REVIEW: Receptors of the PAR Family as a Link between Blood Coagulation and Inflammation
T. N. Dugina, E. V. Kiseleva, I. V. Chistov, B. A. Umarova, and S. M. Strukova*
Department of Human and Animal Physiology, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; E-mail: strukova@mail.ru* To whom correspondence should be addressed.
Received June 11, 2001; Revision received September 11, 2001
Blood coagulation plays a key role among numerous mediating systems that are activated in inflammation. Receptors of the PAR family serve as sensors of serine proteinases of the blood clotting system in the target cells involved in inflammation. Activation of PAR-1 by thrombin and of PAR-2 by factor Xa leads to a rapid expression and exposure on the membrane of endothelial cells of both adhesive proteins that mediate an acute inflammatory reaction and of the tissue factor that initiates the blood coagulation cascade. Certain other receptors (EPR-1, thrombomodulin, etc.), which can modulate responses of the cells activated by proteinases through PAR receptors, are also involved in the association of coagulation and inflammation together with the receptors of the PAR family. The presence of PAR receptors on mast cells is responsible for their reactivity to thrombin and factor Xa and defines their contribution to the association of inflammation and blood clotting processes.
KEY WORDS: protease-activated receptor, thrombin, factor Xa, endothelial cells, mast cells, inflammation, blood clotting
Abbreviations: PAR) protease-activated receptor; EPR-1) protease receptor of effector cells; PAF) platelet activating factor; TRAP) thrombin receptor antagonist peptide; TGF-beta1) transforming growth factor beta1; TNF-alpha and -beta) tumor necrosis factors alpha and beta; MT-SP1) membrane type 1 serine proteinase; MAP-kinases) mitogen-activated protein kinases; PKC) protein kinase C; IL-1) interleukin-1; L-NAME) N-nitro-L-arginine methyl ester; ADP) adenosine diphosphate; NO) nitric oxide; cNOS) constitutive NO-synthase; P and E) platelet and endothelial selectins; ICAM-1) intracellular adhesive molecule 1; VCAM-1) vascular cell adhesive molecule 1; MCP-1) monocyte chemoattractant protein 1; vWf) von Willebrand factor; bFGF) basic fibroblastic growth factor; VEGF) vascular endothelial growth factor; PDGF) platelet-derived growth factor; CTGF) connective tissue growth factor; MMP) matrix metalloproteinases; TFPI) tissue factor pathway inhibitor.
Thrombin plays a key role among serine proteinases of the blood
coagulation system. Together with conversion of fibrinogen to fibrin,
activation of blood coagulation factors V, VIII, XI, and XIII, and
platelet aggregation, thrombin regulates hemostasis by activation of
the anticoagulant system of protein C and inhibition of fibrinolysis
(through activation of TAFI, thrombin-activated fibrinolysis
inhibitor). Thrombin activates various cell types in and outside blood
participating in the processes of development, regulation of vessel
tone, inflammation, tissue repair, atherosclerosis, carcinogenesis, and
many others [1-9].
Thrombin generation from prothrombin occurs in the region of vessel and tissue damage at the initial stage of repair, when a fibrin clot is forming and inflammation is developing. Inflammation leads to the activation of blood cells (platelets, leukocytes) and endothelium and subendothelium of vessels (in particular, of mast cells). Under these conditions, thrombin is involved in the adhesion and recruitment of leukocytes, platelet activation, and cell proliferation [1, 2, 7-10]. The proinflammatory effect of thrombin plays an important role in tissue repair, and also in pathologies that are related to chronic activation of the coagulation cascade like restenosis, atherosclerosis, pneumonic fibroses, glomerulonephritis, and others [2, 11-14].
The kallikrein-kinin and complement systems are the main proteolytic systems that are activated in inflammation. While the mechanisms of functional association of these systems with the development of inflammation have been sufficiently well studied and discussed, the involvement of the enzymes of the hemostatic system in the realization of inflammatory response is now being intensively studied [4, 14-18]. This became possible due to the discovery of the receptors of proteinases of the blood coagulation system on the cells that are involved in hemostatic processes and give a response to the inflammatory stimulus [8, 14, 19-24]. Especially important are endothelial cells, which during inflammation expose on their membrane proteins that are involved in blood coagulation and inflammation [25-27]. At the same time, mediators of inflammation, cytokines, induce the expression of the tissue factor and suppress thrombomodulin expression. These events lead to modification of the pro- and anticoagulant balance of the endothelium [28-30].
Thrombin plays a key role in the activation and regulation of the blood coagulation cascade and is also involved in the stimulation of cellular functions during inflammation and tissue repair [1-4, 7-11, 14, 15].
Certain other coagulation factors also play a crucial role in the interaction of inflammation and coagulation. Especially important is fibrinogen, an adhesive protein that is necessary for platelet aggregation, and fibrin that is a matrix on which cells can migrate, proliferate, and carry out specialized functions due to the presence of a binding locus for the adhesive receptors from integrin (alphaMbeta2 (Mac-1, CD11b/CD18), alphaVbeta3, alpha1beta5) and immunoglobulin (ICAM-1) superfamilies [31, 32]. Endothelial cells, leukocytes, macrophages, fibroblasts, smooth muscle cells, and keratinocytes represent cell types that specifically bind to and migrate on the fibrin matrix.
Receptors of blood coagulation factor Xa (EPR-1), protein C (ECPR, endothelial C protein receptor) and urokinase (u-PAR, urokinase type plasminogen activator receptor, CD87), which are not cleaved by their specific ligands during activation, are identified on endothelial cells [15-18]. In contrast, the receptors of PAR family (proteinase-activated receptors) are subjected to limited proteolysis by trypsin-type serine proteinases [8, 14, 19-24]. The effect of thrombin and factors VIIa and Xa towards the cells is realized through their interaction with membrane receptors including those of the PAR family. In fact, PARs have been identified on endothelial cells, platelets, fibroblasts, smooth muscle cells, monocytes, osteoblasts, neurons, glia, and mast cells [4, 7-10, 19-24, 33-35]. Mast cells are actively involved not only in the immune response of an organism, but in inflammatory and atherosclerotic processes as well, during which thrombin generation and coagulation of blood fibrinogen are always activated. However, the mechanisms of interaction of thrombin and other proteinases of the hemostatic system with mast cells are not yet well understood. The mechanisms of interaction of thrombin and factor Xa with membrane receptors of the cells that are involved in the inflammatory reaction are discussed in this review.
