REVIEW: Thrombin Regulation of Cell Function through Protease-Activated Receptors: Implications for Therapeutic Intervention
C. K. Derian, B. P. Damiano, M. R. D'Andrea, and P. Andrade-Gordon*
The R. W. Johnson Pharmaceutical Research Institute, Welsh and McKean Roads, P.O. Box 776, Spring House, Pennsylvania 19477-0776, USA; fax: 1-(215) 628-3297; E-mail: pandrade@prius.jnj.com* To whom correspondence should be addressed.
Received June 15, 2001; Revision received September 21, 2001
The serine protease thrombin is well recognized as being pivotal to the maintenance of hemostasis under both normal and pathological conditions. Its cellular actions are mediated through a unique family of protease-activated receptors (PARs). These receptors represent a novel family of G protein-coupled receptors that undergo proteolytic cleavage of their amino terminus and subsequent autoactivation by a tethered peptide ligand. This paper reviews the consequences of PAR activation in thrombosis, vascular injury, inflammation, tissue injury, and within the tumor microenvironment.
KEY WORDS: thrombin, platelets, vascular injury, inflammation, tumor microenvironment, fibroblasts, protease-activated receptor
Abbreviations: PAR) protease-activated receptor; GPCR) G-protein coupled receptor; TME) tumor microenvironment.
PROTEASE ACTIVATED RECEPTORS: AN EMERGING FAMILY OF G-PROTEIN
COUPLED RECEPTORS
Classically, cell surface receptors are activated by ligand binding. This interaction induces a conformational change in the receptor that results in a signal being transmitted across the cell membrane. The activation is reversible in that the ligand can dissociate from its receptor. Seven transmembrane domain G-protein coupled receptors (GPCR) are perhaps the most important and well-characterized type of receptor, providing the basis for numerous pharmacological interventions. In recent years, it has become clear that GPCRs mediate the actions of almost all types of chemical messengers from small transmitters (catecholamines, histamine, acetylcholine, etc.), lipid messengers (prostaglandins, leukotrienes, platelet activating factor, etc.), neuropeptides (opiates, neuropeptide Y, tachykinins, etc.), peptide hormones (glucagon, angiotensin, calcitonin, etc.), to large glycoproteins (thyroid-stimulating hormone, luteinizing hormone, etc.).
Within this family of GPCRs, a different and unique mechanism of activation was discovered with the cloning of the thrombin receptor [1]. This novel receptor was termed protease-activated receptor-1 (PAR-1). PAR-1 is activated by proteolytic cleavage at a specific site within the extracellular amino-terminal domain, exposing a new amino terminus, which is then thought to interact with a distal region of the receptor, leading to receptor activation. The discovery of this receptor has provided a framework for understanding how thrombin communicates with cells and provides a new pharmacological target for controlling thrombin's cellular actions. This unusual receptor exemplifies an emerging family of GPCRs that are activated by proteolysis. Given that proteases are very common enzymes in organisms, the catalytic activation of these receptors would seem to be an efficient means to regulate cellular responses. Indeed, many protease-activation mechanisms have been identified in biology, suggesting an important role for protease activated receptors.
PAR-1. PAR-1 exhibits an extended amino terminus containing both a putative thrombin cleavage site (LDPR41/S42) and a hirudin-like domain (K52YEPF) complementary to thrombin's anion binding exosite [2]. Thrombin binds to PAR-1 with increased affinity through this latter site and subsequently cleaves the extended amino terminus. This mechanism of receptor activation is supported by studies utilizing synthetic peptides representing this new amino terminus: SFLLRN for human PAR-1, SFFLRN for hamster and rat PAR-1. Agonist peptides can directly activate recombinant as well as native receptor. Additional structural features of the receptor have been identified from receptor modeling and mutagenesis studies. The second extracellular loop contains several residues important for ligand/receptor interaction [3-6] and thus represents an important site for targeted receptor modulators. The intracellular loops are recognized sites for G-protein coupling and the cytoplasmic tail contains numerous sites for receptor phosphorylation leading to receptor inactivation and internalization [7-9]. PAR-1 from both hamster and rat were also cloned and shown to be highly homologous to human PAR-1 [10, 11]. PAR-1 is widely distributed among cells and tissues, consistent with thrombin's widespread effects described in in vitro studies. Thus, regulation of PAR-1 expression and/or activity presents an important approach to modulating thrombin's actions.
PAR-3 and PAR-4. Since the discovery of PAR-1, three additional PARs have been discovered: PAR-2, PAR-3, and PAR-4 [12-16]. PAR-2 was first identified from a mouse genomic library using hybridization probes derived from the bovine substance K receptor [12]. The human gene was subsequently isolated from a human genomic library and cloned from human kidney cDNA [13, 17]. PAR-2 is the only PAR described to date that is not activated by thrombin. The most widely studied proteolytic activators of PAR-2 are trypsin and tryptase [12, 18-20], however several other serine proteases have recently been shown to activate PAR-2 including membrane-type serine protease-1, factor VIIa, and factor Xa [21, 22]. The cell and tissue distribution of PAR-2, like PAR-1, is widespread with significant expression in pancreas, colon, kidney, small intestine, and liver. PAR-2 is notably absent from platelets.
PAR-3 was first described in PAR-1 deficient mice whose platelets were fully responsive to thrombin [14, 23]. PAR-3 contains a consensus sequence for the thrombin anion-binding site indicating that it is a putative substrate for thrombin cleavage. However, PAR-3 is unique among the PARs in that it is not activated by synthetic peptides representing its tethered peptide ligand. Thus the functional role of PAR-3, other than in murine platelet activation, remains unclear. Human and murine PAR-3 display distinct tissue distributions based on Northern analysis. Human PAR-3 is expressed in several tissues including heart, liver, pancreas, small intestine, stomach, bone marrow, and vascular endothelial cells but is absent from human platelets [14, 24]. In contrast, megakaryocytes in the spleen were the only primary site of PAR-3 expression in mouse tissues [14].
Human PAR-4 was discovered using a bioinformatics approach employing est sequences homologous to other PARs [16]. Concurrently, murine PAR-4 was described in studies with PAR-3 deficient mice [15]. These mice, while devoid of thrombin sensitive responses at low nanomolar thrombin concentrations, were still responsive to higher thrombin concentrations. These observations led to a search for an additional thrombin receptor, culminating in the identification of murine PAR-4. PAR-4, unlike both PAR-1 and PAR-3, does not possess a consensus sequence for binding of thrombin's anionic exosite. Human PAR-4 is activated by approximately 100-fold higher thrombin concentrations than PAR-1. Part of this discrepancy may be attributable to the lack of this binding site on the receptor, which is important for designating thrombin's specificity and potency. The separation in thrombin potency between PAR-1 and PAR-4 implies that they may serve distinct roles in mediating cell function. Alternatively, for human PAR-4 other proteases such as trypsin or cathepsin G may be the primary activators [15, 16, 25].
