REVIEW: Blood Coagulation
S. Butenas and K. G. Mann*
University of Vermont, Department of Biochemistry, Burlington, VT 05405-0068, USA; fax: 802-862-8229; E-mail: kmann@zoo.uvm.edu* To whom correspondence should be addressed.
Received April 2, 2001; Revision received April 23, 2001
The process of tissue factor initiated blood coagulation is discussed. Reactions of the blood coagulation cascade are propagated by complex enzymes containing a vitamin K-dependent serine protease and an accessory cofactor protein that are assembled on a membrane surface in a calcium-dependent manner. These complexes are 105-109-fold more efficient in proteolyses of their natural substrates than enzymes alone. Based upon data acquired using several in vitro models of blood coagulation, tissue factor initiated thrombin generation can be divided into two phases: an initiation phase and a propagation phase. The initiation phase is characterized by the generation of nanomolar amounts of thrombin, femto- to picomolar amounts of factors VIIa, IXa, Xa, and XIa, partial activation of platelets, and almost quantitative activation of procofactors, factors V and VIII. The duration of this phase is primarily influenced by concentrations of tissue factor and TFPI. The characteristic features of the propagation phase are: almost quantitative prothrombin activation at a high rate, completion of platelet activation, and solid clot formation. This phase is primarily regulated by antithrombin III and the protein C system. Thrombin generation during the propagation phase is remarkably suppressed in the absence of factor VIII and IX (hemophilia A and B, respectively) and at platelet counts <5% of mean plasma concentration. The majority of data accumulated in in vitro models and discussed in this review are in good agreement with the results of in vivo observations.
KEY WORDS: blood coagulation, tissue factor, complex enzymes, thrombin generation, platelets, hemophilia, in vitro models
Abbreviations: TF) tissue factor; TFPI) tissue factor pathway inhibitor; CTI) corn trypsin inhibitor.
The blood coagulation cascade is initiated when subendothelial tissue
factor is exposed/expressed to the blood flow following either the
damage or activation of the endothelium. This may occur as a
consequence of the perforation of the vessel wall or activation of
endothelium by chemicals, cytokines, or inflammatory processes [1-6]. Tissue factor binds to a
serine protease, factor VIIa, already present in blood [7, 8] and forms the factor
VIIa-tissue factor complex (extrinsic factor Xase), which
activates the zymogens--factor IX and factor X [9-12]. The limited amounts of the
serine protease factor Xa produced generate picomolar concentrations of
thrombin, which partially activates platelets and cleaves the
procofactors factor V and factor VIII generating the active
cofactors--factor Va and factor VIIIa, respectively [13-15]. Factor VIIIa forms the
intrinsic factor Xase complex with the serine protease, factor
IXa, on a membrane surface provided by platelets, microparticles, and
endothelial and other cells [16-19] and activates factor X at a 50-100-fold higher
rate than the factor VIIa-tissue factor complex [12, 20, 21]. Factor Xa forms the prothrombinase
complex with the cofactor, factor Va, on the membrane surface, which is
the primary activator of prothrombin [22-24]. The thrombin produced further amplifies its own
generation by activating factor XI [25] and
completing activation of platelets and factors V and VIII [15, 26]. Thrombin also cleaves
fibrinogen [27, 28] and
factor XIII [29, 30] to form
the insoluble cross-linked fibrin clot [31, 32].
The procoagulant processes are attenuated by a variety of inhibitors, which inactivate either serine proteases or cofactors. One of the stoichiometric inhibitors, antithrombin III, appears to be the most quantitatively important. Antithrombin III is able to effectively neutralize all serine proteases produced during the blood coagulation process [33]. TFPI, another stoichiometric inhibitor of blood coagulation, which is present at relatively low concentrations in blood [34], is also an important regulator of the serine proteases. The principle targets of TFPI are factor Xa and the factor VIIa-TF-factor Xa complex [35-37]. The dynamic inhibitory system is generated when thrombin binds to its cofactor thrombomodulin, which is constitutively present on the vasculature, and activates protein C to a serine protease activated protein C [38, 39]. Activated protein C attenuates the coagulation reaction by the proteolytic cleavages and consequential inactivation of factor V/Va and factor VIII/VIIIa [40-43]. These inactivating cleavages are somewhat accelerated by protein S [44], although the major function of this protein in the regulation of thrombin generation has not yet been clarified [45-47]. The inactivation of factor VIIIa also occurs rapidly by an activated protein C-independent mechanism through dissociation of the A2 domain [48-51].
Thus, at a minimum, ten plasma proteins (prothrombin, factor V, factor VIII, factor VII, factor VIIa, factor IX, factor X, factor XI, factor XIII, and fibrinogen) and one tissue protein contribute to the solid clot formation. The anticoagulant process is controlled by a minimum of four plasma proteins (antithrombin III, protein C, protein S, and TFPI) and by one membrane-bound protein (thrombomodulin). Thrombin plays essential roles in both pro- and anticoagulant processes. The in vivo concentrations of these proteins vary over a wide range: from subnanomolar (factor VIIa, factor VIII) to micromolar (prothrombin, antithrombin III, fibrinogen) (Table 1) [52]. Moreover, the components of the enzymatic complexes are represented at quite different concentrations. For example, the procofactor of the intrinsic factor Xase, factor VIII, circulates in vivo at 0.7 nM concentration, whereas the precursor of factor IXa, zymogen factor IX, is present at approximately 90 nM concentration.
