 through the Schiff base.
Bacteriorhodopsin is remarkably tolerant to various physical and chemical factors and can be easily isolated in virtually pure state as so-called purple membranes (PM). Purple membranes are fragments of cytoplasm membranes of the extreme halophilic bacterium Halobacterium salinarum. Bacteriorhodopsin is presently the most thoroughly studied retinal-containing protein with promising practical implications [3].
Upon absorbing a light quantum, BR undergoes a cycle of photochemical reactions accompanied by isomerization of the polyene chain of the chromophore and deprotonation and reprotonation of the retinal aldimine moiety. The photocycle is also accompanied by considerable conformational rearrangement of the protein moiety of BR [4]. Determination of the tertiary structure of the initial state of BR and major intermediates of the BR photocycle is an important part of studies of molecular mechanisms of BR functioning and a possible approach to target-directed modification of photocycle parameters [5-10].
Modified retinoids containing various reporter groups (2H-, 3H-, 13C-, and 14C-isotopes; fluorescence, photoaffinity, and spin-probes; fluorine atoms; Br, I, Hg, Fe, and other electron-density labels, etc.) have been widely used over the last 25 years for determining structure of retinal-containing proteins (visual rhodopsin, bacteriorhodopsin, halorhodopsin, and retinochrome) [11, 12]. Reporter groups with high electron density labels are often used in crystallographic research. Engelhard et al. used this approach for labeling the polypeptide moiety of BR [13]. However, there were only a few reports of the use of retinal analogs labeled with heavy atoms [14, 15]. This was due to difficult methods of synthesis and lack of methods of site-directed incorporation of electron-density labels into chromophore molecules.
The goals of this work were to discuss methodological approaches to incorporation of electron-density labels into the chromophore group of bacteriorhodopsin, to consider new methods of synthesis of polyenals containing electron-density labels, and to describe the results of studies of their interaction with bacterioopsin in apomembranes of H. salinarum. Spectral and photochemical characteristics of the resulting BR analogs are also described.
The following factors determined the choice of the target structure of retinal analogs: first, the volume of electron-density label should be minimal to provide maximum similarity of the artificial pigment obtained from BO and the polyenal with natural BR; second, labeled derivatives of retinal should be sufficiently stable during synthesis and interaction with apoprotein (BO); third, initial compounds and reagents should be affordable, whereas methods of synthesis should be versatile and effective.
Molecular structures of all-E-retinal (1), 13Z-11,12-didehydroretinal (acetylenic analog of retinal) (2), cobalt pi-complex of 11,12-acetylenic analog of retinal (3), cobalt pi-complex with terminal label (4), C20-ferrocene analog of retinal (5), and retinal analog containing crown-ether (6) are shown in Scheme 1.
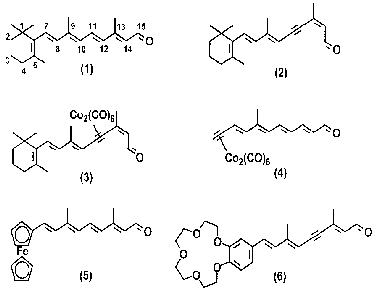
Scheme 1
A new methodological approach to the problem of incorporation of electron-density labels into retinoid molecules has been developed and described in the preceding works [16-18]. This approach is based on synthesis of stable pi-complexes (3, 4) of acetylene derivatives of retinal with Co2(CO)8 [16-18]. Unfortunately, these retinal derivatives were inappropriate for BR studies because neither pigment nor noncovalent complexes are formed by the interaction of BO with the pi-complex (3) [16, 17]. An insignificant increase in the absorption band amplitude at 519 nm in these experiments was due to formation of 11,12-didehydro-BR as a result of slow hydrolysis of complex (3) and formation of 11,12-didehydroretinal (2). Properties of 11,12-didehydroretinal (2) have been thoroughly studied in the preceding works [19-23]. A pigment with an absorption maximum at lambdamax = 450 nm and a shoulder at 500 nm was synthesized within 1.5 h of the reaction of interaction between BO and the complex (4). However, this pigment was unstable and degraded spontaneously within several days of storage in a refrigerator [18]. Methods of synthesis of labeled retinal analogs (3, 4) and results of studies of interaction of these compounds with BO were considered in the preceding works [16-18, 24]. In the present work we consider in more detail the methods of incorporation of electron-density labels into the vicinity of the trimethylcyclohexene ring of the chromophore group (polyenals (5) and (6)). We also suggested methods of substitution of the trimethylcyclohexene ring of natural retinal by a ferrocene fragment or a fragment containing crown-ether.
The ferrocene fragment was chosen for this synthesis, because complex metallocene compounds are stable in aqueous media and the initial compound (formylferrocene) is easily available and can be easily modified [25].
Incorporation of crown-ether in the chromophore group of BR would solve a number of problems of applied and basic research: 1) incorporation of electron-density labels into the BR chromophore (formation of stable complexes between the crown-ether ring and cations [26, 27]) for further detailed studies of the chromophore cavity and elucidation of spatial localization of chromophore group in purple membrane by various crystallographic methods (electron cryomicroscopy, X-ray diffraction, and neutron diffraction); 2) development of various sensor devices and ion-selective electrodes designed to detect various cations (electrochemical or spectrophotometric detection); 3) variation of the crown-ether cycle size and heteroatom structure as an approach to possible selectivity of cation detection in a mixture of cations; 4) selection of the target structure (6) combining a C11-C12-triple bond and a substituted phenyl ring containing crown-ether in one polyenal molecule provides low rate of isomerization of artificial pigments, which is required in some types of photochemical research.
The structure of the polyenal molecule containing crown-ether (6) is similar to the structure of the aromatic analogs of retinal capable of effective incorporation in the apoprotein and formation of photochemically active pigments [28, 29]. It was shown earlier that, in addition to cyclic photochemical activity, these pigments are able to mediate light-dependent proton transport [28, 29].