PARs IN THROMBOSIS
Platelets are the principal cell type participating in the regulation of hemostasis. They are activated by a variety of stimuli including collagen, ADP, thromboxane A2, epinephrine, serotonin, and thrombin, but thrombin is considered the most potent stimulus of platelets. Thrombin induces platelet shape change, degranulation, aggregation, and biosynthesis of lipids, all of which contribute to the generation of thrombi. While these events are important during normal hemostasis to maintain the integrity of the vasculature, increased generation of thrombin and formation of large thrombi that lead to vessel occlusion contribute to the often fatal outcome of myocardial infarction, angina, and stroke. Thus, understanding the mechanism by which thrombin can activate platelets may provide important therapeutic approaches to treating thrombotic related diseases.
The presence of both PAR-3 and PAR-4 in murine platelets led Kahn et al. [15] to propose a dual thrombin receptor activating system in platelets. The thrombin sensitivity in murine platelets in wild type versus PAR-3 deficient mice raised the intriguing possibility that PAR-3 and PAR-4 interacted in some way to signal as PAR-1 does in human platelets. Indeed, recent studies described the cooperativity of the two receptors whereby PAR-3 acted in concert with PAR-4 by localizing thrombin to the cell surface, allowing PAR-4 to be cleaved and activated [26]. The putative role of the PAR-3 tethered peptide ligand remains elusive however. The functionality of PAR-3 alone in murine platelets awaits the generation of a PAR-4 deficient mouse. A dual mechanism of activation has also been proposed for PAR-1 and PAR-4 in human platelets [27]. Interestingly, PAR-4 activation in human platelets has been shown, however PAR-3 has not been identified in human platelets via sensitive methods such as reverse transcriptase-polymerase chain reaction. These observations suggest that PAR pairing seems to be the preferred system by which platelets respond to thrombin, PAR-1/PAR-4 in human platelets and PAR-3/PAR-4 in murine platelets. In other tissues however, the functional role of PAR-3 and PAR-4 remains to be fully characterized. PAR-3 has been described in endothelial cells for example, but PAR-4 has not. Through the use of PAR specific probes and antibodies, the tissue expression patterns for these receptors will be described, ultimately leading to studies that better define the functional role of these additional thrombin responsive receptors.
Clearly, PAR-1 mediates the majority of thrombin's actions on platelets due to its sensitivity to low concentrations of thrombin. Inhibition of PAR-1 in isolated human platelets was obtained with receptor blocking antibodies or the PAR-1 antagonist BMS-200261 [27]. These agents blocked low concentrations of thrombin (1 nM) but were not effective at relatively high concentrations of the enzyme (30 nM). However, total inhibition of platelet aggregation was demonstrated by blocking both PAR-1 and PAR-4 but not either receptor alone. Limitations on the expression of PAR-1 on platelets of different species have hampered our attempts at understanding the significance of PAR-1 relative to other thrombin responsive mechanisms. Several comparative studies demonstrated that among laboratory species, only guinea pig and non-human primates expressed PAR-1 [28-30]. The potential role for thrombin mediated PAR-1 activation in thrombus formation in vivo was first demonstrated with the use of a blocking antibody recognizing the hirudin-like domain in the amino terminal extension of PAR-1 in a non-human primate model of thrombosis [31]. Interruption of thrombin's ability to bind to this region inhibited arterial thrombus formation measured by a decrease in cyclic flow reductions.
More recently, we have identified novel, selective PAR-1 antagonists, represented by RWJ-56110 and RWJ-58259, which effectively inhibit ex vivo platelet aggregation in guinea pigs [32, 33]. These agents represent excellent tools to probe the role of PAR-1 in normal and pathological states. We used RWJ-58259 as a prototype compound to assess the effects of PAR-1 inhibition in platelet dependent thrombosis in vivo. We evaluated RWJ-58259 in two models of thrombosis, the arteriovenous shunt and photoactivation models. Surprisingly, we found that PAR-1 inhibition had no appreciable inhibitory effect on thrombus formation despite significant inhibition of both thrombin and SFLLRN-induced platelet aggregation. Further exploration of the PAR profile on guinea pig platelets revealed a novel triple PAR profile: PAR-1, PAR-3, and PAR-4. Recently, studies exploring the interaction of PAR-3 and PAR-4, as they relate to murine platelet responsiveness to thrombin, were described. Using recombinant systems, PAR-3 was shown to act as a cofactor for thrombin-induced PAR-4 cleavage and activation through binding of thrombin to PAR-3's hirudin-like domain [26]. This unique observation suggests a scenario whereby guinea pig platelets possess the potential for two equivalent thrombin-activating pathways: PAR-1 and PAR-3/PAR-4. A PAR profile as such would limit the usefulness of this species as a model for assessing the role of human PAR-1 in platelet dependent disorders since human platelets do not express PAR-3. Thus, evaluation of the potential antithrombotic efficacy of a PAR-1 antagonist such as RWJ-58259 would require studies that overcome this species issue, such as the use of non-human primate models.
THROMBIN AND PAR-1 IN VASCULAR INJURY
Several lines of evidence suggest that thrombin and, in particular, thrombin activation of PAR-1, plays a pivotal role in vascular injury responses associated with thrombosis, atherosclerosis, balloon angioplasty, and stent implantation [34-36]. The identified vascular actions of thrombin-induced PAR-1 activation comprise the critical processes mediating vascular injury. Although there are many other mediators participating in the injury response, thrombin may act more globally to initiate these responses. Platelet adherence and aggregation, partly the result of PAR-1 activation by thrombin, are critical early events in initiating other vascular responses to injury through the release of other growth factors such as platelet derived growth factor [37]. Thrombin and PAR-1 mediated endothelial cell activation causes increased vascular permeability [38, 39], release of nitric oxide [40], endothelin [41], prostaglandins [42], von Willebrand factor [43], and increased adhesion molecule expression [44]. The latter can lead to neutrophil adhesion at the site of injury. Thrombin-induced vasoconstriction is PAR-1 mediated [40, 45-47]. Acutely, this vasoconstriction may result in underperfusion or ischemia due to narrowing of the vessel lumen [48-50] and potentially increasing the risk of an occlusive thrombus. Vascular smooth muscle cell proliferation in rat [51], rabbit, [52] and human [53] is induced by PAR-1 activation. In addition to cellular proliferation, extracellular matrix protein turnover is an important component of the vascular injury response particularly in mediating restenosis following angioplasty in human vessels [54]. Thrombin activation of PAR-1 causes vascular smooth muscle procollagen synthesis [55], increased synthesis and activation of matrix metalloproteinases in endothelial cells [56], and endothelial cell secretion of extracellular matrix proteins [57].