Table 1. Mean plasma concentrations and
properties of coagulation proteins and inhibitors
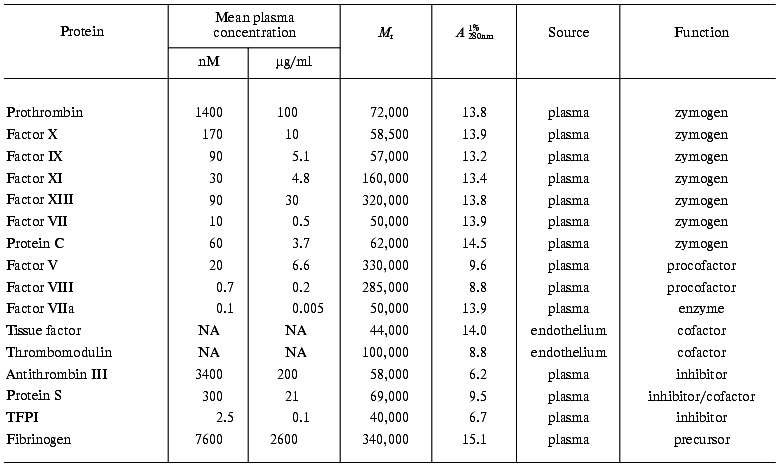
Note: NA, not applicable (membrane protein).
Current literature supports the conclusion that blood coagulation reactions are propagated by the formation of the complex enzymes consisting of the vitamin K-dependent serine protease and a non-enzymatic cofactor protein that are assembled on a membrane surface in a calcium-dependent manner [53, 54]. The significance of the components of such complexes is best illustrated by the alterations in catalytic efficiency that they display in proteolysis of their natural substrates. In case of the extrinsic factor Xase, the proteolytic activity of factor VIIa in the absence of any component of the enzymatic complex is negligible [10, 11]. The additions of calcium and phospholipids, which represent the membrane surface, slightly increase the activity of factor VIIa (Table 2) [10]. However, only the presence of the cofactor, tissue factor, leads to the assembly of a potent enzymatic complex, which hydrolyses factor X at an approximately 2*107-fold higher rate than factor VIIa alone. Similar differences (105-109-fold) in the efficiencies of enzymatic complexes versus those of enzymes alone are observed for the intrinsic factor Xase, prothrombinase, and the alpha-thrombin-thrombomodulin complex, the protein Case (Table 3) [10, 20]. Thus, the absence of a cofactor or membrane binding site is as significant as absence of the serine protease. The significance of the membrane surface in the assembly of these complexes should not to be overlooked, since in the absence of membrane the formation of the complex is either abolished or impaired [20, 23, 55, 56]. Similarly, calcium is also an essential component for efficient complex formation [57].
Table 2. Activation of factor X by factor
VIIa in the presence of various cofactors of the extrinsic factor
Xase
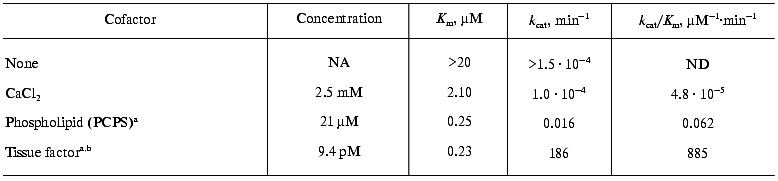
Note: NA, not applicable; ND, not determined; a in the
presence of 5 mM CaCl2; b in the presence of
PCPS.
Table 3. Kinetic properties of the vitamin
K-dependent enzymes and enzymatic complexes
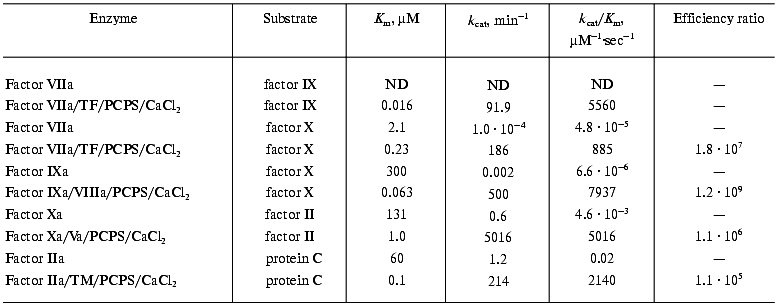
Note: ND, not determined; TM, thrombomodulin.
Although each of the enzymatic complexes involved in blood coagulation and its regulation display unique specificity towards their natural substrates, they share many similarities.
1. The complexes each contain a vitamin K-dependent serine protease and an accessory cofactor protein.
2. In each case, these complexes are composed from structurally similar constituents and exhibit similar requirements for assembly and activity.
3. The complexes are functionally analogous.
4. The assembly of vitamin K-dependent enzymatic complexes leads to substantial increases (105-109-fold) in the proteolytic efficiency of activation of the natural substrates (Table 3).
As a consequence of the formation of vitamin K-dependent enzymatic complexes, the following key events occur.
1. Vitamin K-dependent zymogens are converted to corresponding serine proteases.
2. Either proteolytic activation of plasma procofactors (in case of factor V and factor VIII) or the exposure/expression of the membrane cofactors (tissue factor and thrombomodulin).
3. Activation of the cellular membrane binding sites and, as a consequence, presentation of an appropriate surface for the assembly of the enzymatic complexes.