EARLY CELLULAR RESPONSES DURING INFLAMMATION
During inflammation, the phenotype of endothelial cells changes from thromboresistant to procoagulant and proinflammatory, and the conversion of coagulation proenzymes into the active serine proteinases occurs [15, 36-38]. These processes are accompanied by adhesion and platelet activation on the surface of endothelium due to the expression and exposure of adhesive molecules (vWf, P- and E-selectins, ICAM-1, VCAM-1), PAF and MCP-1. Adhesive proteins and molecules recognize and bind their ligands on monocytes, which adhere and migrate on the surface of endothelium.
The tissue factor (an integral membrane protein that binds factor VII/VIIa and initiates generation of proteinases, coagulation factors IXa, Xa, and thrombin) is exposed on the membrane of the cells activated during inflammation. Simultaneously the anticoagulative systems are inhibited (especially that of protein C, which is activated by thrombin) due to the decrease of thrombomodulin expression (a membrane cofactor of thrombin). The cleavage of glycosaminoglycan chains from glypicans and syndecans of the endothelium reduces the antithrombin III activity. The exposure of the fibrinolysis inhibitor PAI-1 on the endothelium increases, while the exposure of the tissue plasminogen activator (tPA) decreases, leading to the suppression of fibrinolysis [16, 25, 28].
Activated platelets, as well as CD4+ T-lymphocytes activated in the site of inflammation, basophiles, mast, and endothelial cells expose on their surfaces together with other proteins P-selectin and CD40-ligand (CD40L, CD154), a member of the tumor necrosis factor family. CD40-ligand binds to the receptor that is exposed on the activated endothelium (CD40) and stimulates the expression of transcriptional factors (NF-kappaB, AP-1, and others). Expression of the adhesive molecules and the tissue factor and development of an inflammatory response are the consequences of this activation. In addition, platelet CD40-ligand stimulates the tissue factor expression on monocytes [39, 40].
Exposure of the tissue factor on the cells is a key event in the activation of blood coagulation and clot formation during inflammation. The tissue factor serves as a receptor of blood coagulation factors VII/VIIa and initiates coagulation in a complex with them. Antibodies against the tissue factor and its complex with factor VIIa can neutralize the procoagulative blood activity, as demonstrated by injection of endotoxin into experimental animals [18].
The tissue factor pathway inhibitor (TFPI) regulates coagulation at this step. It blocks the activities of factors Xa and VIIa in the complex with tissue factor. Matrix metalloproteinases (MMP) that are released from leukocytes during inflammation, especially MMP-7 (matrilysin) and MMP-9 (gelatinase), rapidly cleave TFPI without influence on the tissue factor and factors VII and Xa. Under these conditions, TFPI disrupts in an atherosclerotic plaque, where it is colocalized with tissue factor and MMP that are activated during inflammation, opening the way for uncontrolled activation of blood clotting by the tissue factor [41].
Generation of thrombin on the surface of monocytes activated during inflammation may be mediated by an additional mechanism that is independent of tissue factor and coagulation factor VII. Monocyte activation leads to the expression of adhesive proteins including those from the beta2-integrin family (alphaMbeta2). In addition to other proteins, they bind coagulation factor X. Cathepsin G that is released from activated leukocytes induces a limited proteolysis of factor X adhered on the monocyte membrane, thus producing an active form. This form can transform prothrombin into thrombin [42].
Macrophages express proteins of the prothrombinase complex (prothrombin and factor Xa) in response to lipopolysaccharides, lymphokines, immune complexes, proteins of complement, viruses, and other inflammatory agents. Some data indicate that thrombin is generated in macrophages, developing muscle, and the nervous system, where prothrombin mRNA has been detected [43, 44].
Interaction between monocytes, endothelium, and activated platelets that expose P-selectin and CD-40 ligand (CD40L) is a key event in the initiation and progression of an atherosclerotic plaque and thrombosis development in a region of plaque lesion [45]. CD40L activates atheroma cells by induction of expression of the adhesive molecules and the tissue factor, production of cytokines, and activation of MMPs, which are then involved in the atherogenesis. These data apparently explain the reduction of manifestation of atherosclerosis in mice injected with CD40L antibodies [45].
Thus, hypercoagulation is an early response of cells to an inflammatory stimulus. It is induced by an increase of blood procoagulative potential and decrease of anticoagulative properties and fibrinolysis. Activation of blood cells, especially platelets and monocytes, and their interaction with activated endothelium stimulate the following adhesion and recruitment of leukocytes giving rise to a new step of blood clotting activation. Regulation of blood coagulation, immune response, and other mechanisms of maintaining homeostasis block the development of the inflammatory process during acute inflammation. In chronic inflammation a vicious cycle of inflammation, clotting, inflammation, clotting, etc. develops, leading to vessel damage, thrombosis, and organic deficiency [28].
THROMBIN RECEPTORS
Specific receptors on the target cells serve as sensors of proteases that appear in blood during coagulation. Endothelial and blood cells express receptors of proteinases of the blood clotting system--factors VIIa, Xa, thrombin, and protein C [14-18].
The action of thrombin and factor Xa on cells is apparently realized through their interaction with several membrane receptors including those of the PAR family. All members of the PAR family are attributed to the superfamily of integral membrane receptors consisting of seven domains and associated with the G-proteins. Receptors of catecholamines, acetylcholine, serotonin, histamine, angiotensins, tachykinins, endothelins, PAF, and others are also attributed to this superfamily [7-9].
Receptors of the PAR family are characterized by their ability to serve as highly specific substrates for the regulatory proteinases, which cleave one peptide bond in the extracellular domain. A new N-terminus of the receptor, referred to as the “tethered ligand”, interacts with the extracellular domain-2 of the cleaved receptor and activates it. Four main members of the PAR family have been described to date: PAR-1, -3, and -4, which are thrombin receptors, and PAR-2, which is a receptor of trypsin and tryptase of mast cells [7-10, 19-23]. Synthetic peptides with structures corresponding to that of the PAR-1, -2, and -4 tethered ligands function as agonists of their own receptors. They are used as a tool to show the involvement of the receptors in the effects of proteinase on cells.