The local elevation of thrombin concentrations at a site of thrombus is well established [58]. The significant incidence of intramural thrombus following angioplasty of atherosclerotic vessels may provide a sustained exposure to thrombin at the vascular injury site [59]. In addition, PAR-1 protein expression and mRNA appear to be upregulated in vascular injury in rat and baboon arteries [60], where PAR-1 expression is associated with proliferating vascular smooth muscle cells. Similar PAR-1 upregulation has been demonstrated following vascular injury in rabbit [61] and mouse [62] carotid arteries and in vein graft remodeling in rats [63]. PAR-1 expression is also increased in human atherosclerotic vessels [64-66]. Thus, the association of elevated thrombin concentrations and increased PAR-1 expression at a site of vascular injury is consistent with a role for thrombin and PAR-1 in vascular injury. Inhibitors of thrombin, such as hirudin, reduce neointimal thickening induced by balloon angioplasty in normal and hypercholesterolemic rabbits and pigs, and in normal rats [67-71]. However, thrombin inhibitors will not only inhibit the activation of PAR-1 but will also inhibit activation of other thrombin receptors such as PAR-3 and PAR-4 and will prevent the other enzymatic actions of thrombin such as coagulation and fibrin polymerization. Thus, the effectiveness of the direct thrombin inhibitors does not prove a role for PAR-1 in vascular injury. Therefore, a number of alternative approaches have been used to assess the specific role of PAR-1. For example, the vascular injury response in mice made genetically deficient in PAR-1 is altered compared to wild type mice [62]. Specifically, intimal area and increased medial area in response to injury tended to be reduced in PAR-1 deficient mice. However, there appeared to be more significant changes in remodeling in response to injury. Although vessel and lumen diameter tended to increase in wild type mice following injury, vessel diameter was unchanged and lumen diameter actually decreased in PAR-1 deficient mice despite the reduced neointimal thickening. This suggests a role for PAR-1 in positive remodeling following vascular injury [72]. Cell density in the neointima was also greater in PAR-1 deficient mice. The increased cell density in normal and injured vessels may have resulted from reduced matrix synthesis and secretion as a result of absence of PAR-1 activation.
An antisense oligodeoxynucleotide to PAR-1 has been used to assess the specific role of PAR-1 in a vascular injury model in rabbits [73]. This oligonucleotide inhibited expression of PAR-1 mRNA in vivo and inhibited the proliferative response to thrombin in vascular smooth muscle cells. However, the degree of intimal thickening was not altered. In the same model, hirudin significantly reduced the intimal thickening response. Thus, the authors concluded that although thrombin may be involved in the injury response, it did not appear that PAR-1 was involved in this animal model.
However, in another study in a rat model, an antibody specific for PAR-1 was shown to inhibit the intimal thickening response at antibody concentrations that effectively inhibited thrombin-induced vascular smooth muscle proliferation in vitro [74]. Hirudin was also effective in this model. More recently, we used a novel small molecule antagonist of PAR-1, RWJ-58259, to assess the role of PAR-1 in vascular injury in rats [33, 75]. RWJ-58259 inhibited thrombin-induced vascular smooth muscle cell proliferation in vitro. RWJ-58259 was placed locally on the adventitial surface of the injured artery using a polymer gel to keep the compound in place. RWJ-58259 treatment significantly inhibited neointimal thickening at 14 days following injury. These findings taken together with the previous studies support an important role for PAR-1 in vascular injury.
PAR-1 IN INFLAMMATION AND TISSUE INJURY
Thrombin contributes to many of the inflammatory and reparative processes accompanying tissue injury including changes in vascular permeability, leukocyte-endothelial interactions, and connective tissue remodeling. Through activation of PAR-1, thrombin mediates several important effects on the vascular endothelium such as stimulation of the synthesis and release of various cytokines and growth factors and induction of adhesion molecule expression. These soluble mediators, which include interleukin-1, interleukin-6, interleukin-8, and monocyte chemoattractant protein-1, act as chemoattractants for and activators of peripheral blood leukocytes [76-78]. The upregulation of several adhesion molecules including CD62P (P-selectin), CD62E (E-selectin), and CD54 (intercellular adhesion molecule-1) participates in a time-dependent coordinated manner to capture rolling leukocytes at sites of injury [79, 80]. PAR-1 dependent increases in vascular contraction and vascular permeability in vitro also indicate a direct effect of thrombin on the vascular response associated with tissue injury. Taken together, these many actions of thrombin on the vascular endothelium indicate a coordinated process that facilitates the transmigration of leukocytes from the blood to the extravascular space.
In addition to thrombin's role in mediating a pro-inflammatory response during tissue injury, thrombin exerts a variety of additional actions on cells of the vascular and extravascular space. Thrombin exhibits in vitro angiogenic activity through induction of endothelial tube formation in Matrigel. as well as promoting neovascularization in vivo [81, 82]. While this thrombin-dependent angiogenic response is important for normal tissue healing, it may contribute to pathological conditions such as in tumor angiogenesis and atherosclerotic plaque formation.
The extravascular actions of thrombin include activation of tissue fibroblasts, monocytes/macrophages, and mast cells. Fibroblasts are important participants during tissue repair by elaborating a variety of cytokines and growth factors for both autocrine and paracrine actions. Secretion of vascular endothelial growth factor, a key angiogenic factor, pro-fibrotic connective tissue growth factor, and the inflammatory cytokine interleukin-8 highlight the importance of PAR-1-induced fibroblast activation in tissue injury [83-85]. In addition to its direct effect on the release of soluble mediators from fibroblasts, thrombin synergizes with interleukin-1beta to secrete prostaglandin E2 from fibroblasts [86]. This interaction is associated with both increased vascular permeability and vasorelaxation, which are contributing factors to tissue edema. The effects on fibroblast secretagogue activity are accompanied by thrombin's strong mitogenic activity toward fibroblasts [87-90].