Our laboratory and others have been studying in vitro the dynamics of the tissue factor-initiated blood coagulation processes under conditions resembling those occurring in vivo. Several models representing coagulation process have been developed for this purpose. The first of these models is the “synthetic plasma” model prepared with highly purified natural and recombinant proteins involved in the coagulation and its regulation and synthetic phospholipid membranes or platelets [13, 19, 58-60]. Proteins and platelets can be mixed at their physiological concentrations (Table 1) and induced to generate thrombin upon the addition of tissue factor or cells bearing tissue factor. When the goal of the experiment is to imitate the deficiencies in blood coagulation protein(s) or other coagulation disorders, concentrations of selected protein(s) can be varied within the desired range. The reaction products and their rates of generation during the tissue factor initiated thrombin formation are evaluated using enzymatic, chromatographic, and immunochemical assays. The second approach involves mathematical models [61, 62] based on the reaction kinetics, binding rates/constants, and stoichiometries of individual coagulation complex and inhibitor reactions. These computer-solved theoretical models are more efficient than empirical models and allow the prediction of the outcome of the reaction at selected parameters. The third model involves the in vitro study of the coagulation of the tissue factor initiated whole blood [14, 63, 64] obtained from healthy individuals and from patients with coagulation abnormalities. The “intrinsic pathway” (contact pathway) in this model is suppressed by a specific inhibitor of factor XIIa, CTI. The CTI enables the exclusive study of tissue factor initiated pathway of blood coagulation with no other anticoagulants added. The convergence of data collected using all three models allows the evaluation and prediction of in vivo responses to alterations in the concentration and functional activity of proteins involved in blood coagulation and its regulation, occurring either naturally or as a consequence of pharmacological treatment [65-74].
Tissue factor initiated thrombin generation in all three models of blood coagulation can be divided into two phases (Fig. 1) [15]. An initiation phase follows the addition of tissue factor and is characterized by the appearance of nanomolar amounts of thrombin generated at a relatively low rate, partial activation of platelets, and almost quantitative proteolysis of procofactors, factor V, and factor VIII [13-15]. During this phase, femto- to picomolar amounts of factor VIIa, factor IXa, factor Xa, and factor XIa are also generated [13, 15]. Subsequently, in the presence of all procoagulant proteins at their mean plasma concentrations, the propagation phase of thrombin generation occurs. In the whole blood, the clot occurs at the inception of this phase [14, 75]. The characteristic features of the propagation phase are an almost quantitative prothrombin conversion at a high rate, completion of platelet activation, and solid clot formation [13-15, 32, 75]. Finally, during the termination phase, thrombin generation is shut down by the protein C pathway (protein C, thrombomodulin, and protein S) and TFPI, and residual serine proteases are inhibited by antithrombin III [33, 35-37, 40-44].
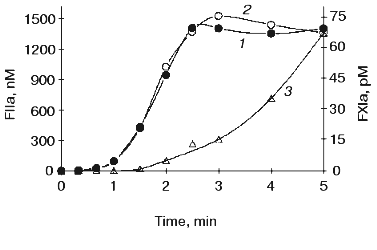
The initiation phase duration (which roughly corresponds to the clotting time) is primarily influenced by the concentration of the initiator, the factor VIIa-tissue factor complex (Fig. 2) [13, 61, 75, 76], which at limited tissue factor is primarily dependent upon the concentration of this protein [75]. The major negative regulator of the initiation phase is TFPI (Fig. 3) [76]. When thrombin generation enters the propagation phase, neither factor VIIa-tissue factor nor TFPI have pronounced effects on the process [13, 61, 75, 76]. Alternatively, the protein C system and antithrombin III have little or no effect on the duration of the initiation phase. However, both of these regulatory systems have prominent effects on the propagation phase of thrombin generation (Fig. 4) [76, 77]. In the “synthetic plasma” model, the limiting reactant controlling this phase in the absence of protein C system is factor Xa [13-15]. However, when the protein C pathway proteins are added to the system, thrombin generation can become dependent upon the presence of active factor Va regulated primarily by the concentration of thrombomodulin [77]. In experiments conducted in the presence of multiple inhibitors of coagulation, synergy is observed with a combination of TFPI and either antithrombin III or protein C system; the two inhibitors taken together produce a more pronounced inhibitory effect (Fig. 4, curve 4) than can be predicted from their individual effects [76, 77] (curves 2 and 3). In the presence of both, TFPI and antithrombin III, the concentration of factor Xa (the limiting reactant of thrombin generation) is decreased by TFPI, which inhibits factor Xa directly and impairs generation of this enzyme by inhibiting factor VIIa-tissue factor complex in a factor Xa-dependent manner [37]. Additionally, antithrombin III also inhibits factor Xa as well as other serine proteases produced in situ during the reaction [33]. In a similar manner, TFPI and the protein C system achieve synergy by the combination of TFPI activity described above and by activation of protein C by thrombin-thrombomodulin followed by the inactivating cleavage of factor Va, an essential component of the prothrombinase complex [40-42, 78, 79]. As a consequence of synergistic effects, thrombin generation becomes a threshold-limited process, influenced by relatively small changes in the concentrations of procoagulants and their inhibitors.Fig. 1. Thrombin generation over time in the “synthetic plasma” mixture. Thrombin generation is initiated with 1.25 pM tissue factor added to the reaction mixture containing mean plasma concentrations of factors V, VII, VIIa, VIII, IX, and X and prothrombin in the presence of 200 µM phospholipids (PCPS) composed of 75% phosphatidylcholine (PC) and 25% phosphatidylserine (PS). Adopted with permission from [15].
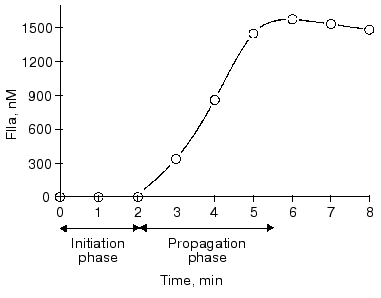
Fig. 2. The initiation phase duration dependence on the factor VIIa-tissue factor concentration in the mathematical model. Thrombin generation is initiated by 5 nM (1), 500 pM (2), 50 pM (3), 10 pM (4), and 5 pM (5) of factor VIIa-tissue factor in the reaction mixture containing mean plasma concentrations of factors V, VIII, IX, and X and prothrombin. Adopted with permission from [61].