MATERIALS AND METHODS
Reagents and equipment. A Bruker MSL-200 NMR-spectrometer (Germany) with a working frequency of 200 MHz was used for detecting 1H-NMR spectra of the compounds of interest dissolved in deuterium-chloroform. The numbering of atoms of the polyene chain of all-E-retinal (1) shown in Scheme 1 was used to attribute the proton signals of synthetic compounds. Ultraviolet absorption spectra of low-molecular-weight compounds and membrane preparations of BR and BR analogs were recorded using a Hitachi-3400 spectrophotometer (Japan). Digital spectral data were processed using a computer.
Mass-spectra were recorded using a Finnigan 4021 mass-spectrometer (USA) with direct input of experimental samples and ionization energy of 70 eV. Silufol UV-254 plates (Kavalier, Czech Republic) and hexane-ether (1 : 1 v/v) (A) or chloroform-methanol (100 : 1 v/v) (B) mixtures of solvents were used for thin-layer chromatography.
Retinoids were synthesized using the following reagents: triphenylphosphine (for synthesis), 80% suspension of sodium hydride in mineral oil, and active manganese dioxide (Merck, Germany); triethylphosphite (purum), chloroacetone (purum), and chloroacetonitrile (purum) (Fluka, Switzerland); 18-crown-6 (purum) (Aldrich, USA); 1 M solution of diisobutylaluminumhydride (DIBAH) in hexane (Acros, USA); benzo-15-crown-5 (pure), triphenylchlorosilane (pure), ferrocene (pure), and inorganic salts and solvents of chemical purity grade (Russia).
2-[N-morpholino]ethanesulfonic acid (Mes) from Sigma (USA), hydroxylamine from Merck, beta-cyclodextrin from Fluka, and inorganic salts of chemical and special purity grades from domestic manufacturers were used in experiments with biological preparations.
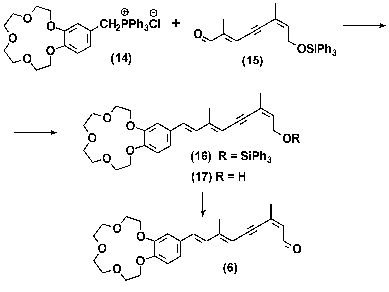
Synthesis of labeled polyenals (5, 6). A system of conjugate double bonds was formed in molecules of analogs (5) and (6) using different methods: polyenal (5) was synthesized by sequential build up of polyene chain by double repetition of olefination of formylferrocene (7) with C5-phosphonate (8) using the Horner-Emmons method [25]; polyenal (6) was synthesized by olefination of C10-aldehyde (15) with ilide by the method of Wittig, the ilide itself being generated from triphenylphosphonium salt (14) (see Schemes 2 and 3).
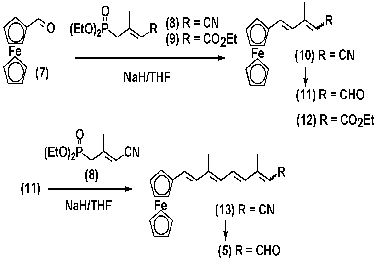
Scheme 2
Scheme 3
The ferrocene analog of retinal (5) was synthesized using C5- phosphonate with a cyanogroup (8) as an olefination agent by the Horner-Emmons method [25]. The terminal nitrile group of principal intermediates (10) and (13) was transformed into an aldehyde using one-stage DIBAH-mediated reduction. In case of phosphonate (8), double olefination by the method of Horner-Emmons produced an isomeric mixture of the target-aldehyde (5). The yield of the target-aldehyde was 61 or 45% as calculated per intermediate polyenal (11) or ferrocene aldehyde (7), respectively [24]. It was shown that the C=C double bond formed as a result of the Horner-Emmons reaction had E-configuration (J7,8 16.0).
The structure of individual all-E-isomer (5) isolated from the isomeric mixture was studied by various physicochemical methods of analysis: UV-spectrophotometry, 1H-NMR-spectroscopy, and mass-spectrometry. The 1H-NMR spectrum of the compound contained signals corresponding to ferrocene protons (delta 4.0-4.5 ppm), polyene chain multiplets (delta 5.8-7.4 ppm), an aldehyde group proton doublet (delta 10.1 ppm), and two proton signals of two methyl groups (delta 2.02 and 2.3 ppm). Therefore, these spectral features are fully consistent with the structure of aldehyde (5). The mass-spectrum of the compound contained molecular ion ([M+] 346.2), which also was consistent with the calculated value.
The E-isomer of the ethyl ester of 3-methyl-5-(ferrocenyl)penta-2,4-dienoic acid (12) was synthesized by the Horner-Emmons method using olefination of formylferrocene (7) with C5-phosphonate (8) containing an ester group. The reaction yield was 77%.
The phosphonium salt (14) used in synthesis of crown-containing analog (6) was prepared by quarternization of triphenylphosphine the using corresponding chloromethyl derivative [30]. The reaction yield was 50%. Aromatic retinal analog containing crown-ether (6) was synthesized by olefination of C10-aldehyde (15) with ilide by the method of Wittig. The ilide molecules used in this reaction were prepared from triphenylphosphonium salt (14) under conditions of phase-transfer catalysis. These conditions have been developed and successfully used in the preceding work for synthesis of a large group of retinal analogs [19]. When the triphenylsilyl protective group had been removed and isomers had been separated from each other, the isolated alcohol (17) was oxidized by MnO2 in CH2Cl2 to 11,12- didehydropolyenal (6) [31-33].