The use of the peptide agonists and knocking out of PAR-1 genes in transgenic mice showed that the action of thrombin in inflammation is mediated by PAR-1 activation. Thrombin cleaves the peptide bond Arg41-Ser42 in the PAR-1 N-terminal domain and exposes the new N-terminal peptide SFLLRN (tethered ligand) that interacts with the receptor and initiates intracellular signalization. PAR-1 activation by thrombin may finally result in the activation of transcription factors that regulate the expression of tissue factor, adhesive proteins, growth factors, cytokines, and other ligands [4, 7-9, 14, 33, 46].
Activating PAR-1, thrombin induces ICAM-1 expression in endothelial cells through the NF-kappaB-site in the promoter. Thrombin also induces the expression of P- and E-selectins, VCAM-1, IL-8 and IL-6, and chemokines [9, 47]. ICAM-1 plays a key role in the development of inflammatory response through stimulation of leukocytes adhesion.
At the same time, thrombin induces the expression of DAF-factor in endothelial cells. This factor accelerates the decay of C3- and C5-convertases of the complement system. DAF is a membrane glycoprotein; it protects endothelial cells from lysis by membrane-attacking complex of the complement system, which is activated during inflammation [48]. Thrombin increases DAF expression several-fold by interaction with PAR-1. This reaction may be blocked by antagonists of PKC and MAP-kinases and NF-kappaB. Thus, thrombin maintains the integrity of a vessel during inflammation and activation of complement and blood coagulation systems.
It is known that thrombin releases NO from the endothelium. NO inhibits platelet aggregation and monocyte adhesion on the endothelium. The anti-inflammatory action of NO may be mediated by inhibition of the expression of the ICAM-1 gene and other adhesive molecules in endothelial cells. NO stabilizes the inactive heterotrimer complex NF-kappaB/IkappaBalpha in the cytoplasm by stimulation of the gene expression of the IkappaBalpha inhibitor [49].
Thus, interacting with PAR-1, thrombin regulates inflammation by two pathways. By one pathway it activates endothelial cells, adhesion and recruitment of monocytes, and increases the permeability of endothelium. By the other, it participates in protection of endothelium from degradation by protease complex of the complement system and also blocks the adhesion of monocytes on the endothelium and platelet aggregation through NO release. In low concentrations, thrombin is a growth factor accelerating proliferation of endothelial cells, fibroblasts, and smooth muscle cells [2, 4, 9, 11]. Thrombin acts as a conductor of cellular responses during inflammation [4].
Association of blood clotting and inflammation is observed in atherosclerosis and vasculitis. In this case PAR-1 gene expression increases in macrophages and smooth muscle cells of atheroma and in fibroblasts of synovial membrane in rheumatoid arthritis [5, 9, 50]. Activation of the complement system on the endothelium during inflammation leads to tissue factor expression and to thrombin generation [48]. The latter activates cells of the vascular wall and induces platelet aggregation.
TGF-beta1 and PDGF are released from aggregating platelets and stimulate the expression of PAR-1 mRNA in smooth muscle cells in a region of a vessel injury, thus providing thrombin mitogenic activity towards these cells [51].
In addition, thrombin stimulates expression of fibroblastic growth factor (CTGF, connective tissue growth factor) through PAR-1 activation [52]. The effect of thrombin on the increase of CTGF mRNA level is manifested early, reaches its maximum within 3 h, and does not depend on the de novo protein synthesis and TGF-beta secretion. The latter was considered until recently as the only CTGF inducer in fibroblasts. Fibroblasts of transgenic mice with PAR-1 deficiency did not respond to thrombin but reacted normally to TGF-beta [53]. These results correlate with the data about a considerable increase of fibroblasts proliferation in the model of wound healing in mice under the effect of immobilized PAR-1 agonists [11].
FACTOR Xa RECEPTORS
Factor Xa as well as thrombin can induce CTGF expression in fibroblasts. This requires the proteolytic activity of the enzyme [52]. The functions of factor Xa are realized through the cleavage of another representative of the PAR family--PAR-2. It is expressed in many cell types involved in inflammation and tissue repair including leukocytes, endothelial cells, keratinocytes, smooth muscle cells, epithelial cells of the gastrointestinal tract, fibroblasts of human lungs, and mast cells [8, 9, 34, 35, 54-59]. PAR-2 is activated by trypsin, tryptase of mast cells, trypsin-like proteinases as, for example, a recently cloned membrane type 1 serine proteinase (MT-SP-1), and also by synthetic peptides--PAR-2 agonists [8, 9, 24, 60, 61]. The primary structure of PAR-2 has 28% of homology with that of PAR-1; the site of specific proteolysis is located at residues Arg36-Ser37 of the N-terminus of the extracellular domain [8, 9, 24].
PAR-2 agonist peptides (murine SLIGRL, human SLIGKV) activate the receptor without its cleavage, and imitate the proteinase action towards endothelial cells, enterocytes, keratinocytes, myocytes, neutrophils, and several tumor cell lines [8, 24, 57]. PAR-2 mediates the progression of an acute inflammatory response characterized by the development of edema and leukocyte infiltration [62-64]. A significant increase of rolling of leukocytes was not observed in mice with PAR-2 deficiency after a surgical trauma [65]. PAR-2 activation in endothelial cells by factor VIIa in the complex with tissue factor and factor Xa leads to the exposure of adhesive proteins (including vWf) and to the decrease of TFPI activity. These events contribute to the reduction of the thromboresistant properties of the endothelium during inflammation [66, 67]. PAR-2 activation in endothelial cells by the peptide agonist SLIGKVD induced a rapid (within 1 h) expression of tissue factor, its exposure on the surface, and activation of blood coagulation [57].
Thus, the presence of PAR-2 on the cells involved in inflammation and their activation by proteinases of the blood clotting system indicates active involvement of these receptors in the association of inflammation and blood coagulation.