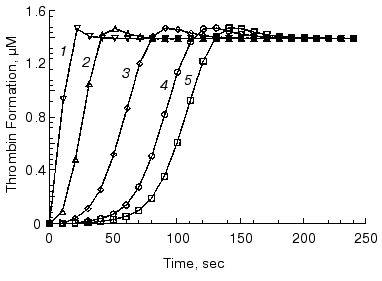
Fig. 3. The influence of TFPI concentration on thrombin generation over time in the “synthetic plasma”. Thrombin generation is initiated by 1.25 pM factor VIIa-tissue factor in the reaction mixture containing mean plasma concentrations of factors V, VIII, IX, and X and prothrombin in the presence of 200 µM PCPS. TFPI concentrations are as follows: 0 (1), 0.5 (2), 1 (3), 2.5 (4), and 5 (5) nM. Adopted with permission from [76].
We have conducted experiments in the tissue factor induced “synthetic plasma” in which concentrations of blood coagulation proteins and their inhibitors are varied from 50 to 150% of their mean plasma values [80], i.e., in the range that is generally considered as being “normal”. In the most extreme situation, when procoagulants are present at 150% of their mean physiological values and all anticoagulants are at 50% (Fig. 5, curve 2), the amount of total thrombin generated in the reaction is 7-fold higher than that observed when all reactants are at 100% (curve 1). At the another extreme of the “normal” situation, when all anticoagulants are present at 150% and procoagulants at 50% (curve 3), the total amount of thrombin produced is decreased to 25% of the value observed in the presence of all proteins at mean plasma concentrations. Thus, the difference in thrombin generation for these two extremes reaches almost 30-fold. The dominant contributors to these extremes are prothrombin and antithrombin III. The addition of purified prothrombin to the whole blood of a normal individual to create a condition in which the prothrombin concentration is 150% of normal (a condition, which can occur in vivo for individuals carrying prothrombin gene mutation G20210A [81]) shortens the clotting time and increases the rate of thrombin generation and the levels achieved. Conversely, the addition of antithrombin III to 150% prolongs the clotting time of whole blood. Individual variation of other coagulation proteins and inhibitors within 50-150% range has little (if any) effect on thrombin generation in the “synthetic plasma” mixtures. Only when factor V, factor VIII, factor IX, and factor X are simultaneously reduced to 50% of their mean plasma concentrations is a substantial decrease in the total thrombin observed.Fig. 4. Synergistic effect of physiological TFPI and antithrombin III concentrations on thrombin generation in the “synthetic plasma”. Thrombin generation is initiated by 1.25 pM factor VIIa-tissue factor in the reaction mixture containing mean plasma concentrations of factors V, VIII, IX, and X and prothrombin in the presence of 200 µM PCPS (1) and in the presence of either TFPI (2) or antithrombin III (3), or both (4). Adopted with permission from [76].
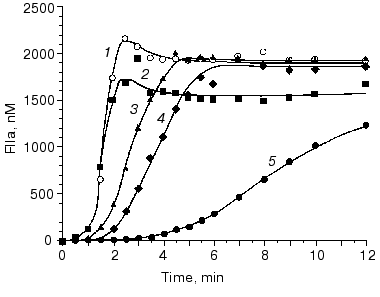
Several disorders of blood coagulation occurring in vivo and caused either by deficiencies of blood coagulation proteins or by their mutations were evaluated in our models. Figure 6 [82] illustrates the influence of factor VLeiden (Arg506-->Gln) on thrombin generation in the “synthetic plasma” model. This mutation abolishes an inactivating cleavage site of factor V(a) at position 506 by the activated protein C [74]. As a consequence of the Arg506-->Gln mutation, factor Va is able to maintain its cofactor activity for a prolonged period of time when compared with the wild-type factor Va [83]. In the absence of the protein C pathway, the thrombin generation profile is almost not altered by the presence of factor VLeiden (Fig. 6, curves 1 and 2). In the presence of protein C at a physiological concentration and thrombomodulin at a relatively high concentration (10 nM), a significant attenuation of thrombin generation (especially during the propagation phase) is observed when the wild-type factor V is present in the reaction mixture (curve 4). In contrast, with homozygous factor VLeiden only a marginal effect on thrombin generation is observed in the presence of the protein C pathway components (curve 6). These data indicate, that the cause of an increased thrombotic risk for the carriers of factor VLeiden is an accelerated and elevated thrombin production during the blood coagulation process.Fig. 5. Thrombin generation in the “synthetic plasma” at extremes of the normal range of procoagulants and coagulation inhibitors. Thrombin generation is initiated by 5 pM factor VIIa-tissue factor in the presence of PCPS at 200 µM and all proteins at mean plasma concentrations (1), in the presence of procoagulants (factors V, VIII, IX, and X and prothrombin) at 150% and coagulation inhibitors (TFPI and antithrombin III) at 50% of their mean plasma concentrations (2), and in the presence of procoagulants at 50% and coagulation inhibitors at 150% of their mean plasma concentrations (3). Adopted with permission from [80].