Mixture of Z- and E-isomers of 3-methyl-5-(ferrocenyl)penta-2,4-dienal (11). A sample of 0.06 g of 80% suspension of NaH in mineral oil was put in a three-neck reactor (50 ml) under an argon atmosphere and washed with absolute hexane (3 × 3 ml). After the resulting preparation was cooled to 0°C, 10 ml of absolute tetrahydrofuran (THF) and 0.34 g (1.56 mmol) of C5-phosphonate (8) was added under intense stirring. While the resulting mixture was stirred for 1 h to provide complete dissolving of NaH, 0.3 g (1.4 mmol) of ferrocene aldehyde (7) solution in absolute THF (10 ml) was gradually added. Once the reaction was over, 5 ml of H2O and 10 ml of Et2O were dropped into the reactor, and the resulting solution was neutralized to pH 6.0 with 1 M HCl. The organic layer was separated and the residue was extracted with Et2O (3 × 30 ml). The organic solutions were combined, washed with water to pH 7, and dried above anhydrous Na2SO4. The solvent was decanted, and the residue was applied to a chromatographic column with 10 g of silica gel. The product was eluted using a hexane-Et2O mixture with an Et2O gradient of 0 to 15% (by volume). The fractions containing nitrile (10) were combined and the solvent was decanted. The residue was vacuum-dried for 1 h at 0.1 mm Hg, dissolved in 10 ml of absolute toluene, and filled in a 100-ml three-neck reactor under an argon atmosphere. When the reaction mixture had been cooled to -70 to -78°C, 1.5 equivalents of 1 M DIBAH solution in hexane were slowly added with a syringe under continuous stirring until the sample temperature reached 20°C. The reaction product was treated with wet silica gel, stirred for 30 min, filtered through a celite layer (1 cm), and washed with Et2O. The resulting filtrate was evaporated to dryness. The residue was applied to a chromatographic column with 10 g of silica gel. The product was eluted using a hexane-Et2O mixture with an Et2O gradient of 0 to 10% (by volume). The fractions containing an isomeric mixture of aldehyde (11) were combined and the solvent was decanted. The net product yield was 0.28 g (71%). According to 1H-NMR spectra, the ratio of isomers 9Z-/9E- was 60 : 40; Rf 0.49 and 0.47 (A), respectively. Pure Å-isomer of compound (11) with Rf 0.47 (A) was isolated by medium-pressure preparative flash-chromatography on silica gel and a hexane-Et2O system with an Et2O gradient of 0 to 10% (by volume).
The UV-spectrum of 9E-isomer (11) (lambdamax, nm, (epsilon), in methanol): 280 (8700), 340 (26700), 502.5 (3100).
1H-NMR-spectrum of 9Z-isomer (11): 1.84 (3H, d, J 1.2, -CH3); 4.14 (5H, s, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 4.40 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 4.49 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C- ]); 5.83 (1H, d, J 8.2, 10-CH); 6.77 (1H, d, J 15.5, 7-CH), 7.38 (1H, d, J 15.5, 8-CH); 10.21 (1H, d, J 8.2, -CHO).
9E-isomer (11): 2.13 (3H, d, J 1.2, -CH3); 4.13 (5H, s, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 4.38 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C- ]); 4.48 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 5.96 (1H, d, J 8.2, 10-CH); 6.75 (1H, d, J 16.0, 7-CH); 6.88 (1H, d, J 16.0, 8-CH); 10.10 (1H, d, J 8.2, -CHO).
all-E-isomer of 3,7-dimethyl-9-(ferrocenyl)nona-2,4,6,8-tetraenal (5). This compound was synthesized from 0.37 g (1.3 mmol) of aldehyde (11) using the method similar to those described above for polyenal (11). The product yield was 0.34 g (75%), Rf 0.45 (A). Pure Å-isomer of compound (5) was isolated by medium-pressure preparative flash-chromatography on silica gel using a hexane-Et2O system with an Et2O gradient of 0 to 10% (by volume).
The UV-spectrum of all-E-isomer (5) (lambdamax, nm, (epsilon), in methanol): 296 (12900); 394 (38700), 508 (6400).
1H-NMR-spectrum of all-E-isomer (5):2.05 (3H, d, J 1.0, 9-CH3); 2.32 (3H, d, J 1.0, 13-CH3); 4.09 (5H, s, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 4.29 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 4.41 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 5.98 (1H, d, J 8.5, 14-CH); 6.23 (1H, d, J 11.0, 10-CH); 6.38 (1H, d, J 15.0, 12-CH); 6.49 (1H, m, 7-CH); 6.49 (1H, m, 8-CH); 7.11 (1H, dd, J 11.0, J 15.0, 11-CH); 10.09 (1H, d, J 8.5, -CHO).
Mass-spectrum [m/z]: 346.2 [M+].
Mixture of Z- and E-isomers of ethyl ether of 3-methyl-5-(ferrocenyl)penta-2,4-dienoic acid (12). A sample of 0.04 g of 60% suspension of NaH in mineral oil was put in a four-neck reactor (volume, 50 ml) under an argon atmosphere and washed three times with absolute hexane (3 ml each time). After the resulting preparation was cooled to 0°C, 10 ml of absolute THF and 0.43 g (1.72 mmol) of C5-phosphonate (9) was added under intense stirring. While the resulting mixture was stirred for 1 h more to provide complete dissolving of NaH, 0.3 g (1.4 mmol) of ferrocene aldehyde (7) solution in 10 ml of THF was added. Once the reaction was over, 5 ml of H2O and 10 ml of Et2O were dropped into the reactor, and the resulting solution was neutralized to pH 6 with 1 M HCl. The organic layer was separated and the residue was extracted three times with Et2O (50 ml each time). Ether extracts were combined with the organic layer, washed with water to pH 7, and dried over anhydrous Na2SO4. The solvent was decanted, and the residue was applied to a chromatographic column packed with 10 g of silica gel. The product was eluted using a hexane-Et2O mixture with an Et2O gradient of 0 to 15% (by volume). The fractions containing ether (12) were combined and the solvent was decanted. The residue was vacuum-dried for 1 h at 0.1 mm Hg. The reaction product yield was 0.34 g (77%). According to 1H-NMR spectra of 9Z- and 9E-isomers, the ratio of 9Z/9E- isomers was 2 : 7. Pure Å-isomer of compound (12) with Rf 0.81 (A) was isolated by medium-pressure preparative flash-chromatography on silica gel and a hexane-Et2O system with an Et2O gradient of 0 to 5% (by volume).