The importance of PAR-1 as the primary thrombin receptor mediating these inflammatory associated events has been explored further using several in vivo models. PAR-1 deficient mice provide a valuable model to explore the direct actions of thrombin on cell and tissue responses. PAR-1 activation by either thrombin or a PAR-1 agonist peptide increased vascular permeability and vasoconstriction in the pulmonary microvasculature [91]. These changes were absent in lung preparations from PAR-1 deficient mice, consistent with the expression of functional PAR-1 on the vasculature. In a model of inflammatory paw edema, PAR-1 expressing mice challenged with either thrombin or a PAR-1 agonist peptide exhibited increased plasma extravasation [92] which was attenuated by either the neurokinin 1 receptor antagonist SR140333 or ablation of sensory nerves by capsaicin. Mice deficient in PAR-1 were protected from the increased edema and leukocyte infiltration confirming that PAR-1 mediates this inflammatory response and does so via a neurogenic mechanism. In a model of rat paw edema, the thrombin inhibitor Hirulog. attenuated carrageenin-stimulated paw edema, implicating thrombin as a central mediator of the response [93]. Additional studies have shown that directly activating PAR-1 via either injection of thrombin or a PAR-1 agonist peptide induces increased extravasation in this model [93, 94]. Degranulation of mast cells by compound 48/80 attenuated the responses to PAR-1 activation, implicating mast cells, either directly or indirectly, as key participants in the inflammatory response.
Mast cell granule constituents play important roles in immediate (type I) hypersensitivity, inflammation, tissue remodeling, and angiogenesis [95-100]. Upon activation, mast cells secrete a range of soluble mediators including histamine, heparin, leukotrienes, cytokines, growth factors, and neutral proteases. Thus, mast cells represent an efficient system through which powerful mediating factors and enzymes are deposited locally upon activation. We recently described the presence of PAR-1 on mast cells in various normal human tissues using immunohistochemical techniques [101]. PAR-1 was distributed on the plasma membrane and on the membranes of the intracellular tryptase-positive granules of the mast cells. Such findings are consistent with earlier observations demonstrating that thrombin can induce mast cell activation [102, 103]. More recently, PAR-1 mRNA has also been identified in mast cells [104]. Taken together, these observations indicate the presence of mast cell receptors for thrombin that initiate or potentiate mast cell activation. Therefore, the function of PAR-1 on the membranes of intracellular granules may be to sustain further mast cell degranulation by replenishing internalized PAR-1 from the plasma membrane upon activation. The discovery of a functional PAR-1 on mast cells suggests a novel mechanism to control mast cell activity through the regulation of PAR-1.
PAR-1 IN THE TUMOR MICROENVIRONMENT (TME)
Of the many factors secreted by tumor cells, thrombin has been correlated with the stage and type of carcinoma and is associated with cell invasion and extracellular matrix degradation [105, 106]. Increased proteolytic activity in the TME can favor capillary sprout elongation and lumen formation during angiogenesis [107]. The numerous cellular actions of thrombin on platelets, vascular cells, mast cells, and leukocytes previously noted may contribute to tumor invasion. Additionally, thrombin-mediated connective tissue deposition and development of tissue fibrosis, characteristics of normal wound healing, are processes also associated with cellular metastasis [108, 109].
In vitro studies have demonstrated increased tumor cell adhesion to endothelium, extracellular matrix and platelets, enhanced metastatic capacity of tumor cells, and activated cell growth and stimulation of angiogenesis in response to thrombin and PAR-1 agonist peptides [110-115]. PAR-1 has been localized in pancreas tumor cells [116], carcinoma, and melanoma cell lines [114]. In breast carcinoma cells, the level of PAR-1 expression has been correlated with the degree of invasiveness [117]. Furthermore, B16F10 melanoma cells transfected with PAR-1, enhanced thrombin-induced tumor cell adhesion to fibronectin 2.5-fold and pulmonary metastasis as high as 39-fold compared to untransfected thrombin-treated tumor cells [118].
The expression of PAR-1 in malignant and benign human tumor tissues was recently described in the context of the surrounding cell types in the TME using immunohistochemistry and in situ hybridization [119]. In this study, PAR-1 was expressed not only on the malignant carcinoma cells, but also on the cell types forming the TME such as the local macrophages, mast cells, reactive stromal fibroblasts, vascular endothelial, and smooth muscle cells. The presence of PAR-1 in these cell types in the TME suggests a role in the actions of thrombin in cell metastasis. The interface between the invading malignant cells and the hosting stromal cells, referred to as the TME, possesses an array of well-orchestrated cell signaling mechanisms which facilitate tumor invasion by stimulating tumor cell proliferation and degradation and remodeling of the extracellular matrix [120].
While PAR-1 expression has previously been shown on endothelial cells, vascular smooth muscle cells and mast cells, little is known about the presence of PAR-1 on macrophages. It has been reported that macrophages can secrete thrombin [121] and that thrombin is localized in pulmonary alveolar macrophages [122]. It is possible that PAR-1 activation might provide a stimulus for macrophages to proliferate, migrate and/or phagocytize degraded stromal proteins, in addition to synthesizing and secreting thrombin into the TME. Additional work will be required to clarify the role of PAR-1 in macrophages as well as their role in cell metastasis.
PAR-1 was also identified on the reactive stromal fibroblasts surrounding the metastatic tumor cells, which were also positive for smooth muscle actin and DNA topoisomerase IIalpha, a proliferation marker [123]. PAR-1 immunoreactivity was not detected on the stromal fibroblasts surrounding the benign, non-metastatic, or normal epithelial cells, which were smooth muscle actin and topoisomerase IIalpha negative in the tissues analyzed. Reactive stromal fibroblasts, which exhibited characteristics of myofibroblasts, are frequently associated with cancers of epithelial origin: a process known as desmoplasia [124]. These cells appear to influence the invasive and metastatic potential of carcinoma cells by an unidentified mechanism [120]. Furthermore, reactive stromal fibroblasts deposit proteins such as collagen types I, II and V in the local matrix in conjunction with tumor invasion, which may facilitate tumor cell adhesion, cell motility and cell proliferation [109, 120]. The expression of tissue factor, a 46-kD transmembrane glycoprotein that serves as a cell surface receptor and an essential co-factor for plasma coagulation factor VII/VIIa, was reported to be consistently observed in stromal cells of invasive breast carcinomas but not in the benign breast tumors [125]. The increased presence of tissue factor/factor VIIa within the TME, which in turn can generate thrombin via the extrinsic coagulation pathway on fibroblasts, parallels our observation of increased PAR-1 expression. Thus, the presence of PAR-1 on stromal fibroblasts raises the possibility that tumor-derived thrombin may activate local stromal fibroblasts.