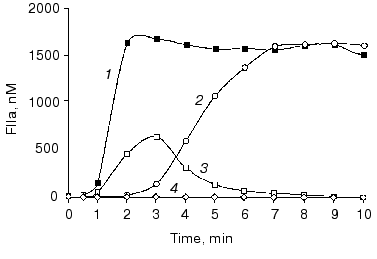
Based upon calculations of prothrombinase activity and the time-course of platelet, factor V and factor X activation (in whole blood and in “synthetic plasma”), it was established that at moderate thrombomodulin concentrations (<1 nM) factor Xa and not factor Va or the membrane prothrombinase binding site concentration is the limiting component of prothrombinase during tissue factor-induced thrombin generation (Table 4) [14, 78]. During the initiation phase, factor Xa and factor IXa are solely generated by the factor VIIa-tissue factor complex [15]. Factor VIII activation by thrombin and the extrinsic factor Xase formation accelerates factor X activation during the propagation phase dramatically [20, 21] (Fig. 7, compare curve 1 and other curves; also see Table 3). As a consequence of this pattern, prothrombin activation during the initiation phase (and the clotting time) is not greatly affected by the absence of precursors of the extrinsic factor Xase (factor VIII and factor IX). However, in the absence of factor VIII or factor IX, thrombin generation during the propagation phase is suppressed [13, 61, 75, 84] (Fig. 8, compare curves 1 and 2). In an extreme situation, when the initiator (factor VIIa-tissue factor complex) is present at relatively low concentrations (<5 pM, below threshold), thrombin generation does not occur [84]. As a consequence of an impaired prothrombin activation in the hemophilia A and B blood, the onsets of platelet activation and clot formation are slightly delayed although they proceed to completion. The addition of factor VIII at physiological concentration (0.7 nM) to hemophilia A blood corrects thrombin generation, platelet activation, and clot formation [75] (Fig. 8, curve 3).Fig. 6. The effect of factor VLeiden on thrombin generation in the “synthetic plasma”. Thrombin generation is initiated by 1.25 pM factor VIIa-tissue factor in the reaction mixture containing mean plasma concentrations of factors V, VIII, IX, and X, TFPI, and prothrombin in the presence of 200 µM PCPS. Closed symbols represent reactions for normal factor V (1) and factor VLeiden (2) in the absence of the protein C pathway proteins. Open symbols represent reactions in the presence of the protein C pathway (65 nM protein C and 10 nM thrombomodulin) with or without protein S (300 nM). Normal factor V with (3) and without protein S (4), factor VLeiden with (5) and without protein S (6). Adopted with permission from [82].
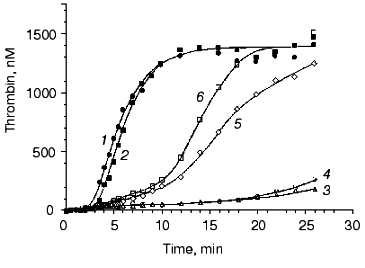
Table 4. Concentrations of
prothrombinase components during whole blood clotting [14]
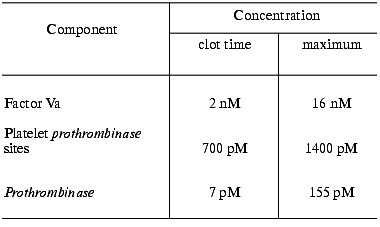
Fig. 7. Factor Xa generation over time in the “synthetic plasma”. Thrombin generation is initiated by 1.25 pM factor VIIa-tissue factor in the reaction mixture containing mean plasma concentrations of factors V, VIII, IX, and X and prothrombin in the presence of 200 µM PCPS (1), in the absence of factor VIII (2), in the absence of prothrombin (3), and in the presence of 3 µM specific thrombin inhibitor hirudin (4). Adopted with permission from [15].
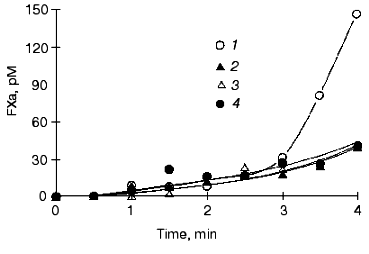
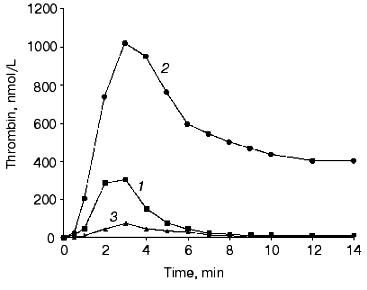
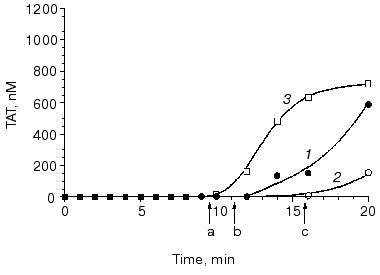
Replacement with factor VIII or factor IX for hemophilia A or B patients who have developed antibodies against these proteins is usually less effective (if at all) and a more aggressive treatment has to be applied [70, 85, 86]. One of possible options, which has been gaining an increasing popularity worldwide, is a replacement with the recombinant factor VIIa [87-90]. Therapeutic concentrations of this enzyme injected/infused during the treatment (~30 nM) are many fold higher than those found in vivo in a normal blood (~0.1 nM) [7, 87, 89, 90]. However, our data obtained in “synthetic plasma” and in hemophilia A and B blood indicate, that factor VIIa at therapeutic concentrations is not able to restore normal thrombin generation. As a consequence, clot formation is little affected by the addition of factor VIIa. Moreover, an increase in factor VIIa concentration above 10 nM (up to 50 nM) has almost no effect on thrombin generation in hemophilia A and B blood [91].Fig. 8. The influence of factor VIII on thrombin generation in whole blood. Thrombin generation (TAT) is initiated by 25 pM tissue factor in normal (1) and hemophilia A blood without replacement (2) and with recombinant factor VIII (3). Arrows indicate clotting time in normal (a) and hemophilia A blood with (b) and without (c) replacement. CTI concentration is 100 µg/ml. Adopted with permission from [75].