The UV-spectrum of E-isomer (12) (lambdamax, nm, (epsilon), in methanol): 270.5 (13500), 320 (28300), 476.5 (2500).
1H-NMR-spectrum of 9E-isomer (12): 1.28 (3H, t, J 7.0, -OCH2CH3); 2.35 (3H, s, -CH3); 4.09 (5H, s, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 4.16 (2H, q, J 7.0, -OCH2CH3); 4.30 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 4.41 (2H, t, J 2.0, [C5H5]Fe[2(betaCH)2(alphaCH)C-]); 5.76 (1H, s, 10-CH); 6.40 (1H, d, J 16.0, 8-CH); 6.73 (1H, d, J 16.0, 7-CH).
Synthesis of 11,12-dehydroretinal analog (6). A 5-mg sample of 18- crown-6 was added under continuous stirring in an argon atmosphere to a suspension containing 1 g K2CO3, 0.69 g (1.19 mmol) phosphonic salt (14), and 0.5 g (1.18 mmol) aldehyde (15) in 30 ml of anhydrous CH2Cl2. The resulting mixture was stirred for 48 h, filtered, and the filtrate was evaporated. The residue was dissolved in 50 ml of methanol and mixed under stirring with NH4F suspension to remove the triphenylsilyl protective groups. The mixture was evaporated and the residue was dissolved in 50 ml of methanol and crystallized at -50°C. The sediment was filtered. Analog of 11,12-dehydroretinol (17) was isolated by adsorption chromatography using 30 g of silica gel and a chloroform-methanol system with a methanol gradient of 0 to 5% (by volume). Fractions with Rf 0.35 (B) were combined and evaporated. Active manganese dioxide (3 g) was added to 0.51 g of solution of compound (17) in 30 ml of CH2Cl2 and stirred for 5 h in an argon atmosphere. The oxidizing agent was removed by filtration through a celite layer, and the filtrate was evaporated. Aromatic analog of retinal (6) was isolated by chromatography on 20 g of silica gel using a chloroform-methanol system with a methanol gradient of 0 to 5% (by volume). Fractions with Rf 0.4 (B) were combined and evaporated. The product yield was 0.45 g (89%). The isolated mixture of isomers of polyenal (6) was subjected to 4-h-long thermal isomerization in boiling benzene in an argon atmosphere. Individual 13Z-isomer (6) was isolated by medium-pressure preparative flash-chromatography.
The UV-spectrum of polyenal (6) (lambdamax, nm, (epsilon), in methanol): 364 (31400).
1H-NMR-spectrum: 2.13 (3H, s, 9-CH3); 2.15 (3H, s, 13-CH3); 3.80, 3.90, 4.20 (16H, 3 m, -CH2O); 5.72 (1H, s, 10-CH); 6.10 (1H, d. q, J 8.0, J 1.5, 14-CH); 6.63 (1H, d, J 16.0, 8-CH); 6.73 (1H, d, J 16.0, 7-CH); 6.78 and 6.98 (3H, m, arom); 10.07 (1H, d, J 8.0, -CHO).
Preparation and analysis of chromoproteins. Purple membranes were isolated from the H. salinarum (strains R1 and ET1001) using differential centrifugation as described in [1, 34]. Apomembranes were obtained by exposure of the PM to light in the cold and in the presence of 1 M hydroxylamine (pH 7.0), centrifugation, and washing with double distilled water six or seven times [12, 29, 35]. Retinaloxime was removed from PM, if marked, by treatment with beta-cyclodextrin [12, 36, 37].
To reconstruct the pigment, an ethanol solution of polyenal (no more than 0.5% by volume) was added with a slight excess to apomembrane suspension in buffer solution (pH 6.0) at room temperature [29]. If the procedure of BR analog formation took more than 1-2 h, the preparation was kept in a refrigerator. Otherwise, the apomembranes gradually denatured, and rates of retinaloxime hydrolysis and partial resynthesis of unmodified BR were increased. The method of continuous illumination has been described earlier [35]. Flash photolysis excited by a neodymium laser pulse (lambda = 532 nm; t1/2 = 15 nsec; pulse energy, 20 mJ) was recorded using a home-made spectrophotometer described in [38]. Apomembranes of the same batch of isolation or BR regenerated from apomembranes were used as control samples for detecting photoreactions of BR analogs.