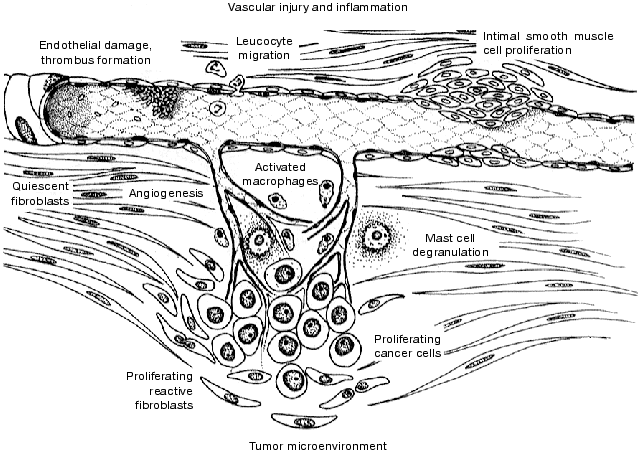
The role of thrombin on cell function is multi-faceted. Induction of the coagulation pathway on the cell surface of platelets, macrophages, vascular cells, or tumor cells in response to tissue injury or inflammation, provides a means by which thrombin is generated and becomes accessible not only intravascularly but also to the resident cells in the subendothelial tissue. The generation of thrombin in response to tissue injury and inflammatory processes results in the coordinated activation of cells within the vascular and extravascular compartments (figure). The identification of a novel proteolytically activated receptor for thrombin, PAR-1, and evidence for its ubiquitous expression on numerous cells types has provided an explanation for thrombin's cellular actions. Accumulating evidence indicates that PAR-1 does indeed mediate many of the cellular actions of thrombin. This includes activation, adhesion, and aggregation of platelets in arterial thrombosis. Within the vascular milieu, thrombin can modulate the interaction of leukocytes and endothelium to initiate a cascade of events leading to extravasation of cells and protein to the extravascular space, important components of inflammation and edema. Thrombin can also initiate or potentiate mast cell activation. In addition, thrombin can stimulate vascular smooth muscle cell and fibroblast proliferation as well as matrix remodeling, critical components of the vascular injury response. The upregulation of PAR-1 in fibroblasts surrounding malignant tumors suggests a possible role in tumor metastasis. These findings suggest that modulation of thrombin's interaction with PAR-1 may represents a novel approach to interfering specifically with the cellular actions of thrombin. Antagonists of PAR-1 may prove to be an effective approach to managing a wide variety of diseases associated with underlying inflammation and tissue injury, including restenosis, atherosclerosis, inflammation, and cancer.
We would like to thank Mr. Robert McBride, University of Medicine and Dentistry of New Jersey, Stratford, New Jersey, for his precise artistic interpretation of our findings.An illustrative representation of PAR-1 mediated cellular effects during vascular injury, inflammation, and within the tumor microenvironment
REFERENCES
1.Vu, T. K. H., Hung, D. T., Wheaton, V. I., and
Coughlin, S. R. (1991) Cell, 64, 1057-1068.
2.Vu, T. K. H., Wheaton, V. I., Hung, D. T., Charo,
I., and Coughlin, S. R. (1991) Nature (London), 353,
674-677.
3.Gerszten, R. E., Chen, J., Ishii, M., Ishii, K.,
Wang, L., Nanevicz, T., Turck, C. W., Vu, T.-K. H., and Coughlin, S. R.
(1994) Nature (London), 368, 648-651.
4.Nanevicz, T., Ishii, M., Wang, L., Chen, M., Chen,
J., Turck, C. W., Cohen, F. E., and Coughlin, S. R. (1995) J. Biol.
Chem., 270, 21619-21625.
5.Beavers, M. P., Maryanoff, B. E., Nguyen, D., and
Blackhart, B. D. (1999) Adv. Med. Chem., 4, 245-271.
6.Blackhart, B. D., Ruslim-Litrus, L., Lu, C.-C.,
Alves, V. L., Teng, W., Scarborough, R. M., Reynolds, E. E., and
Oksenberg, D. (2000) Mol. Pharmacol., 58, 1178-1187.
7.Ishii, K., Chen, J., Ishii, M., Koch, W. J.,
Freedman, N. J., Lefkowitz, R. J., and Coughlin, S. R. (1994) J.
Biol. Chem., 269, 1125-1130.
8.Brass, L. F., Woolkalis, M. J., and Hoxie, J. A.
(1995) Trends Cardiovasc. Med., 5, 123-128.
9.Shapiro, M. J., Trejo, J., Zeng, D., and Coughlin,
S. R. (1996) J. Biol. Chem., 271, 32874-32880.
10.Rasmussen, U. B., Vouret-Craviari, V., Jallat,
S., Schlesinger, Y., Pages, G., Pavirani, A., Lecocq, J. P.,
Pouyssegur, J., and van Obberghen-Schilling, E. (1991) FEBS
Lett., 288, 123-128.
11.Zhong, C., Hayzer, D. J., Corson, M. A., and
Runge, M. S. (1992) J. Biol. Chem., 267, 16975-16979.
12.Nystedt, S., Emilsson, K., Wahlestedt, C., and
Sundelin, J. (1994) Proc. Natl. Acad. Sci. USA, 91,
9208-9212.
13.Nystedt, S., Emilsson, K., Larsson, A.-K.,
Stroembeck, B., and Sundelin, J. (1995) Eur. J. Biochem.,
232, 84-89.
14.Ishihara, H., Connolly, A. J., Zeng, D., Kahn, M.
L., Zheng, Y. W., Timmons, C., Tram, T., and Coughlin, S. R. (1997)
Nature (London), 386, 502-506.
15.Kahn, M. L., Zheng, Y.-W., Huang, W., Bigornia,
V., Zeng, D., Moff, S., Farese, R. V., Jr., Tam, C., and Coughlin, S.
R. (1998) Nature (London), 394, 690-694.
16.Xu, W.-F., Andersen, H., Whitmore, T. E.,
Presnell, S. R., Yee, D. P., Ching, A., Gilbert, T., Davie, E. W., and
Foster, D. C. (1998) Proc. Natl. Acad. Sci. USA, 95,
6642-6646.
17.Bohm, S. K., Kong, W., Bromme, D., Smeekens, S.
P., Anderson, D. C., Connolly, A., Kahn, M., Nelken, N. A., Coughlin,
S. R., Payan, D. G., and Bunnett, N. W. (1996) Biochem. J.,
314, 1009-1016.
18.Molino, M., Barnathan, E. S., Numerof, R., Clark,
J., Dreyer, M., Cumashi, A., Hoxie, J. A., Schechter, N., Woolkalis,
M., and Brass, L. F. (1997) J. Biol. Chem., 272,
4043-4049.
19.Mirza, H., Schmidt, V. A., Derian, C. K., Jesty,
J., and Bahou, W. F. (1997) Blood, 90, 3914-3922.
20.Corvera, C. U., Dery, O., McConalogue, K., Bohm,
S. K., Khitin, L. M., Caughey, G. H., Payan, D. G., and Bunnett, N. W.
(1997) J. Clin. Invest., 100, 1383-1393.