It is established that a deficiency in factor XI (hemophilia C) causes bleeding disorders (most often mild) infrequently, indicating an accessory role of this protein in normal hemostasis [92, 93]. Similarly, in “synthetic plasma” (Fig. 9, compare curves 1 and 2) [15] and in whole blood the absence of, or deficiency in, factor XI has almost no effect on thrombin generation, platelet activation, and clot formation at a relatively high tissue factor concentration [15, 75]. This is due to slow factor XI activation by thrombin and, as a consequence, a relatively late accumulation of factor XIa, i.e., only low subpicomolar amounts of this enzyme are generated when prothrombin activation enters the propagation phase (Fig. 9, curve 3) [15]. However, when the initiation phase of thrombin generation is prolonged either due to low tissue factor concentration or caused by inhibitory effects of elevated concentrations of some proteins involved in blood coagulation, factor XI plays an important role [75, 94]. Thus, when coagulation of normal blood is initiated by 5 pM tissue factor, an approximately 10 min clotting time is observed (Fig. 10, arrow a). The clotting of a hemophilia C patient's blood is prolonged to almost 16 min at 5 pM tissue factor (arrow c). The addition of 25 nM factor XI (physiological concentration) corrects clotting time of the hemophilia C blood (arrow b).
Fig. 9. The influence of factor XI on thrombin generation in the “synthetic plasma”. Thrombin generation is initiated by 1.25 pM factor VIIa-tissue factor in the reaction mixture containing mean plasma concentrations of factors V, VIII, IX, and X and prothrombin in the presence of 200 µM PCPS and in the presence of 100% (1) or absence (2) of plasma factor XI; 3) factor XIa generation over time. Adopted with permission from [15].
Platelets are an essential component of the blood coagulation process in vivo. Aggregated, activated platelets provide procoagulant phospholipid-equivalent surfaces upon which the complex-dependent reactions of the blood coagulation cascade are localized [16, 95]. Additionally, platelets are also a storage compartment of proteins involved in blood coagulation and its regulation [96]. A deficiency in functional platelets, i.e., thrombocytopenia, is associated with bleeding complications [97, 98]. In “synthetic plasma” and in whole blood, thrombin generation profiles observed at platelet concentrations below 0.1*108/ml (<5% of mean plasma value), the thrombin generation profile is similar to that observed in the severe hemophilia blood [75, 99]. These data are consistent with in vivo subjective observations [100, 101]. A strong platelet antagonist, such as prostaglandin E1 used at a pharmacological concentration, can also produce a comparable situation and substantially prolong the clotting time and suppress thrombin generation [102].Fig. 10. Thrombin generation (TAT) in the hemophilia C (factor XI-deficient) blood. Thrombin generation is initiated by 5 pM tissue factor in a normal (1), hemophilia C (2), and hemophilia C with 100% factor XI replacement (3) blood. Arrows indicate clotting time in normal (a) and hemophilia C blood with (b) and without (c) replacement. CTI concentration is 100 µg/ml. Adopted with permission from [75].
The tissue factor-initiated process of blood coagulation based primarily on the knowledge obtained using three in vitro models of blood coagulation is discussed in this review. The majority of data accumulated in these models are in good agreement with the results of the in vivo observations.
This work was supported by Grant HL 46703 from the National Institutes of Health.
REFERENCES
1.Wilcox, J. N., Smith, K. M., Schwartz, S. M., and
Gordon, D. (1989) Proc. Natl. Acad. Sci. USA, 86,
2839-2843.
2.Weiss, H. J., Turitto, V. T., Maumgartner, H. R.,
Nemerson, Y., and Hoffmann, T. (1989) Blood, 73,
968-975.
3.Carlsen, E., Flatmark, A., and Prydz, H. (1988)
Transplantation, 46, 575-580.
4.Bevilaqua, M. P., Pober, J. S., Majeau, G. R., et
al. (1986) Proc. Natl. Acad. Sci. USA, 83, 4533-4537.
5.Yang, H. L., Lu, F. J., Wung, S. L., et al. (1994)
Thromb. Haemost., 71, 325-330.
6.Camera, M., Giesen, P. L., Fallon, J., et al.
(1999) Arterioscler. Thromb. Vasc. Biol., 19,
531-537.
7.Morrissey, J. H., Macik, B. G., Neuenschwander, P.
F., et al. (1993) Blood, 81, 734-744.
8.Eichinger, S., Mannucci, P. M., Tradati, F., et al.
(1995) Blood, 86, 3021-3025.
9.Silverberg, S. A., Nemerson, Y., and Zur, M. (1977)
J. Biol. Chem., 252, 8481-8488.
10.Komiyama, Y., Pedersen, A. H., and Kisiel, W.
(1990) Biochemistry, 29, 9418-9425.
11.Bom, V. J. J., and Bertina, R. M. (1990)
Biochem. J., 265, 327-336.
12.Lawson, J. H., and Mann, K. G. (1991) J. Biol.
Chem., 266, 11317-11327.
13.Lawson, J. H., Kalafatis, M., Stram, S., and
Mann, K. G. (1994) J. Biol. Chem., 269, 23357-23366.
14.Rand, M. D., Lock, J. B., van 't Veer, C.,
Gaffney, D. P., and Mann, K. G. (1996) Blood, 88,
3432-3445.
15.Butenas, S., van 't Veer, C., and Mann, K. G.
(1997) J. Biol. Chem., 272, 21527-21533.
16.Rosing, J., van Rijn, J. L. M. L., Bevers, E. M.,
van Dieijen, G., Comfurius, P., and Zwaal, R. F. (1985) Blood,
65, 319-332.
17.Tans, G., Rosing, J., Thomassen, M. C., Heeb, M.
J., Zwaal, R. F., and Griffin, J. H. (1991) Blood, 77,
2641-2648.
18.Hugel, B., Socie, G., Vu, T., et al. (1999)
Blood, 93, 3451-3456.