MODULATION OF RESPONSES OF CELLS ACTIVATED BY PROTEINASES THROUGH
PAR RECEPTORS
It is known that factor Xa binds and activates endothelial cells and leukocytes through an unsplittable receptor EPR-1 [15, 68]. This interaction triggers the system of signal transduction and generation of intracellular second messengers and modulates the expression of cytokine genes. However, proteolysis is absolutely required for cell activation, since an inhibitor of factor Xa catalytic activity blocks the increase of the intracellular Ca2+ concentration but does not influence the enzyme binding to EPR-1 of endothelial cells of human umbilical vein. Desensitization of PAR-2 receptors by trypsin (or by factor Xa) blocked the cellular response induced by factor Xa. Moreover, factor Xa cleaved a synthetic peptide, an analog of a region that is cleaved by trypsin-like proteinases in the PAR-2 structure. These data provided evidence for the suggestion of a new cascade mechanism of cell activation by factor Xa. This mechanism includes a complementary binding of the enzyme to the EPR-1 receptor followed by a local proteolysis of the PAR-2 receptor [68].
A similar mechanism of cascade activation of PAR-4 by thrombin was observed in murine platelets [69]. It was demonstrated that PAR-3 is not activated by thrombin but binds the enzyme and acts as its cofactor, which is necessary for the cleavage and activation of PAR-4 by thrombin. These data may explain the inefficiency of peptide analogs of PAR-3 agonist in receptor activation.
Biphasic changes of intracellular Ca2+ concentration in human platelets in response to thrombin were observed using agonists and antagonists of PAR-1 and PAR-4: a rapid impulsive response through PAR-1 followed by a slow, long, and powerful one through low-affinitive PAR-4 [70]. Apparently, a valuable cell response that is realized through PAR-4 activation requires the preliminary PAR-1 activation.
The existence of down-regulation of thrombin mitogenic activity by thrombomodulin through PAR-1 of endothelial cells was shown recently [71]. Thrombomodulin is an integral glycoprotein bound to the membrane. It is well known as the thrombin cofactor in protein C activation. Active protein C inhibits thrombin generation by cleavage and inactivation of factors Va and VIIIa. Using antibodies against thrombomodulin and mutated forms of thrombin that are deprived of catalytic activity (and, consequently, of ability to cleave PAR-1) but retaining the ability to bind thrombomodulin, it was demonstrated that thrombomodulin could inhibit the proliferation of endothelial cells. This was caused by the ability of thrombomodulin to prolong significantly the phosphorylation (and retention in the nucleus) of MAP-kinases (namely of ERK, protein kinases activated by extracellular signal). This phosphorylation is induced by thrombin though PAR-1 activation [4, 71]. Thus, thrombomodulin may act like an unsplittable thrombin receptor that modulates the mitogenic signal induced by PAR-1 activation.
INTERACTION OF THROMBIN WITH MAST CELLS
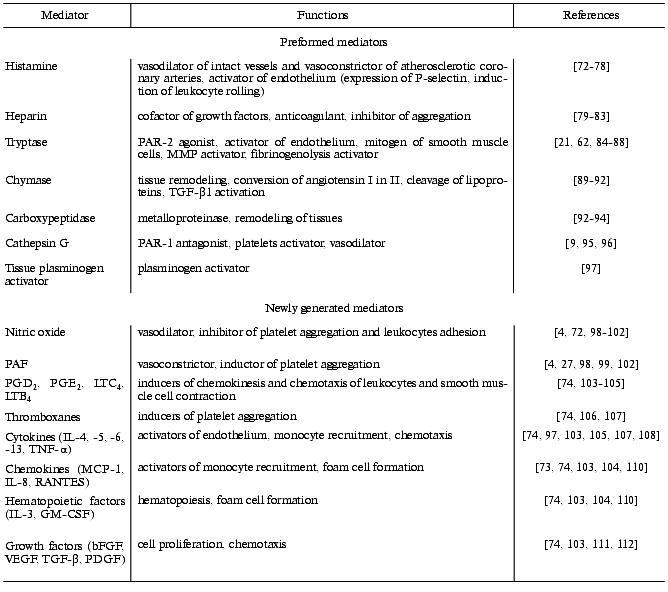
Mast cells play a very important role in cellular “ensembles” that are involved in inflammation, tissue repair, and blood coagulation due to their ability to secrete a broad spectrum of anti-inflammatory, vasoactive mediators, growth factors, and pro- and anticoagulants, which have various biological functions (Table 1) [72-112].
Table 1. Mediators released by mast cells
and several of their functions
It was demonstrated that degranulation of mast cells is one of the major effector links of the anti-inflammatory effect of thrombin in animal models [30, 89]. However, the mechanisms of the non-immune activation of mast cells in a whole organism remain obscure: PAR-1 agonist peptides increase vascular permeability and induce edema like thrombin when injected into animals, but the link of these effects with degranulation of mast cells is questionable [30, 89]. In this connection, investigations of the mechanisms of direct thrombin interaction with mast cells, notably studies of the origin of thrombin receptors and general regularities of the cellular effector reactions, are rapidly developing in recent years. In some publications activation of peritoneal mast cells by thrombin was not detected [34, 113], while the populations of bone marrow and skin mast cells of the same connective tissue type were able to secrete mediators of inflammation [34, 114]. At the same time, the presence of PAR-1 [34, 35] and PAR-2 [35] mRNAs in the peritoneal mast cells suggests the exposure of the receptors on the membrane and the secretory activity of the cells in the presence of serine proteinases--the physiological activators of PAR. Indeed, it was demonstrated in our laboratory that highly purified thrombin induces the release of histamine, beta-hexosaminidase, and heparin by peritoneal mast cells [115, 116, 118]. These data allow attributing of thrombin to the nonimmunologic activators of mast cells together with several peptides, lipids, and exogenous liberators (Table 2).
Table 2. Nonimmunologic activators of mast
cells

Peptide agonists of murine and human PAR-1 (SFFLRN and SFLLRN, respectively) activate dose-dependently mast cells like thrombin. Cathepsin G, a PAR-1 inhibitor, decreases the secretion of the mediators in response to thrombin by cleavage of peptide bonds Phe43-Leu and Phe55-Trp in the N-terminal domain (in addition to Arg41-Ser cleavage by thrombin). Analysis of these data suggests that the thrombin effect on peritoneal mast cells is mediated by PAR-1, although other receptor types might not be excluded.
Blood coagulation factor Xa and peptide agonist of murine PAR-2 [116] induce increased secretion of beta-hexosaminidase. This observation suggests the existence of factor Xa receptors on the membrane of peritoneal mast cells, in particular of PAR-2.