21.Camerer, E., Huang, W., and Coughlin, S. R.
(2000) Proc. Natl. Acad. Sci. USA, 97, 5255-5260.
22.Takeuchi, T., Harris, J. L., Huang, W., Yan, K.
W., Coughlin, S. R., and Craik, C. S. (2000) J. Biol. Chem.,
275, 26333-26342.
23.Connolly, A. J., Ishihara, H., Kahn, M. L.,
Farese, R. V., Jr., and Coughlin, S. R. (1996) Nature (London),
381, 516-519.
24.Schmidt, V. A., Nierman, W. C., Maglott, D. R.,
Cupit, L. D., Moskowitz, K. A., Wainer, J. A., and Bahou, W. F. (1998)
J. Biol. Chem., 273, 15061-15068.
25.Sambrano, G. R., Huang, W., Faruqi, T., Mahrus,
S., Craik, C., and Coughlin, S. R. (2000) J. Biol. Chem.,
275, 6819-6823.
26.Nakanishi-Matsui, M., Zheng, Y.-W., Sulciner, D.
J., Weiss, E. J., Ludeman, M. J., and Coughlin, S. R. (2000) Nature
(London), 404, 609-613.
27.Kahn, M. L., Nakanishi-Matsui, M., Shapiro, M.
J., Ishihara, H., and Coughlin, S. R. (1999) J. Clin. Invest.,
103, 879-887.
28.Connolly, T. M., Condra, C., Feng, D.-M., Cook,
J. J., Stranieri, M. T., Reilly, C. F., Nutt, R. F., and Gould, R. J.
(1994) Thromb. Haemost., 72, 627-633.
29.Derian, C. K., Santulli, R. J., Tomko, K. A.,
Haertlein, B. J., and Andrade-Gordon, P. (1995) Thromb. Res.,
78, 505-519.
30.Chiu, P. J. S., Tetzloff, G. G., Foster, C.,
Chintala, M., and Sybertz, E. J. (1997) Eur. J. Pharmacol.,
321, 129-135.
31.Cook, J. J., Sitko, G. R., Bednar, B., Condra,
C., Mellott, M. J., Feng, D.-M., Nutt, R. F., Shafer, J. A., Gould, R.
J., and Connolly, T. M. (1995) Circulation, 91,
2961-2971.
32.Andrade-Gordon, P., Maryanoff, B. E., Derian, C.
K., Zhang, H.-C., Addo, M. F., Darrow, A. L., Eckardt, A. J., Hoekstra,
W. J., McComsey, D. F., Oksenberg, D., Reynolds, E. E., Santulli, R.
J., Scarborough, R. M., Smith, C. E., and White, K. B. (1999) Proc.
Natl. Acad. Sci. USA, 96, 12257-12262.
33.Andrade-Gordon, P., Derian, C. K., Maryanoff, B.
E., Zhang, H.-Z., Addo, M. F., Cheung, W., Damiano, B. P., D'Andrea, M.
D., Darrow, A. L., de Garavilla, L., Eckardt, A. J., Giardino, E. C.,
Haertlein, B. J., and McComsey, D. F. (2001) J. Pharmacol. Exp.
Ther., 298, 34-42.
34.Stouffer, G. A., Schmedtje, J. F., Gulba, D.,
Huber, K., Bode, C., Aaron, J., and Runge, M. S. (1996) Ann.
Hematol., 73, S39-S41.
35.Harker, L. A., Hanson, S. R., and Runge, M. S.
(1995) Am. J. Cardiol., 75, B12-B17.
36.Stadel, J. M. (1997) Fundam. Clin.
Cardiol., 28, 161-176.
37.Chandrasekar, B., and Tanguay, J.-F. (2000) J.
Am. Coll. Cardiol., 35, 555-562.
38.Malik, A. B., and Fenton, J. W., II (1992)
Semin. Thromb. Hemost., 18, 193-199.
39.Garcia, J. G. N. (1992) J. Lab. Clin.
Med., 120, 513-519.
40.Ku, D. D., and Zaleski, J. K. (1993) J.
Cardiovasc. Pharmacol., 22, 609-616.
41.Emori, T., Hirata, Y., Imai, T., Ohta, K., Kanno,
K., Eguchi, S., and Marumo, F. (1992) Biochem. Pharmacol.,
44, 2409-2411.
42.Garcia, J. G. N., Patterson, C., Bahler, C.,
Aschner, J., Hart, C. M., and English, D. (1993) J. Cell
Physiol., 156, 541-549.
43.Storck, J., Kusters, B., and Zimmermann, E. R.
(1995) Thromb. Res., 77, 249-258.
44.Shankar, R., de la Motte, C. A., Poptic, E. J.,
and Dicorleto, P. E. (1994) J. Biol. Chem., 269,
13936-13941.
45.Muramatsu, I., Laniyonu, A., Moore, G. J., and
Hollenberg, M. D. (1992) Can. J. Physiol. Pharmacol., 70,
996-1003.
46.Antonaccio, M. J., Normandin, D., Serafino, R.,
and Moreland, S. (1993) J. Pharmacol. Exp. Ther., 266,
125-132.
47.Cheung, W., Andrade-Gordon, P., Derian, C. K.,
and Damiano, B. P. (1998) Can. J. Physiol. Pharmacol.,
76, 16-25.
48.Ku, D. D. (1987) J. Pharmacol. Exp. Ther.,
243, 571-576.
49.Damiano, B. P., Mitchell, J. A., Cheung, W. M.,
and Falotico, R. (1996) Am. J. Physiol., 39,
H1585-H1596.
50.Damiano, B. P., Cheung, W. M., Mitchell, J. A.,
and Falotico, R. (1996) J. Pharmacol. Exp. Ther., 279,
1365-1378.
51.McNamara, C. A., Sarembock, I. J., Gimple, L. W.,
Fenton, J. W., II, Coughlin, S. R., and Owens, G. K. (1993) J. Clin.
Invest., 91, 94-98.
52.Herbert, J. M., Lamarche, I., and Dol, F. (1992)
FEBS Lett., 301, 155-158.
53.Kanthou, C., Benzakour, O., Patel, G., Deadman,
J., Kakkar, V. V., and Lupu, F. (1995) Thromb. Haemost.,
74, 1340-1347.
54.Tyagi, S. C., Meyer, L., Schmaltz, R. A., Reddy,
H. K., and Voelker, D. J. (1995) Atherosclerosis, 116,
43-57.
55.Dabbagh, K., Laurent, G. J., McAnulty, R. J., and
Chambers, R. C. (1998) Thromb. Haemost., 79, 405-409.