RESULTS AND DISCUSSION
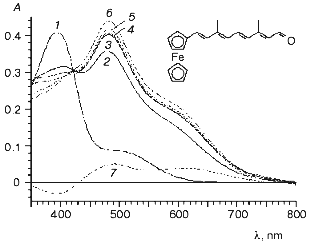
In this work we studied the interaction of BR analogs with apomembranes prepared from PM of two strains of H. salinarum: R1 and ET1001. In the general case, the apomembranes prepared from the strains R1 and 353-P were more suitable for kinetic studies of photocycle of BR analogs than similar preparations from strain ET1001 [12, 37]. Indeed, the rate of relaxation of the M-intermediate in the preparations from strain ET1001 was less than in control (initial BR) even in the case of the pigment regenerated from BO and all-E-retinal (1). Perhaps, these kinetic effects can be regarded as evidence of the existence of slight structural changes similar to those induced by low concentrations of detergents or some other modifiers of lipid-protein interaction. Therefore, in preliminary experiments we studied reactions of BO prepared from strain R1 and reconstructed with ferrocene retinal derivatives (5, 11, 12) with different length of polyene chain and structure of terminal polar group (Figs. 1 and 2). These experiments revealed the following specific features: 1) visible spectrum (>300 nm) of the retinoid derivatives mentioned above had complicated two-band structure (Fig. 1, curves 4 and 5, and Fig. 2, curve 1), the two bands (in case of aldehydes) being observed in the absorption spectra of the resulting protein complexes; 2) carotenoids are significant components of apomembranes isolated from strain R1; 3) a nonlinear shift of the absorption bands of carotenoids is induced by incorporation of aldehydes into BO interior and resulting structural changes of the apomembranes. In other words, carotenoid bands cannot be subtracted completely from the differential spectrum of chromoprotein and apomembranes (Fig. 1, curve 3). It should be noted that an increase in the incorporation duration was accompanied by an increase in the light scattering intensity of the preparation, thereby causing undesirable spectral artifacts.
Fig. 1. Interaction of BO with (a, b) short ferrocenic analog of retinal (11) and (c) corresponding ethyl ester (12) (the time of incubation of the strain R1 apomembranes in the presence of synthetic compounds (11) and (12) was 28 days): 1) apomembranes; 2) apomembranes plus aldehyde (11); 3) difference between curves 2 and 1 (curves 3a and 3b represent the same subtraction curve plotted at different sensitivity scales); 4) ethanol solution of aldehyde (11); 5) ethanol solution of ester (12); 6) product of interaction between ester (12) and apomembranes relative to membranes. The medium: 5 mM Mes (pH 6).
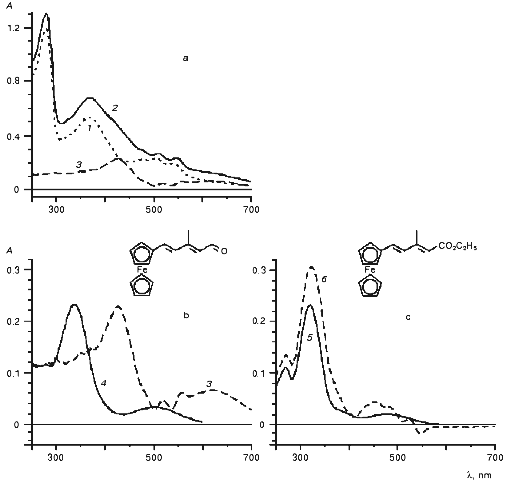
To obtain a clearer spectral pattern of interaction between the ferrocene retinal analog (5) and BO, apomembranes were prepared from the carotenoidless strain ET1001 and additionally treated with beta-cyclodextrin to remove retinaloxime. It is seen from Fig. 2 that within the first 8 min of the reaction a large part of the absorption band of added aldehyde (Fig. 2, curve 1, lambdamax = 394 nm) is 85 nm shifted toward longer wavelength (Fig. 2, curve 2). This shift is obviously due to formation of a complex between the retinal analog and BO. Further formation of BR analog proceeded rather slowly and was completed within four days of incubation (Fig. 2, curves 3-6). The position of the absorption band of the pigment during this process was shifted only insignificantly (by several nanometers) toward longer wavelength (to 483 nm). It should be noted that this absorption maximum falls within the wavelength range typical of BR analogs in which trimethylcyclohexene ring is replaced by aromatic rings of various structure (460-530 nm) [28, 29]. The calculated protein shift (OS) values of pigments relative to model compound (protonated polyenal (5) aldimine with n-butylamine) are given in the table.Fig. 2. Formation of chromoproteins from ferrocene analog of retinal (5) and apomembranes (strain ET1001, retinaloxime was removed using beta-cyclodextrin): 1) ethanol solution of aldehyde (5); 2-6) incubation of this polyenal in water suspension of apomembranes for 8 (2), 25 (3), 60 min (4), and 27 (5), 93 h (6) (light scattering and apomembrane spectra were subtracted); 7) difference between curves 4 and 2. During one month of storage the spectrum of the chromoprotein remained virtually unchanged and coincided with curve 6. The medium: 100 mM NaCl, 5 mM Mes (pH 6).
Spectral properties of retinoid derivatives (5), (6), (11), and (12),
their aldimine complexes with n-butylamine, and complexes with
BO
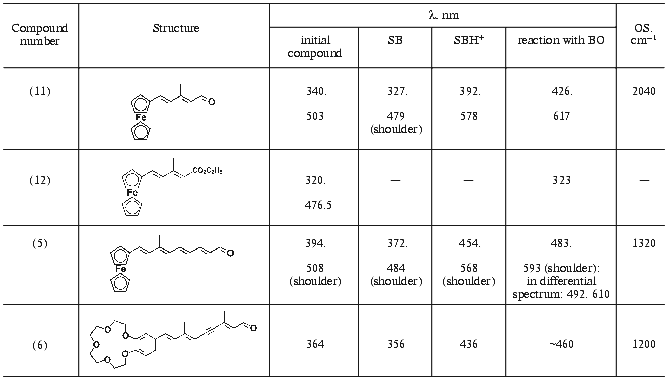
Note: SB) Schiff base in methanol prepared from retinal analog and
n-butylamine; SBH+) protonated form obtained by
treatment (with trifluoroacetic acid) of Schiff base in methanol
prepared from retinal analog and n-butylamine; OS) opsin shift
calculated as OS = 1/lambdamax(SBH+) -
1/lambdamax(pigment).