19.Stern, D., Nawroth, P., Handley, D., and Kisiel,
W. (1985) Proc. Natl. Acad. Sci. USA, 82, 2523-2527.
20.Mann, K. G., Krishnaswamy, S., and Lawson, J. H.
(1992) Sem. Hematol., 29, 213-226.
21.Ahmad, S. S., Rawala-Sheikh, R., and Walsh, P. N.
(1992) Sem. Thromb. Hemost., 18, 311-323.
22.Papahadjopoulos, D., and Hanahan, D. J. (1964)
Biochim. Biophys. Acta, 90, 436-439.
23.Nesheim, M. E., Taswell, J. B., and Mann, K. G.
(1979) J. Biol. Chem., 254, 10952-10962.
24.Kalafatis, M., Swords, N. A., Rand, M. D., et al.
(1994) Biochim. Biophys. Acta, 1227, 113-129.
25.Gailani, D., and Broze, G. J., Jr. (1991)
Science, 253, 909-912.
26.Pieters, J., Lindhout, T., and Hemker, H. C.
(1989) Blood, 74, 1021-1024.
27.Mosesson, M. W. (1992) Sem. Hematol.,
29, 177-188.
28.Bailey, K., Bettelheim, F. R., Lorand, L., et al.
(1951) Nature, 167, 233-234.
29.Lorand, L., and Konishi, K. (1964) Arch.
Biochem. Biophys., 105, 58-67.
30.Naski, M. C., Lorand, L., and Shafer, J. A.
(1991) Biochemistry, 30, 934-941.
31.Shen, L., and Lorand, L. (1983) J. Clin.
Invest., 71, 1336-1341.
32.Brummel, K. E., Butenas, S., and Mann, K. G.
(1999) J. Biol. Chem., 274, 22862-22870.
33.Olson, S. T., Bjork, I., and Shore, J. D. (1993)
Meth. Enzymol., 222, 525-559.
34.Nowotny, W. F., Brown, S. G., Miletich, J. P., et
al. (1991) Blood, 78, 387-393.
35.Girard, T. J., Warren, L. A., Novotny, W. F., et
al. (1989) Nature, 338, 518-520.
36.Rapaport, S. I. (1991) Thromb. Haemost.,
66, 6-15.
37.Broze, G. J., Jr., Warren, L. A., Novotny, W. F.,
et al. (1988) Blood, 71, 335-343.
38.Esmon, N. L., Owen, W. G., and Esmon, C. T.
(1982) J. Biol. Chem., 257, 859-864.
39.Esmon, C. T., Esmon, N. L., Bonniec, B. F., et
al. (1993) Meth. Enzymol., 222, 359-385.
40.Walker, F. J., Sexton, P. W., and Esmon, C. T.
(1979) Biochim. Biophys. Acta, 571, 333-342.
41.Suzuki, K., Stenflo, J., Dahlback, B., et al.
(1983) J. Biol. Chem., 258, 1914-1920.
42.Hockin, M. F., Kalafatis, M., Shatos, M., and
Mann, K. G. (1997) Arterioscler. Thromb. Vasc. Biol., 17,
2765-2775.
43.Eaton, D., Rodriguez, H., and Vehar, G. A. (1986)
Biochemistry, 25, 505-512.
44.Esmon, C. T. (1987) Science, 235,
1348-1352.
45.Hackeng, T. M., van 't Veer, C., Meijers, J. C.
M., and Bouma, B. N. (1994) J. Biol. Chem., 269,
21051-21058.
46.Koppelman, S. J., van 't Veer, C., Sixma, J. J.,
and Bouma, B. N. (1995) Blood, 86, 2653-2660.
47.Van 't Veer, C., Butenas, S., Golden, N. J., and
Mann, K. G. (1999) Thromb. Haemost., 82, 80-87.
48.Lollar, P., and Parker, C. G. (1990) J. Biol.
Chem., 265, 1688-1692.
49.Fay, P. J., Haidaris, P. J., and Smudzin, T. M.
(1991) J. Biol. Chem., 266, 8957-8962.
50.Lu, D., Kalafatis, M., Mann, K. G., et al. (1997)
Blood, 87, 4708-4717.
51.Pipe, S. W., Saenko, E. L., Eickhorst, A. N.,
Kemball-Cook, G., and Kaufman, R. J. (2001) Blood, 97,
685-691.
52.Kalafatis, M., Egan, J. O., van 't Veer, C.,
Cawthern, K. M., and Mann, K. G. (1997) Crit. Rev. Eukaryot. Gene
Expr., 7, 241-280.
53.Seegers, W. H. (1949) Proc. Soc. Exptl. Biol.
Med., 72, 677-680.
54.Mann, K. G., Nesheim, M., Church, W., Haley, P.,
and Krishnaswamy, S. (1990) Blood, 76, 1-16.
55.Tracy, P. B., Nesheim, M. E., and Mann, K. G.
(1981) J. Biol. Chem., 256, 743-751.
56.Lamphear, B. J., and Fay, P. J. (1992) J.
Biol. Chem., 267, 3725-3730.
57.Nelsestuen, G. L., Kisiel, W., and DiScipio, R.
G. (1978) Biochemistry, 17, 2134-2138.
58.Hoffman, M., Monroe, D. M., Oliver, J. A., and
Roberts, H. R. (1995) Blood, 86, 1794-1801.
59.Kjalke, M., Oliver, J. A., Monroe, D. M., et al.
(1997) Thromb. Haemost., 78, 1202-1208.
60.Kjalke, M., Monroe, D., Hoffman, M., et al.
(1998) Thromb. Haemost., 80, 578-584.
61.Jones, K. C., and Mann, K. G. (1994) J. Biol.