56.Duhamel-Clerin, E., Orvain, C., Lanza, F.,
Cazenave, J.-P., and Klein-Soyer, C. (1997) Arterioscler.,
Thromb., Vasc. Biol., 17, 1931-1938.
57.Papadimitriou, E., Manolopoulos, V. G., Hayman,
G. T., Maragoudakis, M. E., Unsworth, B. R., Fenton, J. W., II, and
Lelkes, P. I. (1997) Am. J. Physiol., 272,
C1112-C1122.
58.Walz, D. A., Anderson, G. F., Ciaglowski, R. E.,
Aiken, M., and Fenton, J. W., II (1985) Proc. Soc. Exp. Biol.
Med., 180, 518-526.
59.Uchida, Y., Hasegawa, K., Kawamura, K., and
Shibuya, I. (1989) Am. Heart J., 117, 769-776.
60.Wilcox, J. N., Rodriguez, J., Subramanian, R.,
Ollerenshaw, J., Zhong, C., Hayzer, D. J., Horaist, C., Hanson, S. R.,
and Lumsden, A. (1994) Circ. Res., 75, 1029-1038.
61.Guitteny, A.-F., and Herbert, J.-M. (1997)
Eur. J. Pharmacol., 327, 157-162.
62.Cheung, W.-M., D'Andrea, M. R., Andrade-Gordon,
P., and Damiano, B. P. (1999) Arterioscler., Thromb.,
Vasc. Biol., 19, 3014-3024.
63.Maloney, J. D., Stark, V. K., and Hoch, J. R.
(1999) Surg. Forum, 50, 477-479.
64.Nelken, N. A., Soifer, S. J., O'Keefe, J., Vu,
T.-K. H., Charo, I. F., and Coughlin, S. R. (1992) J. Clin.
Invest., 90, 1614-1621.
65.Dechend, R., Mo, X., Schulz, W., Gross, M.,
Praus, M., Dietz, R., and Gulba, D. C. (1996) Fibrinolysis,
10, 41-45.
66.Hanbin, W., Yinkui, S., Chengwen, S., Longbin,
L., Rui, L., Fumeng, L., Haitao, W., and Wanrong, C. (1998) Immunol.
J., 14, 71-74.
67.Buchwald, A. B., Sandrock, D., Unterberg, C.,
Ebbecke, M., Nebendahl, K., Luders, S., Munz, D. L., and Wiegand, V.
(1993) J. Am. Coll. Cardiol., 21, 249-254.
68.Heras, M., Chesebro, J., Webster, M., Mruk, J.,
Grill, D., Penny, W., Bowie, E., Badimon, L., and Fuster, V. (1990)
Circulation, 82, 1476-1484.
69.Abendschein, D. R., Recchia, D., Meng, Y. Y.,
Oltrona, L., Wickline, S. A., and Eisenberg, P. R. (1996) J. Am.
Coll. Cardiol., 28, 1849-1855.
70.Barry, W. L., Gimple, L. W., Humphries, J. E.,
Powers, E. R., McCoy, K. W., Sanders, J. M., Owens, G. K., and
Sarembock, I. J. (1996) Circulation, 94, 88-93.
71.Gerdes, C., Faber-Steinfeld, V., Yalkinoglu, O.,
and Wohlfeil, S. (1996) Arterioscler., Thromb., Vasc.
Biol., 16, 1306-1311.
72.Schwartz, R. S. (1997) Sem. Interv.
Cardiol., 2, 83-88.
73.Herbert, J. M., Guy, A. F., Lamarche, I., Mares,
A. M., Savi, P., and Dol, F. (1997) J. Cell. Physiol.,
170, 106-114.
74.Takada, M., Tanaka, H., Yamada, T., Ito, O.,
Kogushi, M., Yanagimachi, M., Kawamura, T., Musha, T., Yoshida, F.,
Ito, M., Kobayashi, H., Yoshitake, S., and Saito, I. (1998) Circ.
Res., 82, 980-987.
75.Zhang, H.-C., Derian, C. K., Andrade-Gordon, P.,
Hoekstra, W. J., McComsey, D. F., White, K. B., Poulter, B. L., Addo,
M. F., Cheung, W.-M., Damiano, B. P., Oksenberg, D., Reynolds, E. E.,
Pandey, A., Scarborough, R. M., and Maryanoff, B. E. (2001) J. Med.
Chem., 44, 1021-1024.
76.Colotta, F., Sciacca, F. L., Sironi, M., Luini,
W., Rabiet, M. J., and Mantovani, A. (1994) Am. J. Pathol.,
144, 975-985.
77.Eckardt, A. J., Andrade-Gordon, P., and Derian,
C. K. (1999) FASEB J., 13, A38.
78.Chi, L., Li, Y., Stehno-Bittel, L., Gao, J.,
Morrison, D. C., Stechschulte, D. J., and Dileepan, K. N. (2001) J.
Interferon Cytokine Res., 21, 231-240.
79.Sugama, Y., Tiruppathi, C., Janakidevi, K.,
Andersen, T. T., Fenton, J. W., II, and Malik, A. B. (1992) J. Cell
Biol., 119, 935-944.
80.Shankar, R., De la Motte, C. A., and di Corleto,
P. E. (1992) J. Biol. Chem., 267, 9376-9382.
81.Tsopanoglou, N. E., Pipili-Synetos, E., and
Maragoudakis, M. E. (1993) Am. J. Physiol., 264,
C1302-C1307.
82.Haralabopoulos, G. C., Grant, D. S., Kleinman, H.
K., and Maragoudakis, M. E. (1997) Am. J. Physiol., 273,
C239-C245.
83.Chambers, R. C., Leoni, P., Blanc-Brude, O. P.,
Wembridge, D. E., and Laurent, G. J. (2000) J. Biol. Chem.,
275, 35584-35591.
84.Ludwicka-Bradley, A., Tourkina, E., Suzuki, S.,
Tyson, E., Bonner, M., Fenton, J. W., II, Hoffman, S., and Silver, R.
M. (2000) Am. J. Respir. Cell Mol. Biol., 22,
235-243.
85.Ollivier, V., Chabbat, J., Herbert, J. M., Hakim,
J., and De Prost, D. (2000) Arterioscler. Thromb. Vasc. Biol.,
20, 1374-1381.
86.Derian, C. K., and Eckardt, A. J. (1997) Exp.
Cell Res., 232, 1-7.
87.Hung, D. T., Vu, T. K. H., Nelken, N. A., and
Coughlin, S. R. (1992) J. Cell Biol., 116, 827-832.