The absorption spectrum of the chromoprotein, like the absorption spectrum of aldehyde (5) contains a long-wavelength shoulder (table), the absorption ratio in minor and main bands of BR analog being increased approximately 2.5-fold. The formation of BR complexes can also be monitored using differential spectra of the same preparation as measured at different times. The absorption change dynamics over 27 h in this preparation is shown in Fig. 2, curve 7. The minimum of the curve (at 391.5 nm) is very close to the absorption maximum of aldehyde (5), whereas the short- wavelength maximum of the curve (near 492 nm) is close to the main absorption maximum of BR analog (lambdamax = 483 nm) and the second maximum of curve 7 (at 610 nm) is characteristic of the long-wavelength absorption maximum of the protein complex.
The rate of reconstruction between BO and short ferrocene analog (11) was lower and the extinction coefficient of the resulting complex was smaller than in case of reconstruction with polyenal (5). Therefore, it was reasonable to compare the interaction of apomembranes with ferrocene analog (11) and with retinal derivative (12) in which an ester group substitutes the terminal aldehyde group, because such substitution prevented formation of a covalent complex with BO [39]. It follows from Fig. 1 that reconstruction of polyenal (11) with apomembranes was accompanied by a large-magnitude bathochromic shift (table), which was virtually absent in the case of apomembrane reconstruction with ester (12).
The BR preparations containing ferrocene derivatives (5, 11) were stable at 4-5°C for more than 3 months. This conclusion was supported by invariability of their spectral characteristics.
According to absorption flash-spectroscopy, the photocycle of these BR analogs is either absent or its efficiency is less than 1%.
The interaction of apomembranes with retinal analog (6) containing crown-ether gave rise to formation of a pigment with absorption maximum near 460 nm (Fig. 3a). The rate of formation of the pigment was close to the rate of reconstruction of purple BR complex from BO and all-E-retinal. At 25°C the onset of the crown-ether-BR absorption band formation was detected within the first several minutes after the aldehyde addition to apomembranes. The formation of the band was completed within 1 h.
Exposure to laser light flashes caused reversible bleaching of the main absorption band of the pigment (Fig. 3, curves 2 and 6). The photocycle of the pigment includes an M-like intermediate with the differential absorption maximum at 380 nm (Fig. 3d). According to the relaxation kinetics of the M- like intermediate, the photocycle rate of the crown-ether-containing BR analog was significantly less than in control BR (Fig. 3c). The quantum efficiency of the crown-ether-containing BR photocycle was about 3% that value in control BR. This estimate was obtained from analysis of photoinduced response amplitude in the main absorption band as normalized to the absorption value in this band: DeltaA460/A460 (crown-ether BR) and DeltaA570/A570 (control BR) [35, 40]. Measurements at equal protein concentration or equal absorption level at the actinic light wavelength (532 nm) in control and experimental samples gave similar results.Fig. 3. Optical properties of chromoprotein prepared from BO and retinal analog (6) containing crown-ether. a) Absorption spectrum of product of interaction between aldehyde (6) and apomembranes (strain ET1001, retinaloxime was removed using beta-cyclodextrin, reaction medium contained 80 mM NaCl, 5 mM Mes (pH 6)). Suspension of apomembranes was used as a reference sample. b-d) Absorption changes induced by laser flash (lambda = 532 nm, t1/2 = 15 nsec) in suspension of membrane preparations at 21°C. b) Photoresponse of crown-ether-BR (spectrum (a), dotted (1) and solid (2) lines) and apomembranes (dashed line (3)) at 460 nm. Curve 1 was recorded after 1.5 h of BO incubation in the presence of aldehyde at room temperature; curve 2 was the same sample after 19 h of storage in a refrigerator. c-d) Comparison between photoresponse kinetics: c) crown-ether-BR (4) and BR (5) at the wavelength of 380 nm; d) crown-ether-BR at 380 nm (4) and 460 nm (6); (duration of interaction between strain R1 apomembranes with aldehyde in samples (4) and (6) was 3.5 h). Computer structural models of all-E-isomers of polyenal (6) (crown-ret), retinal (1) (ret), and acetylene analog of retinal (acet-ret) were built using the HyperChem v.4.5 program (carbon backbone and oxygen bonds are shown by thin and bold lines, respectively).
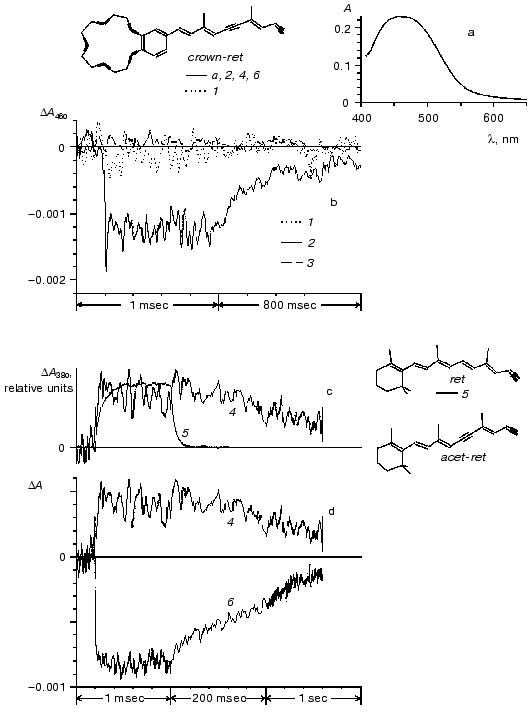
A high rate of covalent complex formation by polyenal (6) is an interesting feature of this analog (Fig. 3). It was shown earlier that substitution of the ionone ring by a phenyl ring is accompanied by a significant decrease in the rate of pigment formation, photocycle kinetics, and decrease in the quantum efficiency. It should be noted that such a significant decrease in the photocycle rate and quantum efficiency in crown-ether BR is due to a combination of ring modification and triple bond formation at C11-C12 position. Indeed, the rate of relaxation of the M-intermediate with differential absorption maximum at 400 nm in all-E-acetylene analog of BR was close to control, whereas the quantum efficiency of proton transport and photocycle was about half as high as in control BR [12, 19]. It should be noted that similar changes in the rate of relaxation of M-intermediate and photocycle efficiency were observed in BR analog with beta-ionone ring substituted by phenyl ring [29, 40].