PARs are characterized by rapid desensitization after their activation by proteinases. In our experiments PAR desensitization by thrombin in mast cells led to the decrease of the secretory response to the PAR-1 agonist peptide [116]. This result provides additional evidence for PAR-1 involvement in the effector response to thrombin. At the same time, preliminary treatment of mast cells with thrombin was accompanied by an augment of beta-hexosaminidase secretion in response to the PAR-2 peptide agonist. The observed effect may be explained by an increase of PAR-2 quantity on the membrane of a mast cell due to an additional exposure of preformed intracellular pool. The existence of the latter was demonstrated recently [35].
The results obtained in our study that PAR-1 activation may stimulate PAR-2 exposure on the membrane suggest the existence of cooperative relationships between the receptors of mast cells including those from the PAR family. The proposed hypothesis is in agreement with published data. Thus, it was demonstrated that PAR-3 stimulates thrombin interaction with PAR-4 on platelets [52]. Gordon et al. [108] observed not only the activation in response to thrombin on murine peritoneal mast cells, but also detected a selective modulation of cell reactivity by the enzyme. It was revealed that thrombin induces dose-dependently (similar to immune liberator, allergen) the interleukin-6 synthesis in the cells and its subsequent secretion. The response of mast cells to under-threshold concentrations of allergen increased more than 100-fold in the presence of threshold thrombin concentrations. The observed permissive effect of low concentrations of immune liberator on the synthesis and secretion of cytokine in response to the non-immune activator of mast cells, thrombin, to our mind clearly indicates the cooperative relationships between receptors on the mast cell and the possibility of thrombin involvement in the generation of cytokines during inflammation.
However, the possibility of the presence of other types of thrombin receptor on mast cells together with PARs should not be excluded. Our early observation about the influence of different thrombin forms and concentrations on varying the membrane ionic permeability of mast cells, Na+/H+ exchange, and intracellular Ca2+ concentration [115, 119], and data about a significantly higher IL-6 concentration released by thrombin from murine mast cells in comparison to the secretion on the PAR-1 peptide agonist [108] provide evidence for this suggestion.
Nitric oxide is an endogenous regulator of the activity of mast cells; it activates guanylate cyclase and inhibits histamine and PAF secretion during activation by cytokines [99, 120]. The possibility of modulation of the reactivity of mast cells mediated by PAR-1 was demonstrated in the study of the receptor agonist influence on NO release [98]. The PAR-1 agonist peptide stimulated NO release in mast cells; the effect was blocked in the presence of L-NAME (an inhibitor of NO generation) or calmidasolium (an inhibitor of calmodulin and Ca-dependent constitutive isoform of NO-synthase (c-NOS)). It was demonstrated that NO donors inhibit degranulation of peritoneal mast cells [121] and histamine release by mast cells activated by lipopolysaccharides and prevent leukocyte activation and the increase in endothelium permeability induced by histamine [100-102].
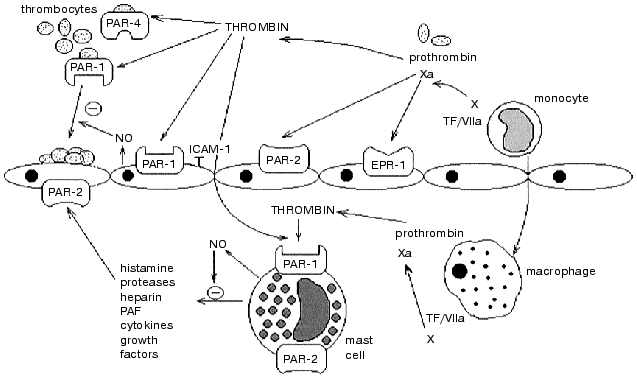
Thus, blood coagulation activation during inflammation leads to the generation of thrombin, factor Xa and other proteinases. Interacting with their receptors (of the PAR family and/or others), these proteinases are involved in the regulation of the early stages of inflammation through NO release and blockage of the expression of the adhesive molecules, as well as in the stimulation of inflammation and tissue repair through activation of mast and endothelial cells, fibroblasts, smooth muscle cells, and others (see figure). Receptors of proteinases of the blood clotting system serve as their sensors on the target cells and are involved in the association of blood coagulation and inflammation.
This work was supported by the Russian Foundation for Basic Research (project No. 01-04-48624) and special federal technical scientific program “Investigations in the priority branches of science and technology with civilian purposes” (program code “New methods in bioengineering”, project “Biocatalytic technologies” No. 04-202/0).Involvement of PAR-family receptors in the regulation of function of cells participating in blood coagulation and inflammation. PAR-1 is expressed on platelets and on endothelial and mast cells. Thrombin is generated from prothrombin in blood vessels and tissues (macrophages). Thrombin activates: 1) endothelial PAR-1 leading to the secretion and expression of the mediators of inflammation and anticoagulants (NO, ICAM-1, and others); 2) PAR-1 and PAR-4 of platelets stimulating adhesion and aggregation followed by the activation of blood clotting cascade; 3) PAR-1 of mast cells inducing the secretion of pro-inflammatory (histamine, PAF, cytokines, and others) and anti-inflammatory (NO, heparin) mediators. Factor Xa activates prothrombin conversion to thrombin, and also interacts with endothelium through EPR-1 and PAR-2 receptors, with mast cells through PAR-2 (TF is tissue factor)
REFERENCES
1.Fenton, J. W., II. (1995) Thromb.
Haemost.,74, 493-498.
2.Carney, D. H., Redin, W., and McCroskey, L. (1992)
Sem. Thromb. Hemost., 18, 91-103.
3.Strukova, S. M., Kireeva, E. G., and Dugina, T. N.
(1997) Vestnik MGU, Ser. 16. Biologiya, 8-14.
4.Strukova, S. M. (2001) Biochemistry
(Moscow), 66, 8-18.
5.Henrikson, K. P., Salazar, S. L., Fenton, J. W.,
and Pentecost, B. T. (1999) Br. J. Cancer, 79,
401-406.
6.Glusa, E., Paintz, M., and Bretschneider, E. (1996)
Sem. Thromb. Hemost., 22, 261-266.