88.Vouret-Craviari, V., van Obberghen-Schilling, E.,
Rasmussen, U. B., Pavirani, A., Lecocq, J. P., and Pouyssegur, J.
(1992) Mol. Biol. Cell, 3, 95-102.
89.Reilly, C. F., Connolly, T. M., Feng, D. M.,
Nutt, R. F., and Mayer, E. J. (1993) Biochem. Biophys. Res.
Commun., 190, 1001-1008.
90.Dawes, K. E., Gray, A. J., and Laurent, G. J.
(1993) Eur. J. Cell Biol., 61, 126-130.
91.Vogel, S., Gao, X., Mehta, D., Ye, R. D., John,
T. A., Andrade-Gordon, P., Tiruppathi, C., and Malik, A. B. (2000)
Physiol. Genomics, 4, 137-145.
92.De Garavilla, L., Vergnolle, N., Young, S. H.,
Ennes, H., Steinhoff, M., Ossovskaya, V. S., D'Andrea, M. R., Mayer, E.
A., Wallace, J. L., Hollenberg, M. D., Andrade-Gordon, P., and Bunnett,
N. W. (2001) Br. J. Pharmacol., 133, 975-987.
93.Cirino, G., Cicala, C., Bucci, M. R., Sorrentino,
L., Maraganore, J. M., and Stone, S. R. (1996) J. Exp. Med.,
183, 821-827.
94.Vergnolle, N., Hollenberg, M. D., and Wallace, J.
L. (1999) Br. J. Pharmacol., 126, 1262-1268.
95.Galli, S. J., Wershil, B. K., Gordon, J. R., and
Martin, T. R. (1989) Ciba Found. Symp., 147, 53-73.
96.Gordon, J. R., Burd, P. R., and Galli, S. J.
(1990) Immunol. Today, 11, 458-464.
97.Irani, A. A. (1995) in Mast Cell Proteases in
Immunology and Biology (Caughey, G., ed.) Marcel Decker, New York,
pp. 127-143.
98.He, S., and Walls, A. F. (1997) Eur. J.
Pharmacol., 328, 89-97.
99.Laine, P., Kaartinen, M., Penttila, A., Panula,
P., Paavonen, T., and Kovanen, P. T. (1999) Circulation,
99, 361-369.
100.Galli, S. J. (2000) Curr. Opin.
Hematol., 7, 32-39.
101.D'Andrea, M. R., Rogahn, C. J., and
Andrade-Gordon, P. (2000) Biotech. Histochem., 75,
85-90.
102.Razin, E., and Marx, G. (1984) J.
Immunol., 133, 3282-3285.
103.Strukova, S. M., Dugina, T. N., Khlgatian, S.
V., Redkozubov, A. E., Redkozubova, G. P., and Pinelis, V. G. (1996)
Semin. Thromb. Hemost., 22, 145-150.
104.Nishikawa, H., Kawabata, A., Kuroda, R.,
Nishida, M., and Kawai, K. (2000) Jpn. J. Pharmacol., 82,
74-77.
105.Koivunen, E., Saksela, O., Itkonen, O., Osman,
S., Huhtala, M. L., and Stenman, U. H. (1991) Int. J. Cancer,
47, 592-596.
106.Walz, D. A., and Fenton, J. W. (1995)
Invasion Metastasis, 14, 303-308.
107.Liotta, L. A., Steeg, P. S., and
Stetler-Stevenson, W. G. (1991) Cell, 64, 327-336.
108.Carney, D. H., Mann, R., Redin, W. R., Pernia,
S. D., Berry, D., Heggers, J. P., Hayward, P. G., Robson, M. C.,
Christie, J., and Annable, C. (1992) J. Clin. Invest.,
89, 1469-1477.
109.Chambers, R. C., Dabbagh, K., Mcanulty, R. J.,
Gray, A. J., Blanc-Brude, O. P., and Laurent, G. J. (1998) Biochem.
J., 333, 121-127.
110.Nierodzik, M. L. R., Kajumo, F., and Karpatkin,
S. (1992) Cancer Res., 52, 3267-3272.
111.Nierodzik, M. L. R., Bain, R. M., Liu, L.-X.,
Shivji, M., Takeshita, K., and Karpatkin, S. (1996) Br. J.
Haematol., 92, 452-457.
112.Klementsen, B., and Jorgensen, L. (1997)
APMIS, 105, 546-558.
113.Wojtukiewicz, M. Z., Tang, D. G., Ciarelli, J.
J., Nelson, K. K., Walz, D. A., Diglio, C. A., Mammen, E. F., and Honn,
K. V. (1993) Int. J. Cancer, 54, 793-803.
114.Wojtukiewicz, M. Z., Tang, D. G., Ben-Josef,
E., Renaud, C., Walz, D. A., and Honn, K. V. (1995) Cancer Res.,
55, 698-704.
115.Tsopanoglou, N. E., and Maragoudakis, M. E.
(1997) Angiogenesis, 1, 192-200.
116.Rudroff, C., Schafberg, H., Nowak, G., Weinel,
R., Scheele, J., and Kaufmann, R. (1998) Pancreas, 16,
189-194.
117.Even-Ram, S., Uziely, B., Cohen, P.,
Grisaru-Granovsky, S., Maoz, M., Ginzburg, Y., Reich, R., Vlodavsky,
I., and Bar-Shavit, R. (1998) Nat. Med., 4, 909-914.
118.Nierodzik, M. L., Chen, K., Takeshita, K., Li,
J.-J., Huang, Y.-Q., Feng, X.-S., D'Andrea, M. R., Andrade-Gordon, P.,
and Karpatkin, S. (1998) Blood, 92, 3694-3700.
119.D'Andrea, M. R., Derian, C. K., Santulli, R.
J., and Andrade-Gordon, P. (2001) Am. J. Pathol., 158,
2031-2041.
120.Gregoire, M., and Lieubeau, B. (1995) Cancer
Metast. Rev., 14, 339-350.
121.Lindahl, U., Pejler, G., Boegwald, J., and
Seljelid, R. (1989) Arch. Biochem. Biophys., 273,
180-188.
122.Zacharski, L. R., Memoli, V. A., Morain, W. D.,
Schlaeppi, J.-M., and Rousseau, S. M. (1995) Thromb. Haemost.,
73, 793-797.
123.D'Andrea, M. R., Farber, P., and Foglesong, P.
D. (1994) Appl. Immunohistochem., 2, 177-185.
124.Schmitt-Graff, A., Desmouliere, A., and
Gabbiani, G. (1994) Virchows Arch., 425, 3-24.
125.Vrana, J. A., Stang, M. T., Grande, J. P., and
Getz, M. J. (1996) Cancer Res., 56, 5063-5070.