Although the photoinduced response of crown-ether BR was relatively small, three experimental factors facilitated its detection. First, the amplitude of photoinduced changes of unmodified BR at 460 nm is very small. Second, there is a considerable kinetic difference between photocycles of modified chromoprotein and unmodified BR. Third, high rate of crown-ether reconstruction allowed the apomembrane preparations to be obtained with comparable concentrations of modified chromoprotein and residual BR.
It has been shown in many studies that freshly prepared complexes of 13Z-11,12-didehydro-BR had virtually no photocycle activity. For example, the amplitude of optical changes in the red spectral region (560-670 nm) induced by laser light flash in freshly prepared complexes was found to be at least an order of magnitude lower than after one day of storage. The isomeric composition of the preparation stored for one day at the temperature of ~2-5°C remained virtually unchanged [19, 21, 22]. The absorption band of the preparation has been also formed virtually completely by the time of the first testing of optical photoresponse. Further changes in the absorption maximum position did not exceed the normal range of variability of preparations obtained at different time (several nanometers). We failed to detect the transition between spectrally similar but functionally different forms of this BR analog because of methodological difficulty of separation of individual isomeric forms, low amplitude of the absorption changes of interest, and considerable between-sample variability of the time of formation of the functionally active form of 11,12-didehydro-BR. Perhaps, this variability was caused by minor methodological variations in methods of bacterial growth, isolation of apomembranes, and, what is more important, ambient temperature. Nevertheless, the fact of the existence of these forms is of considerable interest in itself because it demonstrates that certain functionally important structural modifications have only insignificant spectral manifestations. Similar effects were observed in case of 11,12-didehydrocrown-retinal analog. Immediately after reconstruction of some of these preparations, their photocycle was virtually indistinguishable from that in apomembranes (Fig. 3b). Although we did not study the statistical characteristics of the effect at length, comparison between Figs. 3b and 3d shows that different samples were characterized by significantly different rates of formation of functionally active complex.
Thus, it was shown in this work that ferrocene analog of retinal (5) is capable of slow formation of a stable pigment. This pigment is appropriate for further crystallographic studies. It was also shown that retinal analog (6) containing crown-ether can be, in principle, incorporated into BO. After additional studies of complex formation between this retinal analog and metal cations, we plan to incorporate the resulting complexes into the chromophore cavity of BR.
We are grateful to N. V. Khitrin for assistance in preparation of figures.
This study was supported in part by the Russian Foundation for Basic Research, project Nos. 01-03-32078 and 00-04-48588.
REFERENCES
1.Oesterhelt, D., and Stoekenius, W. (1971)
Nature, New Biol., 233, 149-152.
2.Ovchinnikov, Yu. A., Abdullaev, N. G., Feigina, M.
Yu., Kiselev, A. V., Lobanov, N. A., and Nazimov, I. V. (1978)
Bioorg. Khim., 4, 1573-1574.
3.Hampp, N. (2000) Chem. Rev., 100,
1755-1776.
4.Kaulen, A. D. (2000) Biochim. Biophys. Acta,
1460, 204-219.
5.Subramaniam, S., and Henderson, R. (2000)
Nature, 406, 653-657.
6.Mitsuoka, K., Hirai, T., Murata, K., Miyazawa, A.,
Kidera, A., Kimura, Y., and Fujiyoshi, Y. (1999) J. Mol. Biol.,
286, 861-882.
7.Luecke, H., Schobert, B., Richter, H., Cartailler,
J., and Lanyi, J. K. (1999) J. Mol. Biol., 291,
899-911.
8.Lanyi, J. (2000) Biochim. Biophys. Acta,
1460, 339-345.
9.Essen, L. O., Siegert, R., Lehmann, W. D., and
Oesterhelt, D. (1998) Proc. Natl. Acad. Sci. USA, 95,
11673-11678.
10.Pebay-Peyrola, E., Neutze, R., and Landau, E. M.
(2000) Biochim. Biophys. Acta, 1460, 119-132.
11.Mitsner, B. I., Khodonov, A. A., Zvonkova, E. N.,
and Evstigneeva, R. P. (1986) Bioorg. Khim., 12,
5-53.
12.Khodonov, A. A., Eremin, S. V., Lockshin, J. L.,
Shvets, V. I., Demina, O. V., Khitrina, L. V., and Kaulen, A. D. (1996)
Bioorg. Khim., 22, 745-776.
13.Engelhard, M., Pevec, B., and Hess, B. (1989)
Biochemistry, 28, 5432-5438.
14.Buldt, G., Konno, K., Nakanishi, K., Plohn,
H.-J., Rao, B. N., and Dencher, N. A. (1991) Photochem.
Photobiol., 54, 873-879.
15.Seiff, F., Westerhauser, J., Wallat, I., and
Heyn, M. P. (1986) Proc. Natl. Acad. Sci. USA, 83,
7746-7750.
16.Khodonov, A. A., and Khitrina, L. V. (1994)
Abst. VI Int. Conf. Retinal Proteins, Leiden, Netherlands, p.
118.
17.Khodonov, A. A., Demina, O. V., Shevyakov, S. V.,
Alexeeva, S. G., and Khitrina, L. V. (1996) Abst. XI Int.
Symp. Carotenoids, Leiden, Netherlands, p. 52.
18.Shevyakov, S. V., Khodonov, A. A., Mironova, E.