7.Grand, R., Turnell, A., and Grabham, P. (1996)
Biochem. J., 313, 353-358.
8.Coughlin, S. (1999) Proc. Natl. Acad. Sci.
USA, 96, 11023-11027.
9.Dery, O., Corvery, C. U., Steinhorff, M., and
Bunnett, N. W. (1998) Am. J. Physiol., 274,
C1429-C1452.
10.Brass, L. (1997) Coronary Artery Disease,
8, 49-58.
11.Strukova, S. M., Dugina, T. N., Chistov, I. V.,
Markvicheva, E. A., Kuptsova, S. V., Kolokolchikova, E. G., Rumsh, L.
D., Zybov, V. P., and Gluza, E. (1998) Bioorg. Khim., 24,
288-292.
12.Chambers, R. C., Leoni, P., Blanc-Brude, O. P.,
Wembridge, D. E., and Laurent, G. J. (2000) J. Biol. Chem.,
275, 35584-35591.
13.Cunningham, M. A., Rondeau, E., Chen, X.,
Coughlin, S. R., Holdsworth, S. R., and Tipping, P. G. (2000) J.
Exp. Med., 191, 455-462.
14.Coughlin, S. R. (2000) Nature, 407,
258-264.
15.Cirino, G., Napoli, C., Bucci, M., and Cicala, C.
(2000) TIPS, 21, 170-172.
16.Preissner, K. T., Nawroth, P. P., and Kanse, S.
M. (2000) J. Pathol., 190, 360-372.
17.Cirino, G., Cicala, C., Bucci, M., Sorrentino,
A., Ambrosini, G., de Dominicis, G., and Altieri, D. (1997) J. Clin.
Invest., 99, 2446-2451.
18.Esmon, C., Xu, J., Gu, J.-M., Qu, D., Laszik, Z.,
Ferrell, G., Stearns-Kurosawa, D., Kurosawa, S., Taylor, F., and Esmon,
N. (1999) Thromb. Haemost., 82, 251-258.
19.Vu, T.-K., Hung, D., Wheaton, V., and Coughlin,
S. (1991) Cell, 64, 1057-1068.
20.Rasmussen, U., Vouret-Craviari, V., Jallot, S.,
Schlesinger, Y., Pages, G., Parirani, A., Lecocq, J.-P., Pouyssegur J.,
and van Obberghen-Schilling, E. (1991) FEBS Lett., 288,
123-128.
21.Molino, M., Barnathan, E., Numerof, M., Clark,
J., Dreyer, M., Cumashi, A., Hoxi, J., Schechter, N., Woolkalis, M.,
and Brass, L. (1997) J. Biol. Chem., 272, 4043-4049.
22.Xu, W. F., Andersen, H., Whitmore, T. E.,
Presnell, S. R., Yee, D. P., Ching, A., Gilbert, T., Davie, E. W., and
Foster, D. C. (1998) Proc. Natl. Acad. Sci. USA, 95,
6642-6646.
23.Ishihara, H., Connolly, A. J., Zeng, D., Kahn, M.
L., Zheng, Y. W., Timmons, C., Tram, T., and Coughlin, S. R.
(1998)Nature, 394, 690-694.
24.Nistedt, S., Emilsson, K., Wahlestedt, C., and
Sundelin, J. (1994) Proc. Natl. Acad. Sci. USA, 91,
9208-9212.
25.Cicala, C., and Cirino, G. (1998) Life
Sci., 62, 1817-1824.
26.Alpin, A. E., Howe, A., Alahari, S. K., and
Juliano, R. L. (1998) Pharmacol. Rev., 50, 197-263.
27.Strukova, S. M. (1995) in Inflammation
(Serov, V. V., and Paukov, V. S., eds.) [in Russian], Meditsina,
Moscow, pp. 52-81.
28.Esmon, C. T. (2000) Haematologica,
84, 254-259.
29.Esmon, C. T. (1999) Bailliere's Clin.
Haematol., 12, 343-359.
30.Cirino, G., Cicala, C., Bucci, M., Sorrentino,
L., Maraganore, J., and Stone, S. (1996) J. Exp. Med.,
183, 821-827.
31.Degen, J. L. (1999) Thromb. Haemost.,
82, 858-864.
32.Altieri, D. C. (1999) Thromb. Haemost.,
82, 781-786.
33.Coughlin, S. (1999) Thromb. Haemost.,
82, 353-356.
34.Nishikawa, H., Kawabata, A., Kuroda, R., Nishida,
M., and Kawai, K. (2000) Jap. J. Pharmacol., 82,
74-77.
35.D'Andrea, M., Derian, C., Leturcq, D., Baker, S.,
Brunmark, A., Ling, P., Darrow, A., Santulli, R. J., Brass, L. F., and
Andrade-Gordon, P. (1998) J. Histochem. Cytochem., 46,
157-164.
36.Libby, P., Egan, D., and Skarlatos, S. (1998)
Circulation, 96, 4095-4103.
37.Kol, A., and Libby, P. (1998) Trends
Cardiovasc. Med., 8, 191-199.
38.Schwartz, U. R., Kobsar, A. L., Koksch, M.,
Walter, U., and Eigenthaler, M. (2000) Biochem. Pharmacol.,
60, 1399-1407.
39.Bierhaus, A., Chen, J., Liliensiek, B., and
Nawroth, P. P. (2000) Sem. Thromb. Hemost., 26,
571-587.
40.Lindmark, E., Tenno, T., and Siegbahn, A. (2000)
Arterioscler. Thromb. Vasc. Biol., 20, 2322-2328.
41.Belaaouaj, A. A., Li, A., Wun, T. C., Welgus, H.
G., and Shapiro, S. D. (2000) J. Biol. Chem., 275,
27123-27128.
42.Plescia, J., and Altieri, D. (1996) Biochem.
J., 319, 873-879.
43.Lindahl, U., Peiler, G., Bozgwald, J., and
Seljelid, R. (1989) Arch. Biochem. Biophys., 273,
180-188.
44.Zoubine, M. N., Ma, J. Y., Smirnova, I. V.,
Citron, B. A., and Festoff, B. W. (1996) Develop. Biol.,
179, 447-457.
45.Mach, F., Schonbeck, U., Sukhova, G. K.,
Atkinson, E., and Libby, P. (1998) Nature, 394,
200-203.