Chem., 269, 23367-23373.
62.Beltrami, E., and Jesty, J. (1995) Proc. Natl.
Acad. Sci. USA, 92, 8744-8748.
63.Kirchhofer, D., Tschopp, T. B., and Baumgartner,
H. R. (1995) Arterioscler. Thromb. Vasc. Biol., 15,
1098-1106.
64.Peyrou, V., Lormeau, J. C., Herault, J. P., et
al. (1999) Thromb. Haemost., 81, 400-406.
65.Doggen, C. J., Cats, V. M., Bertina, R. M., and
Rosendaal, F. R. (1998) Circulation, 97, 1037-1041.
66.Chiu, H. C., Whitaker, E., and Colman, R. W.
(1983) J. Clin. Invest., 72, 493-503.
67.Poort, S. R., Michiels, J. J., Reitsma, P. H.,
and Bertina, R. M. (1994) Thromb. Haemost., 72,
819-824.
68.Ragni, M. V., Lewis, J. H., Spero, J. A., and
Hasiba, U. (1981) Am. J. Hematol., 10, 79-88.
69.Hoyer, L. W. (1994) N. Engl. J. Med.,
330, 38-47.
70.Ozsoylu, S., and Ozer, F. L. (1973) Acta
Haematol., 50, 305-314.
71.Coller, B. S., Owen, J., Jesty, J., et al. (1987)
Arteriosclerosis, 7, 456-462.
72.Woodward, M., Lowe, G. D., Rumley, A., et al.
(1997) Br. J. Haematol., 97, 785-797.
73.Harrison, L., Johnston, M., Massicotte, P., et
al. (1997) Ann. Int. Med., 126, 133-136.
74.Bertina, R. M., Koeleman, B. P., Koster, T., et
al. (1994) Nature, 369, 64-67.
75.Cawthern, K. M., van 't Veer, C., Lock, J. B.,
DiLorenzo, M. E., Branda, R. F., and Mann, K. G. (1998) Blood,
91, 4581-4592.
76.Van 't Veer, C., and Mann, K. G. (1997) J.
Biol. Chem., 272, 4367-4377.
77.Van 't Veer, C., Golden, N. J., Kalafatis, M.,
and Mann, K. G. (1997) J. Biol. Chem., 272,
7983-7994.
78.Kalafatis, M., Rand, M. D., and Mann, K. G.
(1994) J. Biol. Chem., 269, 31869-31880.
79.Hockin, M. F., Cawthern, K. M., Kalafatis, M.,
and Mann, K. G. (1999) Biochemistry, 38, 6918-6934.
80.Butenas, S., van 't Veer, C., and Mann, K. G.
(1999) Blood, 94, 2169-2178.
81.Poort, S. R., Rosendaal, F. R., Reitsma, P. H.,
and Bertina, R. M. (1996) Blood, 88, 3698-3703.
82.Van 't Veer, C., Kalafatis, M., Bertina, R. M.,
Simioni, P., and Mann, K. G. (1997) J. Biol. Chem., 272,
20721-20729.
83.Kalafatis, M., Bertina, R. M., Rand, M. D., and
Mann, K. G. (1995) J. Biol. Chem., 270, 4053-4057.
84.Van 't Veer, C., Golden, N. J., Kalafatis, M.,
Simioni, P., Bertina, R. M., and Mann, K. G. (1997) Blood,
90, 3067-3072.
85.Shapiro, S. S., and Hultin, M. (1975) Sem.
Thromb. Hemost., 1, 336-385.
86.Lusher, J. M., Arkin, S., Abildgaard, C. F., and
Schwartz, R. S. (1993) N. Engl. J. Med., 328,
453-459.
87.Nicolaisen, E. M., Hansen, L. L., Poulsen, F.,
Glazer, S., and Hedner, U. (1996) Thromb. Haemost., 76,
200-204.
88.Hedner, U. (2000) Sem. Thromb. Hemost.,
26, 363-366.
89.Shapiro, A. D., Gilchrist, G. S., Hoots, W. K.,
Cooper, H. A., and Gastineau, D. A. (1998) Thromb. Haemost.,
80, 773-778.
90.Ingerslev, J. (2000) Sem. Thromb. Hemost.,
26, 425-432.
91.Butenas, S., Brummel, K. E., Branda, R. F., and
Mann, K. G. (2000) Blood, 96, 2290 (abstract).
92.Walsh, P. N. (1992) Sem. Hematol.,
29, 189-201.
93.Bolton-Maggs, P. H. B. (1996) Baillieres Clin.
Hematol., 9, 355-368.
94.Butenas, S., Brummel, K. E., and Mann, K. G.
(2001) Abst. XVIII Congr. of the ISTH.
95.Walsh, P. N. (1974) Blood, 43,
597-605.
96.Hartwig, J. H. (1998) Thrombosis and
Hemorrhage (Loscalzo, J., and Schafer, A. I., eds.) Williams and
Wilkins, Baltimore, MD, pp. 207-228.
97.Rutherford, C. J., and Frenkel, E. P. (1994)
Med. Clin. North. Am., 78, 555-575.
98.Miescher, P. A. (1973) Sem. Hematol.,
10, 311-325.
99.Butenas, S., Branda, R. F., van 't Veer, C.,
Cawthern, K. M., and Mann, K. G. (2001) Thromb. Haemost.,
86, 660-667.
100.Beutler, E. (1993) Blood, 81,
1411-1413.
101.Roy, A. J., Jaffe, N., and Djerassi, I. (1973)
Transfusion, 13, 283-290.
102.Butenas, S., Cawthern, K. M., van 't Veer, C.,
DiLorenzo, M., Lock, J. B., and Mann, K. G. (2001) Blood,
97, 2314-2322.