V., Alexeeva, S. G., Shvets, V. I., Demina, O. V., Khitrina, L. V., and
Kaulen, A. D. (2000) Abst. IX Int. Conf. Retinal Proteins, IBRC
Szeged, Hungary, p. 115.
19.Drachev, L. A., Drachev, A. L., Kaulen, A. D.,
Khitrina, L. V., Khodonov, A. A., and Mitsner, B. I. (1989) Bioorg.
Khim., 15, 307-312.
20.Balashov, S. P., Sineshchekov, V. A., Mitsner, B.
I., Khodonov, A. A., Khitrina, L. V., and Kurella, E. G. (1988)
Proc. Yamada XXI Conf.: Molecular Physiology of Retinal Proteins
(Kyoto) (Hara, T., ed.) Yamada Foundation, Osaka, Japan, pp.
345-346.
21.Khitrina, L. V., Drachev, L. A., Eremin, S. V.,
Kaulen, A. D., and Khodonov, A. A. (1992) Proc. V Int. Conf.:
Structures and Functions of Retinal Proteins (Dourdan) (Rigaud, J.
L., ed.), Vol. 221, in: Collogue INSERM, John Libbey Eurotext
Ltd., Montrouge, France, pp. 167-170.
22.Drachev, L. A., Kaulen, A. D., Khitrina, L. V.,
Eremin, S. V., Khodonov, A. A., Shvets, V. I., and Chekulaeva, L. N.
(1993) Biochemistry (Moscow), 58, 819-826 (Russ.).
23.Khitrina, L. V., Kaulen, A. D., Khodonov, A. A.,
and Eremin, S. V. (1995) Mol. Biol. (Moscow), 29,
1398-1407.
24.Khodonov, A. A., Shevyakov, S. V., Lockshin, J.
L., Alexeeva, S. G., Demina, O. V., Khitrina, L. V., Terpugov, E. L.,
and Degtyareva, O. V. (1996) Abst. VII Int. Conf. Retinal
Proteins, Zichron-Yaacov, Israel, p. 58.
25.Sato, M., Cono, H., and Shiga, M. (1968) Bull.
Chem. Soc. Japan, 41, 252-261.
26.Gromov, S. P., and Alfimov, M. V. (1997) Izv.
Akad. Nauk, Ser. Khim., No. 4, 641-665.
27.Gromov, S. P., Fedorov, O. A., Vedernikov, A. I.,
Samoshin, V. V., and Alfimov, M. V. (1993) Izv. Akad. Nauk, Ser.
Khim., No. 5,996-997.
28.Khodonov, A. A., Mitsner, B. I., Zvonkova, E. N.,
and Evstigneeva, R. P. (1987) Bioorg. Khim., 13,
238-251.
29.Drachev, L. A., Zorina, V. V., Khitrina, L. V.,
Mitsner, B. I., Khodonov, A. A., and Chekulaeva, L. N. (1987)
Biokhimiya, 52, 1559-1569.
30.Tashmukhamedova, I. A., Stempnevskaya, I. Yu.,
Morozova, E. G., and Sirotenko, E. G. (1989) Khim. Geterotsikl.
Soedin., 4, 470-474.
31.Khodonov, A. A., Lukin, A. Yu., Shevyakov, S. V.,
Mironova, E. V., Shvets, V. I., Alexeeva, S. G., Demina, O. V.,
Terpugov, E. L., and Degtyareva, O. V. (1999) Abst. III Int. Symp.
Organic Photochromism, Fukuoka, Japan, p. 99.
32.Khodonov, A. A., Lukin, A. Yu., Shevyakov, S. V.,
Mironova, E. V., Shvets, V. I., Alexeeva, S. G., Demina, O. V.,
Terpugov, E. L., Degtyareva, O. V., Khitrina, L. V., and Kaulen, A. D.
(2000) Mol. Cryst. Liq. Cryst., 345, 15-20.
33.Khodonov, A. A., Lukin, A. Yu., Shevyakov, S. V.,
Mironova, E. V., Shvets, V. I., Alexeeva, S. G., Demina, O. V.,
Terpugov, E. L., Degtyareva, O. V., Khitrina, L. V., and Kaulen, A. D.
(2000) Abst. IX Int. Conf. Retinal Proteins, IBRC Szeged,
Hungary, p. 94.
34.Oesterhelt, D., and Stoeskenius, W. (1974)
Meth. Enzymol., 31, 667-678.
35.Drachev, A. L., Drachev, L. A., Evstigneeva, R.
P., Kaulen, A. D., Lazarova, Z. R., Laikhter, A. L., Mitsner, B. I.,
Skulachev, V. P., Khitrina, L. V., and Chekulaeva, L. N. (1984)
Biol. Membr. (Moscow), 1, 1125-1142.
36.Mitsner, B. I., Varga, M., Shvets, V. I.,
Khitrina L. V., Drachev, L. A., Skulachev, V. P., Danshina, S. V., and
Chekulaeva, L. N. (1988) Biol. Membr. (Moscow), 5,
135-138.
37.Danshina, S. V., Drachev, A. L., Drachev, L. A.,
Eremin, S. V., Kaulen, A. D., Khitrina, L. V., and Mitsner, B. I.
(1990) Arch. Biochem. Biophys., 279, 225-231.
38.Kalaidzidis, I. V., Kalaidzidis, Ya. L., and
Kaulen, A. D. (1998) FEBS Lett., 427, 59-63.
39.Shkrob, A. M., Rodionov, A. V., and Ovchinnikov,
Yu. A. (1981) Bioorg. Khim., 7, 1169-1194.
40.Drachev, L. A., Drachev, A. L., Chekulaeva, L.
N., Evstigneeva, R. P., Kaulen, A. D., Khitrina, L. V., Khodonov, A.
A., Lazarova, Z. R., and Mitsner, B. I. (1989) Arch. Biochem.
Biophys., 270, 184-197.