46.Nelken, N. A., Soifer, S. J., O'Keefe, J., Vu, T.
K., Charo, I. F., and Coughlin, S. R. (1992) J. Clin. Invest.,
90, 1614-1621.
47.Kaplanski, G., Marin, V., Fabrigoule, M., Boulay,
V., Benoliel, A. M., Bongrand, P., Kaplanski, S., and Farnarier, C.
(1998) Blood, 92, 1259-1267.
48.Lindington, E. A., Haskard, D. O., and Mason, J.
C. (2000) Blood, 96, 2784-2792.
49.Libby, P., Sukhova, G., Lee, R., and Liao, J.
(1997) Int. J. Cardiol., 62, S23-S29.
50.Morris, R.,Winjard, P., Blake, D., and Morris, C.
(1994) Ann. Reum. Dis., 53, 72-79.
51.Schini-Kerth, V., Bassus, S., and Fissithaler, B.
(1997) Circulation, 96, 3888-3896.
52.Chambers, R. C., Leoni, P., Blanc-Brude, O. P.,
Wembridge, D. E., and Laurent, G. J. (2000) J. Biol. Chem.,
275, 35584-35591.
53.Pendurthi, U. R., Allen, K. E., Ezban, M., and
Rao, L. V. M. (2000) J. Biol. Chem., 275,
14632-14641.
54.Howells, G. L., Macey, M., Chinni, C., Hou, L.,
Fox, M. T., Harriott, P., and Stone, S. (1997) J. Cell Sci.,
110, 881-887.
55.Akers, I. A., Parsons, M., Hill, M. R.,
Hollenberg, M. D., Sanjar, S., Laurent, G. J., and McAnulty, R. J.
(2000) Am. J. Physiol. Lung Cell. Mol. Physiol., 278,
L193-L201.
56.Steinhoff, M., Corvera, C. U., Thoma, M. S.,
Kong, W., McAlpine, B. E., Caughey, G. H., Ansel, J. C., and Bunnett,
N. W. (1999) Exp. Dermatol., 8, 282-294.
57.Alm, A. K., Norstrom, E., Sundelin, J., and
Nystedt, S. (1999) Thromb. Haemost., 81, 984-988.
58.Hamilton, J. R., Chow, J. M., and Cocks, T. M.
(1999) Br. J. Pharmacol., 127, 617-622.
59.Damiano, B. P., D'Andrea, M. R., de Garavilla,
L., Cheung, W. M., and Andrade-Gordon, P. (1999) Thromb.
Haemost., 81, 808-814.
60.Alm, A. K., Gagnemo-Persson, R., Sorsa, T., and
Sundelin, J. (2000) Biochem. Biophys. Res. Commun., 275,
77-83.
61.Takeuchi, T., Harris, J. L., Huang, W., Yan, K.,
Coughlin, S. R., and Craikc, S. (2000) J. Biol. Chem.,
275, 26333-26342.
62.Steinhoff, M., Vergnolle, N., Young, S. H.,
Tognetto, M., Amadesi, S., Ennes, H. S., Trevisani, M., Hollenberg, M.
D., Wallace, J. L., Caughey, G. H., Mitchell, S. E., Williams, L. M.,
Geppetti, P., Mayer, E. A., and Bunnett, N. W. (2000) Nat. Med.,
6, 51-158.
63.Vergnolle, N. (1999) J. Immunol.,
163, 5064-5069.
64.Vergnolle, N., Hollenberg, M. D., Sharkey, K. A.,
and Wallace, J. L. (1999) Br. J. Pharmacol., 127,
1083-1090.
65.Lindner, J. R., Kahn, M. L., Coughlin, S. R.,
Sambrono, G. R., Schauble, E., Bernstein, D., Foy, D., Afezi-Mogham,
A., and Ley, K. (2000) J. Immunol., 165, 6504-6510.
66.Camerer, E., Huang, W., and Coughlin, S. R.
(2000) Proc. Natl. Acad. Sci. USA, 97, 5255-5260.
67.Lager, F., Morys-Wortmann, C., Kusters, B., and
Storck, J. (1999) Br. J. Haematol., 105, 542-550.
68.Bono, F., Schaeffer, P., Herault, J. P., Michaux,
C., Nestor, A. L., Guillemot, J. C., and Herbert, J. M. (2000)
Arterioscler. Thromb. Vasc. Biol., 20, E107-E112.
69.Nakanishi-Matsui, M., Zheng, Y. W., Sulciner, D.
J., Weiss, E. J., Ludeman, M. J., and Coughlin, S. R. (2000)
Nature, 404, 609-613.
70.Covic, L., Gresser, A. L., and Kuliopulos, A.
(2000) Biochemistry, 39, 5458-5467.
71.Olivot, J.-M., Estebanell, E., Lafay, M.,
Brohard, B., Aiach, M., and Rendu, F. (2001) Circ. Res.,
88, 681-687.
72.Kubes, P., and Granger, D. N. (1996)
Cardiovasc. Res., 32, 699-708.
73.Kelley, J. L., Chi, D. S., Abou-Auda, W., Smith,
K., and Krishnaswamy, G. (2000) Mol. Med. Today, 6,
304-308.
74.Metcalfe, D. D., Baram, D., and Mekori, Y. (1997)
Physiol. Rev., 77, 1033-1079.
75.Schwartz, L. B. (1997) Textbook of
Rheumatology (Kelley, W. N., et al., eds.) W. B. Saunders Comp.,
Philadelphia, pp. 161-175.
76.Holgate, S. T. (2000) Clin. Exp. Allergy
(Suppl.), 1, 28-32.
77.White, M. (1999) J. Allergy Clin.
Immunol., 103, 378-381.
78.Laine, P., Kaartinen, M., Penttila, A., Panula,
P., Paavonen, T., and Kovanen, P. (1999) Circulation, 99,
361-369.
79.Bankl, H. C., Grosschmidt, K., Pikula, B., Bankl,
H., Lechner, K., and Valent, P. (1999) Hum. Pathol., 30,
188-194.
80.Lassila, R., Lindstedt, K., and Kovanen, P.
(1997) Arterioscler. Thromb. Vasc. Biol., 17,
3578-3587.
81.Day, R., and Forbes, A. (1999) Lancet,
354, 62-65.
82.Lindahl, U. (1999) Haemostasis, 29,
38-47.
83.Fareed, J., Hoppensteadt, D. A., and Bick, R. L.
(2000) Sem. Thromb. Hemost., 26, 5-21.
84.Schechter, N. M., Brass, L. F., Lavker, R. M.,
and Jensen, P. J. (1998) J. Cell Physiol., 176,
365-373.
85.Corvera, C. U., Dery, O., McConalogue, K., Bohm,
S. K., Khitin, L. M., Caughey, G. H., Payan, D. G., and Bunnett, N. W.
(1997) J. Clin. Invest., 100, 1383-1393.
86.Jonson, J. L. (1998) Arterioscler. Thromb.
Vasc. Biol., 18, 1707-1715.
87.Mirza, H., Schmidt, V. A., Derian, C. K., Jesty,
J., and Bahou, W. F. (1997) Blood, 90, 3914-3922.
88.Thomas, V. A., Wheeless, C. J., Stack, M. S., and
Johnson, D. A. (1998) Biochemistry, 37, 2291-2298.
89.Kovanen, P. (1997) Heart Vessels (Suppl.),
12, 125-127.
90.Muramatsu, M., Katada, J., Hayashi, I., and
Majima, M. (2000) J. Biol. Chem., 275, 5545-5552.
91.Lindstedt, K., Wang, Y., Shiota, N., Hyytiainen,
M., Kokkonen, J., Keski-Oja, J., and Kovanen, P. (2001) FASEB
J., 15, 1377-1388.
92.Huang, C., Sali, A., and Stevens, R. (1998) J.
Clin. Immunol., 18, 169-183.
93.Goldstein, S. M., Kaempfer, B. S., and Wintroub,
B. U. (1989) Progr. Clin. Biol. Res., 297, 145-154.
94.Abe, M., Kurosawa, M., Ishikawa, O., and Miyachi,
Y. (2000) J. Allergy Clin. Immunol., 106, S78-S84.
95.Kinlough-Rathbone, R. L., Perry, D. W., Rand, M.
L., and Packham, M. A. (1999) Thromb. Res., 95,
315-323.
96.Glusa, E., and Adam, C. (2001) Br. J.
Pharmacol., 133, 422-428.
97.Valent, P., Sillaber, C., Baghestanian, M.,
Bankl, H. C., Kiener, H. P., Lechner, K., and Binder, B. R. (1998)
Int. Arch. Allerg. Immunol., 115, 2-8.
98.Strukova, S. M., Chistov, I. V., Umarova, B. A.,
Dugina, T. N., Storozhevykh, T. P., Pinelis, V. G., and Glusa, E.
(1999) Biochemistry (Moscow), 64,658-664.
99.Hogaboam, C., Befus, A., and Wallace, J. (1993)
J. Immunol., 151, 3767-3774.
100.Salvemini, D., Masini, E., Pistlli, A.,
Mannaioni, P. F., and Vane, J. (1991) Cardiovasc. Pharmacol.,
17, S258.
101.Gaboury, J. P., Niu, X., and Kubes, P. (1996)
Circulation, 93, 318-324.
102.Al-Naemi, H., and Baldwin, A. L. (1999) Am.
J. Physiol., 277, H2010-H2016.
103.Costa, J. J., Weller, P. F., and Galli, S. J.
(1997) J. Am. Med. Assoc., 278,1815-1822.
104.Galli, S. J. (2000) Curr. Opin.
Hematol., 7,32-39.
105.O'Sullivan, S. (1999) Acta Physiol.
Scand., 644, 1-74.
106.Morrow, J. D., Oates, J. A., Roberts, L. J.,
II, Zackert, W. E., Mitchell, T. A., Lazarus, G., and Guzzo, C. (1999)
J. Invest. Dermatol., 113, 93-97.
107.Wedemeyer, J., Tsai, M., and Galli, S. J.
(2000) Curr. Opin. Hematol., 12, 624-631.
108.Gordon, J. R., Zhang, X., Stevenson, K., and
Cosford, K. (2000) Cell Immunol., 205,128-135.
109.Olsson, N., Piek, E., ten Dijke, P., and
Nilsson, G. (2000) J. Leukoc. Biol., 67, 350-356.
110.Kaplan, A. P. (2001) Int. Arch. Allergy
Immunol., 124, 423-431.
111.Youn, B. S., Mantel, C., and Broxmeyer, H. E.
(2000) Immunol. Rev., 177, 150-174.
112.Artuc, M., Hermes, B., Steckelings, U. M.,
Grutzkau, A., and Henz, B. M. (1999) Exp. Dermatol., 8,
1-16.
113.Razin, E., and Marx, G. (1984) J.
Immunol., 133, 3282-3285.
114.Razin, E., Baranes, D., and Marx, G. (1985)
Exp. Cell Res., 160, 380-386.
115.Strukova, S. M., Dugina, T. N., Khlgatian, S.
V., Redkozubov, A. E., Redkozubova, G. P., and Pinelis, V. G. (1996)
Sem. Thromb. Hemost., 22, 145-150.
116.Kisseleva, E. V., Dugina, T. N., Gluza, E., and
Strukova, S. M. (2001) Fiziol. Zh. im. Sechenova, in press.
117.Befus, A. D., Mowat, C., Gilchrist, M., Hu, J.,
Solomon, S., and Bateman, A. (1999) J. Immunol.,
163,947-953.
118.Umarova, B. A., Dugina, T. N., Shestakova, E.
V., Gluza, E., and Strukova, S. M. (2000) Byul. Eksp. Biol.
Med., 129, 370-373.
119.Strukova, S. M., Dugina, T. N., and Khlgatian,
S. V. (1997) Thromb. Haemost. (Suppl.), Abst. XVI Congr. of
ISTH, Florence, Italy, pp. 345-346.
120.Iikira, M., Takaishi, T., Hirai, K., Yamada,
H., Iida, M., Koshino, T., and Morita, Y. (1998) Int. Arch. Allergy
Immunol., 115, 129-136.
121.Koranteng, R. D., Dearman, R. J., Kimber, I.,
and Coleman, J. W. (2000) Inflamm. Res., 49,240-246.